
Mood Stabilizers in Psychiatric Disorders and Mechanisms Learnt from In Vitro Model Systems

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Psychiatric Disorders
2.1. Bipolar Disorder
2.1.1. Neuroimaging Studies Associated with BD
2.1.2. Genome-Wide Association Studies in BD
2.1.3. Human Cellular Models of BD
2.2. Schizophrenia
2.2.1. Neuroimaging Studies Associated with Schizophrenia
2.2.2. GWAS Studies in Schizophrenia
2.2.3. Human Cellular Models of Schizophrenia
3. Mood Stabilizers
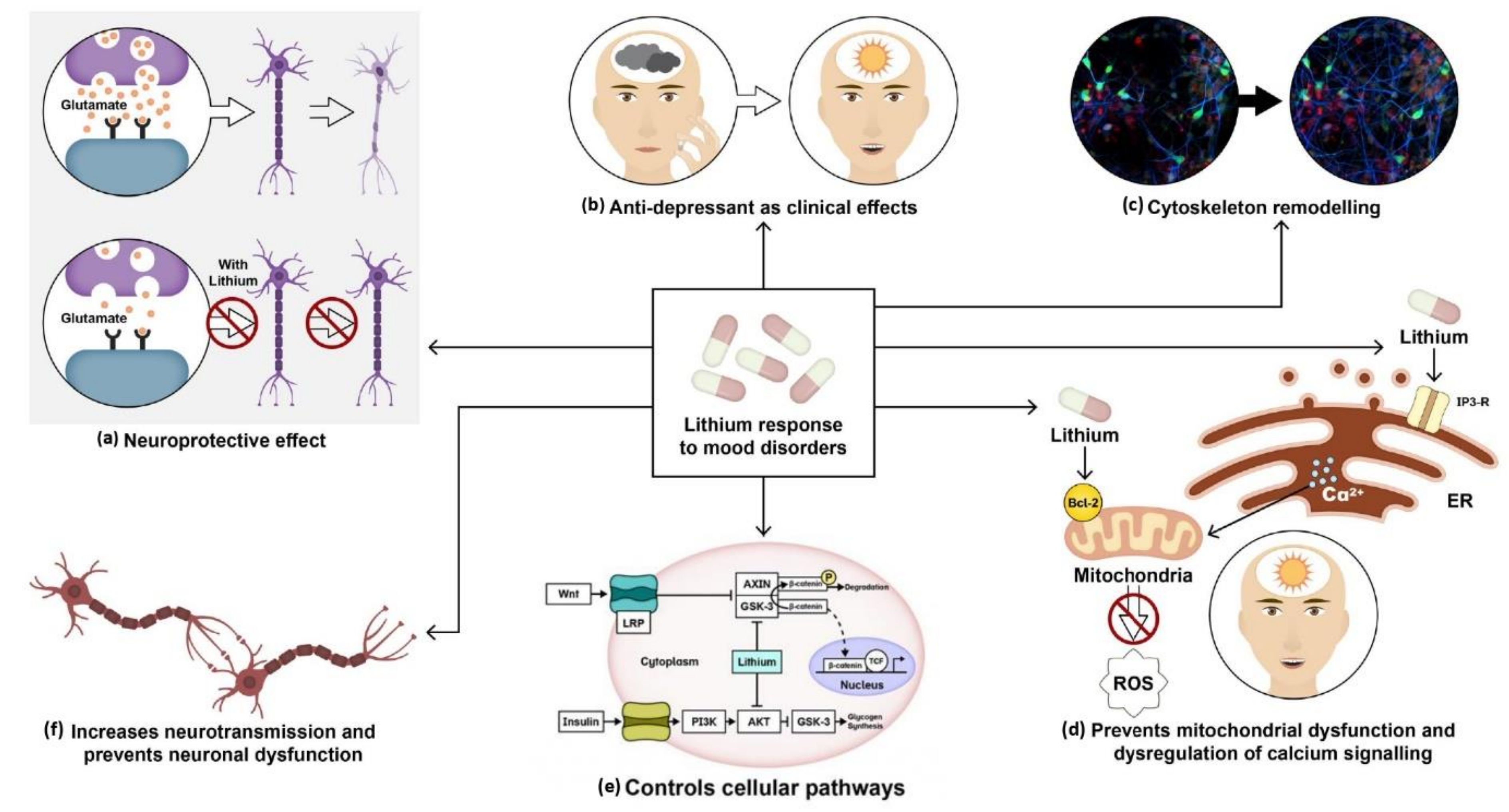
3.1. Lithium
3.1.1. Lithium Treatment in BD Human Studies
3.1.2. Lithium Treatment in Cellular Models of BD
3.1.3. Lithium Treatment in Schizophrenia Human Studies
3.2. Valproic Acid (VPA)
3.2.1. VPA Treatment in BD Human Studies
3.2.2. VPA Treatment in Cellular Models of BD
3.2.3. VPA Treatment in Schizophrenia Human Studies
4. Limitations in the Use of iPSCs for Modeling Psychiatric Disorders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ngui, E.M.; Khasakhala, L.; Ndetei, D.; Roberts, L.W. Mental disorders, health inequalities and ethics: A global perspective. Int. Rev. Psychiatry 2010, 22, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar]
- WHO. Mental Disorders; WHO: Geneva, Switzerland; Available online: https://www.who.int/news-room/fact-sheets/detail/mental-disorders (accessed on 28 November 2020).
