
Implication of N-Methyl-d-Aspartate Receptor in Homocysteine-Induced Age-Related Macular Degeneration


 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
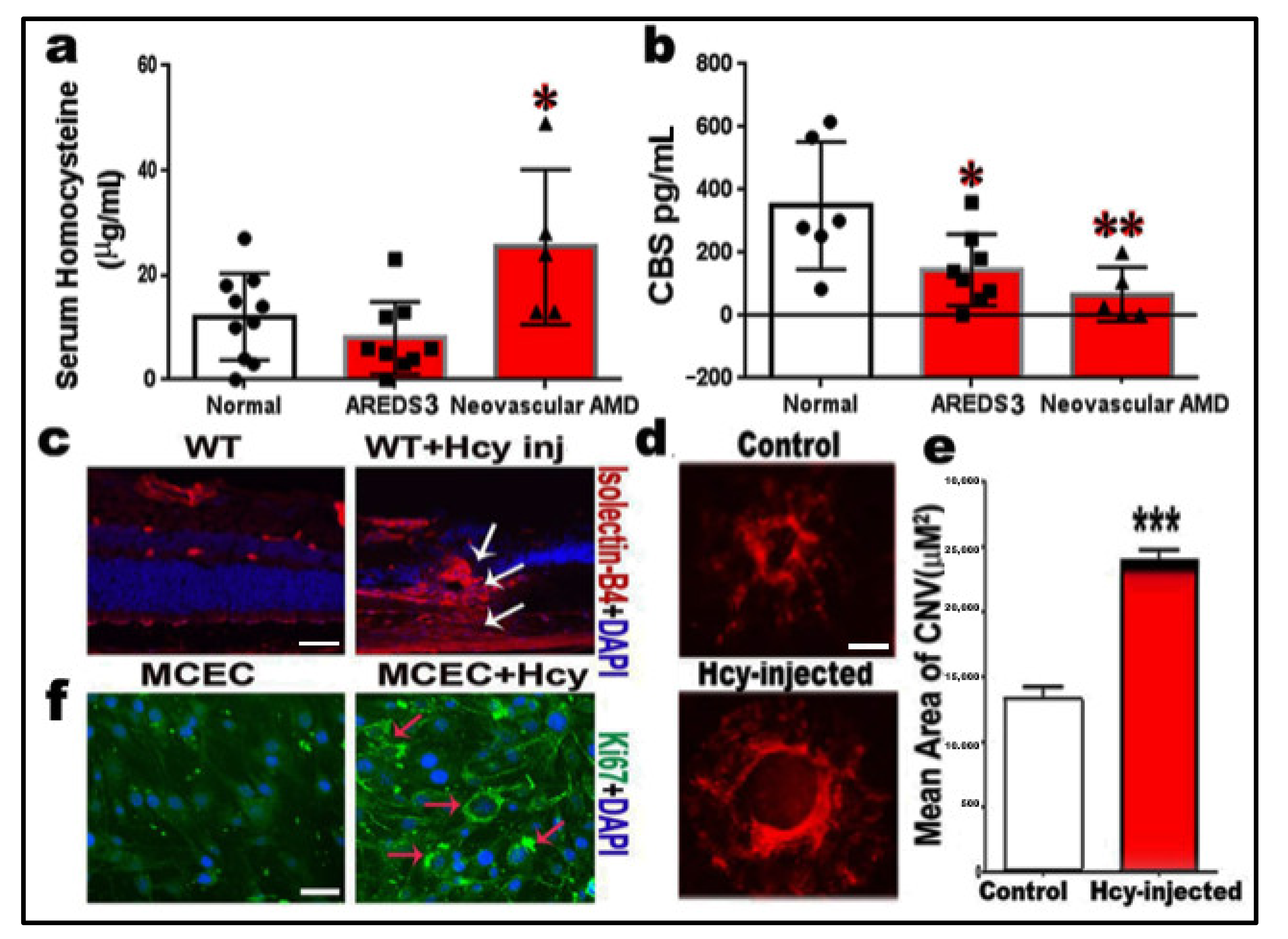
2.1. Measurements of Homocysteine and CBS Enzyme Levels
2.2. Homocysteine Promotes Angiogenesis and Induction of Choroidal Neovascularization (CNV)
2.3. Homocysteine Activates HIF-1α and VEGF in RPE Cells
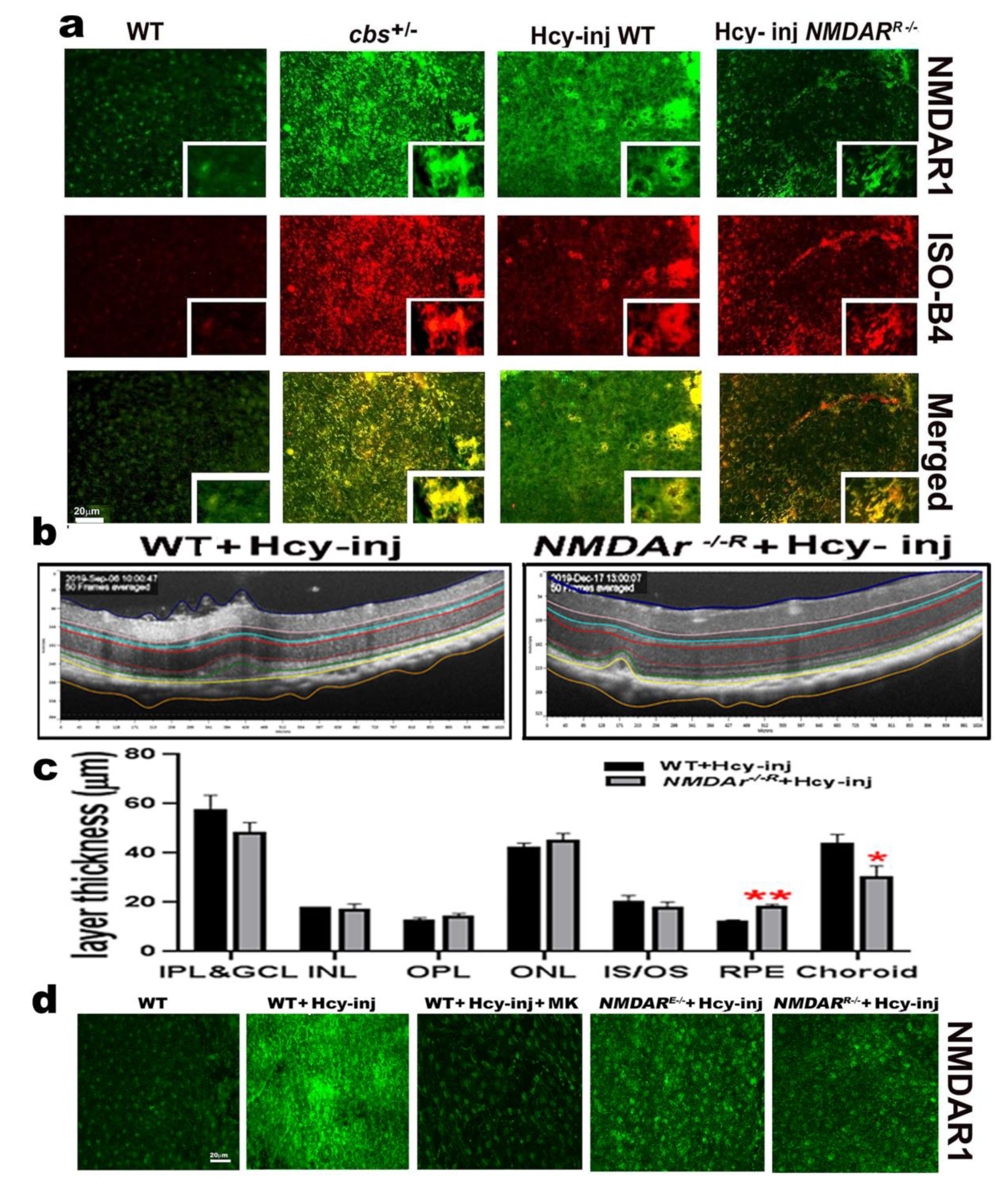
2.4. Homocysteine Activates NMDA Receptors in RPE Cells
2.5. Mouse with Deletion/Inhibition of NMDAR in Retinal Pigmented Epithelia (NMDARR −/−)
2.6. Effect of NMDAR Deletion in RPE Cells on Hcy-Induced BRB Dysfunction
2.7. Effect of NMDAR Deletion in RPE Cells on Hcy-Induced CNV and Retinal Thickness
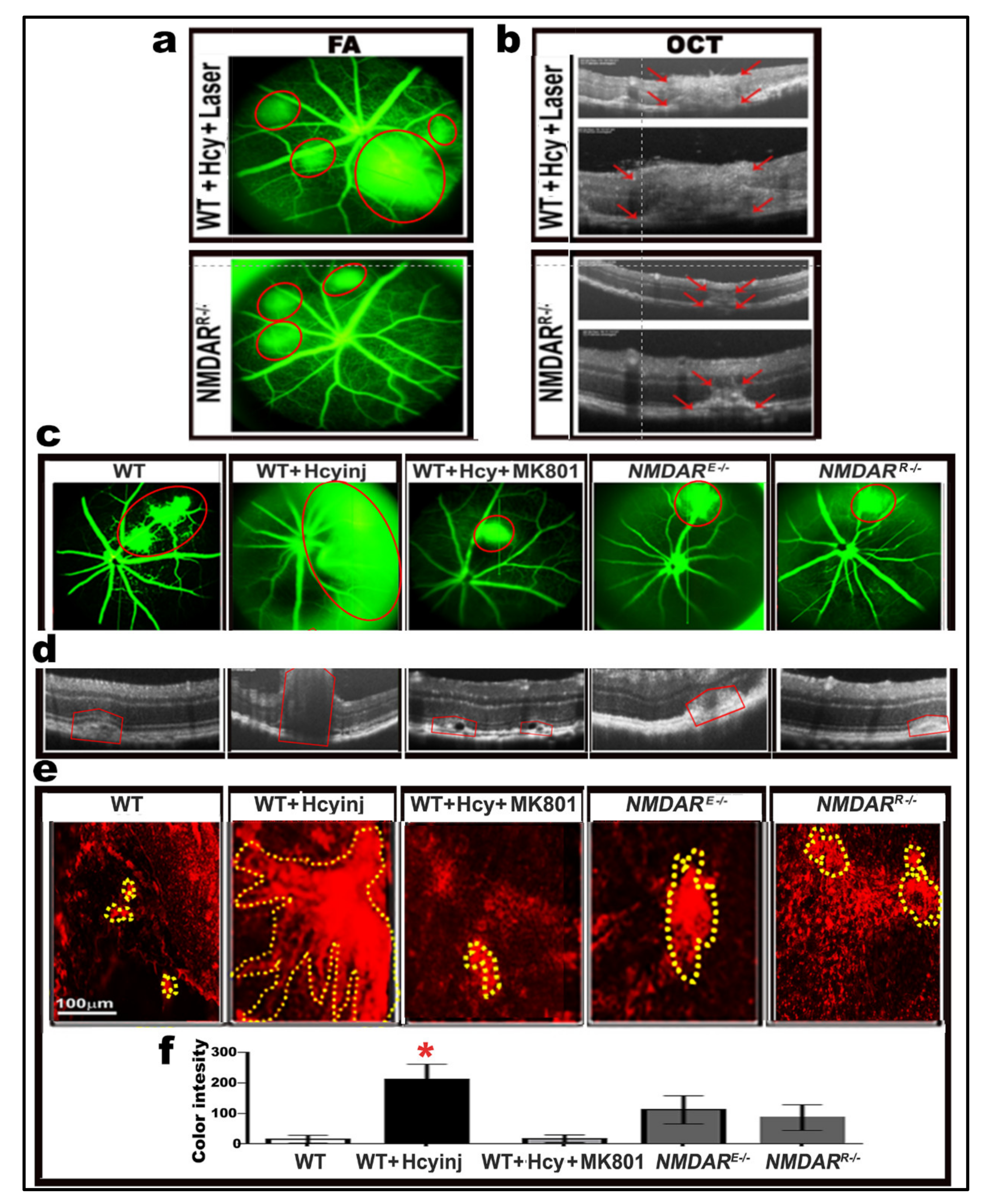
2.8. Effect of Pharmacological and Genetic Intervention of NMDAR on Retinal Morphology and Vasculature after Laser Induction of Choroidal Neovascularization (CNV)
3. Discussion
4. Materials and Methods
4.1. Animals
4.1.1. Mouse with Deletion or Inhibition of NMDAR
4.1.2. Mouse with HHcy
4.2. Measurement of Homocysteine and Cystathionine Beta-Synthase (CBS) Enzyme Levels
4.3. Cell Culture
4.4. Isolation and Culture of Primary Retinal Pigment Epithelium (RPE)
4.5. Isolation and Culture of Mouse Choroidal Endothelial Cells (MCEC)
4.6. Fluorescein Isothiocyanate (FITC)-Dextran Permeability Assay
4.7. Quantitative Reverse-Transcriptase Polymerase Chain Reaction (RT-q PCR) for NMDAR1
4.8. Western Blot Analysis
4.9. Enzyme-Linked Immunosorbent Assay (ELISA)
4.10. Immuno-Fluorescent Assessment
4.11. Optical Coherence Tomography (OCT) and Fluorescein Angiography (FA)
4.12. Laser Induction Using Phoenix MICRON Image-Guided Laser System
4.13. Measuring Retinal Thickness
4.14. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schmidt, K.G.; Bergert, H.; Funk, R.H. Neurodegenerative diseases of the retina and potential for protection and recovery. Curr. Neuropharmacol. 2008, 6, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Rein, D.B.; Wittenborn, J.S.; Zhang, X.; Honeycutt, A.A.; Lesesne, S.B.; Saaddine, J.; Vision Health Cost-Effectiveness Study Group. Forecasting age-related macular degeneration through the year 2050: The potential impact of new treatments. Arch. Ophthalmol. 2009, 127, 533–540. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, M.M.; Owen, L.A.; Morrison, M.A.; Morgan, D.J.; Li, M.; Shakoor, A.; Vitale, A.; Iyengar, S.; Stambolian, D.; Kim, I.K.; et al. Genetics of age-related macular degeneration (AMD). Hum. Mol. Genet. 2017, 26, R45–R50. [Google Scholar] [CrossRef]
- Obeid, R.; Ninios, K.; Loew, U.; Gatzioufas, Z.; Hoffmann, S.; Seitz, B.; Geisel, J.; Herrmann, W. Aqueous humor glycation marker and plasma homocysteine in macular degeneration. Clin. Chem. Lab. Med. 2013, 51, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Kamburoglu, G.; Gumus, K.; Kadayifcilar, S.; Eldem, B. Plasma homocysteine, vitamin B12 and folate levels in age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2006, 244, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Gensler, G.; Klein, M.L.; Milton, R.C. Evaluation of plasma homocysteine and risk of age-related macular degeneration. Am. J. Ophthalmol. 2006, 141, 201–203. [Google Scholar] [CrossRef]
- Nowak, M.; Swietochowska, E.; Wielkoszynski, T.; Marek, B.; Kos-Kudla, B.; Szapska, B.; Kajdaniuk, D.; Glogowska-Szelag, J.; Sieminska, L.; Ostrowska, Z.; et al. Homocysteine, vitamin B12, and folic acid in age-related macular degeneration. Eur. J. Ophthalmol. 2005, 15, 764–767. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Mander, S.; Hussein, K.A.; Elsherbiny, N.M.; Smith, S.B.; Al-Shabrawey, M.; Tawfik, A. Hyperhomocysteinemia disrupts retinal pigment epithelial structure and function with features of age-related macular degeneration. Oncotarget 2016, 7, 8532–8545. [Google Scholar] [CrossRef]
- Tawfik, A.; Markand, S.; Al-Shabrawey, M.; Mayo, J.N.; Reynolds, J.; Bearden, S.E.; Ganapathy, V.; Smith, S.B. Alterations of retinal vasculature in cystathionine-beta-synthase heterozygous mice: A model of mild to moderate hyperhomocysteinemia. Am. J. Pathol. 2014, 184, 2573–2585. [Google Scholar] [CrossRef]