- Wittchen, H.; Jacobi, F.; Rehm, J.; Gustavsson, A.; Svensson, M.; Jönsson, B.; Olesen, J.; Allgulander, C.; Alonso, J.; Faravelli, C.; et al. The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 655–679. [Google Scholar] [CrossRef] [Green Version]
- Rehm, J.; Shield, K.D. Global Burden of Disease and the Impact of Mental and Addictive Disorders. Curr. Psychiatry Rep. 2019, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.M. Gender differences in bipolar disorder. Psychiatr. Clin. North Am. 2003, 26, 595–620. [Google Scholar] [CrossRef]
- Negash, A.; Alem, A.; Kebede, D.; Deyessa, N.; Shibre, T.; Kullgren, G. Prevalence and clinical characteristics of bipolar I disorder in Butajira, Ethiopia: A community-based study. J. Affect. Disord. 2005, 87, 193–201. [Google Scholar] [CrossRef]
- Diflorio, A.; Jones, I. Is sex important? Gender differences in bipolar disorder. Int. Rev. Psychiatry 2010, 22, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Baldassano, C.F.; Marangell, L.B.; Gyulai, L.; Ghaemi, S.N.; Joffe, H.; Kim, D.R.; Sagduyu, K.; Truman, C.J.; Wisniewski, S.; Sachs, G.S.; et al. Gender differences in bipolar disorder: Retrospective data from the first 500 STEP-BD participants. Bipolar Disord. 2005, 7, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, S.; Usall, J.; Cobo, J.; Labad, J.; Kulkarni, J. Gender Differences in Schizophrenia and First-Episode Psychosis: A Comprehensive Literature Review. Schizophr. Res. Treat. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Craighead, W.E.; Craighead, L.W. The role of psychotherapy in treating psychiatric disorders. Med. Clin. North Am. 2001, 85, 617–629. [Google Scholar] [CrossRef]
- Holtzheimer, P.E.; Mayberg, H.S. Deep Brain Stimulation for Psychiatric Disorders. Annu. Rev. Neurosci. 2011, 34, 289–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moncrieff, J.; Cohen, D.; Porter, S. The Psychoactive Effects of Psychiatric Medication: The Elephant in the Room. J. Psychoact. Drugs 2013, 45, 409–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakofsky, J.; Rapaport, M. Mood Disorders. Contin. Minneap Minn. 2018, 24, 804–827. [Google Scholar] [CrossRef]
- Chiu, C.-T.; Wang, Z.; Hunsberger, J.G.; Chuang, D.-M. Therapeutic Potential of Mood Stabilizers Lithium and Valproic Acid: Beyond Bipolar Disorder. Pharmacol. Rev. 2013, 65, 105–142. [Google Scholar] [CrossRef] [Green Version]
- Leucht, S.; Helfer, B.; Dold, M.; Kissling, W.; McGrath, J. Lithium for schizophrenia. Cochrane Database Syst. Rev. 2015, 10, CD003834. [Google Scholar] [CrossRef]
- Citrome, L.; Levine, J.; Allingham, B. Changes in use of valproate and other mood stabilizers for patients with schizophrenia from 1994 to 1998. Psychiatr. Serv. 2000, 51, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Hyman, S.E. The daunting polygenicity of mental illness: Making a new map. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170031. [Google Scholar] [CrossRef]