- Mohamed, R.; Sharma, I.; Ibrahim, A.S.; Saleh, H.; Elsherbiny, N.M.; Fulzele, S.; Elmasry, K.; Smith, S.B.; Al-Shabrawey, M.; Tawfik, A. Hyperhomocysteinemia Alters Retinal Endothelial Cells Barrier Function and Angiogenic Potential via Activation of Oxidative Stress. Sci. Rep. 2017, 7, 11952. [Google Scholar] [CrossRef]
- Diederen, R.M.; La Heij, E.C.; Deutz, N.E.; Kijlstra, A.; Kessels, A.G.; van Eijk, H.M.; Liem, A.T.; Dieudonne, S.; Hendrikse, F. Increased glutamate levels in the vitreous of patients with retinal detachment. Exp. Res. 2006, 83, 45–50. [Google Scholar] [CrossRef]
- Ola, M.S.; Hosoya, K.; LaNoue, K.F. Regulation of glutamate metabolism by hydrocortisone and branched chain keto acids in cultured rat retinal Muller cells (TR-MUL). Neurochem. Int. 2011, 59, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Zhang, H.; Wang, Y.H.; Liu, L.J.; Teng, Y.; Liu, P. Time-dependent reduction of glutamine synthetase in retina of diabetic rats. Exp. Eye Res. 2009, 89, 967–971. [Google Scholar]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D’Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hunter, C.; Weiss, H.R.; Chi, O.Z. Effects of blockade of ionotropic glutamate receptors on blood-brain barrier disruption in focal cerebral ischemia. Neurol. Sci. 2010, 31, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Andras, I.E.; Deli, M.A.; Veszelka, S.; Hayashi, K.; Hennig, B.; Toborek, M. The NMDA and AMPA/KA receptors are involved in glutamate-induced alterations of occludin expression and phosphorylation in brain endothelial cells. J. Cereb. Blood Flow Metab. 2007, 27, 1431–1443. [Google Scholar] [CrossRef]
- Tawfik, A.; Mohamed, R.; Kira, D.; Alhusban, S.; Al-Shabrawey, M. N-Methyl-D-aspartate receptor activation, novel mechanism of homocysteine-induced blood-retinal barrier dysfunction. J. Mol. Med. 2021, 99, 119–130. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Archiv Eur. J. Physiol. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Hardingham, G.E. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem. Soc. Trans. 2009, 37, 1147–1160. [Google Scholar] [CrossRef]