- Zhou, S.; Szczesna, K.; Ochalek, A.; Kobolák, J.; Varga, E.; Nemes, C.; Chandrasekaran, A.; Rasmussen, M.; Cirera, S.; Hyttel, P.; et al. Neurosphere Based Differentiation of Human iPSC Improves Astrocyte Differentiation. Stem Cells Int. 2016, 2016, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Fang, K.-H.; Cao, S.-Y.; Qu, Z.-Y.; Li, Q.; Krencik, R.; Xu, M.; Bhattacharyya, A.; Su, Y.-W.; Zhu, D.-Y.; et al. Efficient generation of region-specific forebrain neurons from human pluripotent stem cells under highly defined condition. Sci. Rep. 2015, 5, 18550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krencik, R.; Zhang, S.-C. Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nat. Protoc. 2011, 6, 1710–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.-Y.; Du, Z.-W.; Zhang, S.-C. Differentiation of human oligodendrocytes from pluripotent stem cells. Nat. Protoc. 2009, 4, 1614–1622. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.D.; Göke, J.; Ng, H.H. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Miura, Y.; Li, M.-Y.; Birey, F.; Ikeda, K.; Revah, O.; Thete, M.V.; Park, J.-Y.; Puno, A.; Lee, S.H.; Porteus, M.H.; et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 1421–1430. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Kadoshima, T.; Soen, M.; Narii, N.; Ishida, Y.; Ohgushi, M.; Takahashi, J.; Eiraku, M.; Sasai, Y. Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat. Commun. 2015, 6, 8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.; Quadrato, G.; Arlotta, P. Studying the Brain in a Dish: 3D Cell Culture Models of Human Brain Development and Disease. Curr. Top. Dev. Biol. 2018, 129, 99–122. [Google Scholar] [PubMed]
- Manji, H.K.; Henter, I.D.; Zarate, C.A. Bipolar Disorder: A Neurobiological Synthesis. Curr. Top Behav. Neurosci. 2010, 5, 331–340. [Google Scholar]
- Miller, T.H. Bipolar Disorder. Prim. Care 2016, 43, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Whiteford, H.A.; Degenhardt, L.; Rehm, J.; Baxter, A.J.; Ferrari, A.J.; Erskine, H.E.; Vos, T.; Murray, J.L.; Burstein, R.; Johns, N.; et al. Global burden of disease attributable to mental and substance use disorders: Findings from the Global Burden of Disease Study 2010. Lancet 2013, 382, 1575–1586. [Google Scholar] [CrossRef]
- Falret, J.P. About hallucinations (1854). Vertex 2015, 26, 234–238. [Google Scholar] [PubMed]
- Angst, J.; Sellaro, R. Historical perspectives and natural history of bipolar disorder. Biol. Psychiatry 2000, 48, 445–457. [Google Scholar] [CrossRef]
- McCormick, U.; Murray, B.; McNew, B. Diagnosis and treatment of patients with bipolar disorder: A review for advanced practice nurses. J. Am. Assoc. Nurse Pr. 2015, 27, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.L.; Kupfer, D.J. Bipolar disorder diagnosis: Challenges and future directions. Lancet 2013, 381, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Association, A.P. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®); American Psychiatric Pub: Washington, DC, USA, 2013. [Google Scholar]
- Merikangas, K.R.; Akiskal, H.S.; Angst, J.; Greenberg, P.E.; Hirschfeld, R.M.A.; Petukhova, M.; Kessler, R.C. Lifetime and 12-Month Prevalence of Bipolar Spectrum Disorder in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 2007, 64, 543–552. [Google Scholar] [CrossRef]
- Hibar, D.P.; ENIGMA Bipolar Disorder Working Group; Westlye, L.T.; Doan, N.T.; Jahanshad, N.; Cheung, J.W.; Ching, C.R.K.; Versace, A.; Bilderbeck, A.C.; Uhlmann, A.; et al. Cortical abnormalities in bipolar disorder: An MRI analysis of 6503 individuals from the ENIGMA Bipolar Disorder Working Group. Mol. Psychiatry 2018, 23, 932–942. [Google Scholar] [CrossRef]
- Drevets, W.C. Subgenual prefrontal cortex abnormalities in mood disorders. Nature 1997, 386, 824–827. [Google Scholar] [CrossRef]