- Uchida, N.; Kiuchi, Y.; Miyamoto, K.; Uchida, J.; Tobe, T.; Tomita, M.; Shioda, S.; Nakai, Y.; Koide, R.; Oguchi, K. Glutamate-stimulated proliferation of rat retinal pigment epithelial cells. Eur. J. Pharmacol. 1998, 343, 265–273. [Google Scholar] [CrossRef]
- Forooghian, F.; Razavi, R.; Timms, L. Hypoxia-inducible factor expression in human RPE cells. Br. J. Ophthalmol. 2007, 91, 1406–1410. [Google Scholar] [CrossRef]
- Elmasry, K.; Mohamed, R.; Sharma, I.; Elsherbiny, N.M.; Liu, Y.; Al-Shabrawey, M.; Tawfik, A. Epigenetic modifications in hyperhomocysteinemia: Potential role in diabetic retinopathy and age-related macular degeneration. Oncotarget 2018, 9, 12562–12590. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, A.; Mohamed, R.; Elsherbiny, N.M.; DeAngelis, M.M.; Bartoli, M.; Al-Shabrawey, M. Homocysteine: A Potential Biomarker for Diabetic Retinopathy. J. Clin. Med. 2019, 8, 121. [Google Scholar] [CrossRef]
- Bird, A.C. Therapeutic targets in age-related macular disease. J. Clin. Investig. 2010, 120, 3033–3041. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.C.; Bressler, N.M.; Bressler, S.B.; Chisholm, I.H.; Coscas, G.; Davis, M.D.; de Jong, P.T.; Klaver, C.C.; Klein, B.E.; Klein, R.; et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. The International ARM Epidemiological Study Group. Surv. Ophthalmol. 1995, 39, 367–374. [Google Scholar] [CrossRef]
- Chew, E.Y.; Clemons, T.E.; Agron, E.; Sperduto, R.D.; Sangiovanni, J.P.; Kurinij, N.; Davis, M.D.; Age-Related Eye Disease Study Research Group. Long-term effects of vitamins C and E, beta-carotene, and zinc on age-related macular degeneration: AREDS report no. Ophthalmology 2013, 120, 1604–1611.e4. [Google Scholar] [CrossRef] [PubMed]
- Ferris, F.L., 3rd; Fine, S.L.; Hyman, L. Age-related macular degeneration and blindness due to neovascular maculopathy. Arch. Ophthalmol. 1984, 102, 1640–1642. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Patel, M.; Chan, C.C. Molecular pathology of age-related macular degeneration. Prog. Retin. Eye Res. 2009, 28, 1–18. [Google Scholar] [CrossRef]