- Bouras, C.; Kövari, E.; Hof, P.R.; Riederer, B.M.; Giannakopoulos, P. Anterior cingulate cortex pathology in schizophrenia and bipolar disorder. Acta Neuropathol. 2001, 102, 373–379. [Google Scholar] [CrossRef]
- Guidotti, A.; Auta, J.; Davis, J.M.; Gerevini, V.D.; Dwivedi, Y.; Grayson, D.R.; Impagnatiello, F.; Pandey, G.; Pesold, C.; Sharma, R.; et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: A postmortem brain study. Arch. Gen. Psychiatry 2000, 57, 1061–1069. [Google Scholar] [CrossRef] [Green Version]
- Rajkowska, G.; Halaris, A.; Selemon, L.D. Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol. Psychiatry 2001, 49, 741–752. [Google Scholar] [CrossRef]
- Berretta, S.; Pantazopoulos, H.; Lange, N. Neuron Numbers and Volume of the Amygdala in Subjects Diagnosed with Bipolar Disorder or Schizophrenia. Biol. Psychiatry 2007, 62, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Edvardsen, J.; Torgersen, S.; Roysamb, E.; Lygren, S.; Skre, I.; Onstad, S.; Øien, P.A. Heritability of bipolar spectrum disorders. Unity or heterogeneity? J. Affect. Disord. 2008, 106, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuffin, P.; Rijsdijk, F.; Andrew, M.; Sham, P.; Katz, R.; Cardno, A. The Heritability of Bipolar Affective Disorder and the Genetic Relationship to Unipolar Depression. Arch. Gen. Psychiatry 2003, 60, 497–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baum, E.A.; NIMH Genetics Initiative Bipolar Disorder Consortium; Akula, N.; Cabanero, M.; Cardona, I.; Corona, W.; Klemens, B.; Schulze, T.G.; Cichon, S.; Rietschel, M.; et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol. Psychiatry 2007, 13, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Sklar, P.O.A.A.; Cichon, S.; Psychiatric, GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 2011, 43, 977–983. [Google Scholar]
- Djurovic, S.; Tesli, M.; Hope, S.; Mattingsdal, M.; Michelsen, A.; Ueland, T. Up-regulation of NOTCH4 gene expression in bipolar disorder. Am. J. Psychiatry 2012, 169, 1292–1300. [Google Scholar]
- Lee, K.W.; San Woon, P.; Teo, Y.Y.; Sim, K. Genome wide association studies (GWAS) and copy number variation (CNV) studies of the major psychoses: What have we learnt? Neurosci. Biobehav. Rev. 2012, 36, 556–571. [Google Scholar] [CrossRef]
- Dushlaine, C.; Rossin, L.; Lee, P.; Duncan, L.; Parikshak, N.; Newhouse, S.; Ripke, S.; The Network and Pathway Analysis Subgroup of the Psychiatric Genomics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. 2015, 18, 199–209. [Google Scholar]
- Chang, H.; The Swedish Bipolar Study Group; Li, L.; Peng, T.; Grigoroiu-Serbanescu, M.; Bergen, S.E.; Landén, M.; Hultman, C.M.; Forstner, A.J.; Strohmaier, J.; et al. Identification of a Bipolar Disorder Vulnerable Gene CHDH at 3p21.1. Mol. Neurobiol. 2016, 54, 5166–5176. [Google Scholar] [CrossRef]
- Forstner, A.J.; Hecker, J.; Hofmann, A.; Maaser, A.; Reinbold, C.S.; Mühleisen, T.W.; Leber, M.; Strohmaier, J.; Degenhardt, F.; Treutlein, J.; et al. Identification of shared risk loci and pathways for bipolar disorder and schizophrenia. PLoS ONE 2017, 12, e0171595. [Google Scholar] [CrossRef] [Green Version]
- Stahl, E.A.; eQTLGen Consortium; Breen, G.; Forstner, A.J.; McQuillin, A.; Ripke, S.; Trubetskoy, V.; Mattheisen, M.; Wang, Y.; Coleman, J.R.I.; et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 2019, 51, 793–803. [Google Scholar] [CrossRef]