- Ates, O.; Azizi, S.; Alp, H.H.; Kiziltunc, A.; Beydemir, S.; Cinici, E.; Kocer, I.; Baykal, O. Decreased serum paraoxonase 1 activity and increased serum homocysteine and malondialdehyde levels in age-related macular degeneration. Tohoku J. Exp. Med. 2009, 217, 17–22. [Google Scholar] [CrossRef]
- Huang, P.; Wang, F.; Sah, B.K.; Jiang, J.; Ni, Z.; Wang, J.; Sun, X. Homocysteine and the risk of age-related macular degeneration: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 10585. [Google Scholar] [CrossRef] [PubMed]
- Javadzadeh, A.; Ghorbanihaghjo, A.; Bahreini, E.; Rashtchizadeh, N.; Argani, H.; Alizadeh, S. Plasma oxidized LDL and thiol-containing molecules in patients with exudative age-related macular degeneration. Mol. Vis. 2010, 16, 2578–2584. [Google Scholar] [PubMed]
- Ghosh, S.; Saha, M.; Das, D. A study on plasma homocysteine level in age-related macular degeneration. Nepal J. Ophthalmol. 2013, 5, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, A.; Samra, Y.A.; Elsherbiny, N.M.; Al-Shabrawey, M. Implication of Hyperhomocysteinemia in Blood Retinal Barrier (BRB) Dysfunction. Biomolecules 2020, 10, 1119. [Google Scholar] [CrossRef]
- Beard, R.S., Jr.; Reynolds, J.J.; Bearden, S.E. Hyperhomocysteinemia increases permeability of the blood-brain barrier by NMDA receptor-dependent regulation of adherens and tight junctions. Blood 2011, 118, 2007–2014. [Google Scholar] [CrossRef]
- Sharp, C.D.; Hines, I.; Houghton, J.; Warren, A.; Jackson, T.H.T.; Jawahar, A.; Nanda, A.; Elrod, J.W.; Long, A.; Chi, A.; et al. Glutamate causes a loss in human cerebral endothelial barrier integrity through activation of NMDA receptor. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2592–H2598. [Google Scholar] [CrossRef]
- Kalani, A.; Kamat, P.K.; Tyagi, S.C.; Tyagi, N. Synergy of homocysteine, microRNA, and epigenetics: A novel therapeutic approach for stroke. Mol. Neurobiol. 2013, 48, 157–168. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef]
- Andre, H.; Tunik, S.; Aronsson, M.; Kvanta, A. Hypoxia-Inducible Factor-1alpha Is Associated with Sprouting Angiogenesis in the Murine Laser-Induced Choroidal Neovascularization Model. Invest. Ophthalmol. Vis. Sci. 2015, 56, 6591–6604. [Google Scholar] [CrossRef]
- Mowat, F.M.; Luhmann, U.F.; Smith, A.J.; Lange, C.; Duran, Y.; Harten, S.; Shukla, D.; Maxwell, P.H.; Ali, R.R.; Bainbridge, J.W. HIF-1alpha and HIF-2alpha are differentially activated in distinct cell populations in retinal ischaemia. PLoS ONE 2010, 5, e11103. [Google Scholar] [CrossRef]
- Barchitta, M.; Maugeri, A. Association between Vascular Endothelial Growth Factor Polymorphisms and Age-Related Macular Degeneration: An Updated Meta-Analysis. Dis. Markers 2016, 2016, 8486406. [Google Scholar] [CrossRef]
- Kwak, N.; Okamoto, N.; Wood, J.M.; Campochiaro, P.A. VEGF is major stimulator in model of choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3158–3164. [Google Scholar]
- Aiello, L.P.; Pierce, E.A.; Foley, E.D.; Takagi, H.; Chen, H.; Riddle, L.; Ferrara, N.; King, G.L.; Smith, L.E. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 10457–10461. [Google Scholar] [CrossRef]
- Ozaki, H.; Hayashi, H.; Vinores, S.A.; Moromizato, Y.; Campochiaro, P.A.; Oshima, K. Intravitreal sustained release of VEGF causes retinal neovascularization in rabbits and breakdown of the blood-retinal barrier in rabbits and primates. Exp. Eye Res. 1997, 64, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, P.S.; Perry, R.L.; Tawfik, A.; Smith, R.M.; Perry, E.; Roon, P.; Bozard, B.R.; Ha, Y.; Smith, S.B. Homocysteine-mediated modulation of mitochondrial dynamics in retinal ganglion cells. Invest. Ophthalmol. Vis. Sci. 2011, 52, 5551–5558. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Homocysteine, vitamins, and vascular disease prevention. Am. J. Clin. Nutr. 2007, 86, 1563S–1568S. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Wang, W.; Xiao, Z.; Hong, Y. Multi-Vitamin B Supplementation Reverses Hypoxia-Induced Tau Hyperphosphorylation and Improves Memory Function in Adult Mice. J. Alzheimers Dis. 2016, 54, 297–306. [Google Scholar] [CrossRef]
- Garcia, S.; Lopez, E.; Lopez-Colome, A.M. Glutamate accelerates RPE cell proliferation through ERK1/2 activation via distinct receptor-specific mechanisms. J. Cell Biochem. 2008, 104, 377–390. [Google Scholar] [CrossRef]
- Reigada, D.; Lu, W.; Mitchell, C.H. Glutamate acts at NMDA receptors on fresh bovine and on cultured human retinal pigment epithelial cells to trigger release of ATP. J. Physiol. 2006, 575, 707–720. [Google Scholar] [CrossRef]
- Sawicki, A. Calcium absorption in the digestive tract and calcium and phosphorus metabolism in postoperative hypoparathyroidism. Pol. Arch. Med. Wewn. 1986, 75, 417–423. [Google Scholar]
- Henney, C.S. The specificity of the early immune response to dinitrophenylated human gamma-G-globulin. Immunochemistry 1970, 7, 275–287. [Google Scholar] [CrossRef]
- Tawfik, A.; Smith, S.B. Increased ER stress as a mechanism of retinal neurovasculopathy in mice with severe hyperhomocysteinemia. Austin J. Clin. Ophthalmol. 2014, 1, 1023. [Google Scholar] [PubMed]
- Elsherbiny, N.M.; Sharma, I.; Kira, D.; Alhusban, S.; Samra, Y.A.; Jadeja, R.; Martin, P.; Al-Shabrawey, M.; Tawfik, A. Homocysteine Induces Inflammation in Retina and Brain. Biomolecules 2020, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Nita, M.; Grzybowski, A.; Ascaso, F.J.; Huerva, V. Age-related macular degeneration in the aspect of chronic low-grade inflammation (pathophysiological parainflammation). Mediat. Inflamm. 2014, 2014, 930671. [Google Scholar] [CrossRef] [PubMed]
- Owen, L.A.; Shakoor, A.; Morgan, D.J.; Hejazi, A.A.; McEntire, M.W.; Brown, J.J.; Farrer, L.A.; Kim, I.; Vitale, A.; DeAngelis, M.M. The Utah Protocol for Postmortem Eye Phenotyping and Molecular Biochemical Analysis. Invest. Ophthalmol Vis. Sci. 2019, 60, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Age-Related Eye Disease Study Research Group. The Age-Related Eye Disease Study system for classifying age-related macular degeneration from stereoscopic color fundus photographs: The Age-Related Eye Disease Study Report Number. Am. J. Ophthalmol. 2001, 132, 668–681. [Google Scholar]
- Martin, P.M.; Ananth, S.; Cresci, G.; Roon, P.; Smith, S.; Ganapathy, V. Expression and localization of GPR109A (PUMA-G/HM74A) mRNA and protein in mammalian retinal pigment epithelium. Mol. Vis. 2009, 15, 362–372. [Google Scholar]
- Fei, P.; Zaitoun, I.; Farnoodian, M.; Fisk, D.L.; Wang, S.; Sorenson, C.M.; Sheibani, N. Expression of thrombospondin-1 modulates the angioinflammatory phenotype of choroidal endothelial cells. PLoS ONE 2014, 9, e116423. [Google Scholar]
- Murakami, T.; Felinski, E.A.; Antonetti, D.A. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J. Biol. Chem. 2009, 284, 21036–21046. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samra, Y.A.; Kira, D.; Rajpurohit, P.; Mohamed, R.; Owen, L.A.; Shakoor, A.; Kim, I.K.; DeAngelis, M.M.; Sheibani, N.; Al-Shabrawey, M.; et al. Implication of N-Methyl-d-Aspartate Receptor in Homocysteine-Induced Age-Related Macular Degeneration. Int. J. Mol. Sci. 2021, 22, 9356. https://doi.org/10.3390/ijms22179356
Samra YA, Kira D, Rajpurohit P, Mohamed R, Owen LA, Shakoor A, Kim IK, DeAngelis MM, Sheibani N, Al-Shabrawey M, et al. Implication of N-Methyl-d-Aspartate Receptor in Homocysteine-Induced Age-Related Macular Degeneration. International Journal of Molecular Sciences. 2021; 22(17):9356. https://doi.org/10.3390/ijms22179356
Chicago/Turabian StyleSamra, Yara A., Dina Kira, Pragya Rajpurohit, Riyaz Mohamed, Leah A. Owen, Akbar Shakoor, Ivana K. Kim, Margaret M. DeAngelis, Nader Sheibani, Mohamed Al-Shabrawey, and et al. 2021. "Implication of N-Methyl-d-Aspartate Receptor in Homocysteine-Induced Age-Related Macular Degeneration" International Journal of Molecular Sciences 22, no. 17: 9356. https://doi.org/10.3390/ijms22179356
APA StyleSamra, Y. A., Kira, D., Rajpurohit, P., Mohamed, R., Owen, L. A., Shakoor, A., Kim, I. K., DeAngelis, M. M., Sheibani, N., Al-Shabrawey, M., & Tawfik, A. (2021). Implication of N-Methyl-d-Aspartate Receptor in Homocysteine-Induced Age-Related Macular Degeneration. International Journal of Molecular Sciences, 22(17), 9356. https://doi.org/10.3390/ijms22179356