- Mullins, N.; Forstner, A.; Connell, K.; Coleman, J.; Qiao, Z.; Als, T.; Bigdeli, T.; Borte, S.; Bryois, J.; Charney, A.; et al. Genome-wide association study of over 40,000 bipolar disorder cases provides novel biological insights. medRxiv 2021, 53, 817–829. [Google Scholar]
- Coleman, J.R.; Gaspar, H.A.; Bryois, J.; Breen, G.; Byrne, E.M.; Forstner, A.J.; Holmans, P.A.; de Leeuw, C.A.; Mattheisen, M.; McQuillin, A.; et al. The Genetics of the Mood Disorder Spectrum: Genome-wide Association Analyses of More Than 185,000 Cases and 439,000 Controls. Biol. Psychiatry 2020, 88, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Emamghoreishi, M.; Schlichter, L.; Li, P.P.; Parikh, S.; Sen, J.; Kamble, A.; Warsh, J.J. High intracellular calcium concentrations in transformed lymphoblasts from subjects with bipolar I disorder. Am. J. Psychiatry 1997, 154, 976–982. [Google Scholar]
- Solís-Chagoyán, H.; Calixto, E.; Figueroa, A.; Montaño, L.; Berlanga, C.; Rodríguez-Verdugo, M.; Romo, F.; Jiménez, M.; Gurrola, C.Z.; Riquelme, A.; et al. Microtubule organization and L-type voltage-activated calcium current in olfactory neuronal cells obtained from patients with schizophrenia and bipolar disorder. Schizophr. Res. 2013, 143, 384–389. [Google Scholar] [CrossRef]
- Chen, H.M.; Delong, C.J.; Bame, M.; Rajapakse, I.; Herron, T.J.; McInnis, M.G.; O’Shea, K.S. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Transl. Psychiatry 2014, 4, e375. [Google Scholar] [CrossRef] [Green Version]
- Madison, J.M.; Zhou, F.; Nigam, A.; Hussain, A.; Barker, D.D.; Nehme, R.; Van Der Ven, K.; Hsu, J.; Wolf, P.A.; Fleishman, M.; et al. Characterization of bipolar disorder patient-specific induced pluripotent stem cells from a family reveals neurodevelopmental and mRNA expression abnormalities. Mol. Psychiatry 2015, 20, 703–717. [Google Scholar] [CrossRef]
- Tandon, R.; Gaebel, W.; Barch, D.M.; Bustillo, J.; Gur, R.E.; Heckers, S.; Malaspina, D.; Owen, M.J.; Schultz, S.; Tsuang, M.; et al. Definition and description of schizophrenia in the DSM-5. Schizophr. Res. 2013, 150, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Charlson, F.J.; Ferrari, A.J.; Santomauro, D.; Diminic, S.; Stockings, E.; Scott, J.G.; McGrath, J.; Whiteford, H.A. Global Epidemiology and Burden of Schizophrenia: Findings From the Global Burden of Disease Study 2016. Schizophr. Bull. 2018, 44, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Iacono, W.G.; Beiser, M. Are males more likely than females to develop schizophrenia? Am. J. Psychiatry 1992, 149, 1070–1074. [Google Scholar]
- Grossman, L.S.; Harrow, M.; Rosen, C.; Faull, R.; Strauss, G.P. Sex differences in schizophrenia and other psychotic disorders: A 20-year longitudinal study of psychosis and recovery. Compr. Psychiatry 2008, 49, 523–529. [Google Scholar] [CrossRef] [Green Version]
- Andreasen, N.C. The evolving concept of schizophrenia: From Kraepelin to the present and future. Schizophr. Res. 1997, 28, 105–109. [Google Scholar] [CrossRef]
- Maatz, A.; Hoff, P. The birth of schizophrenia or a very modern Bleuler: A close reading of Eugen Bleuler’s ‘Die Prognose der Dementia praecox’ and a re-consideration of his contribution to psychiatry. Hist. Psychiatry 2014, 25, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Berrios, G.E. Positive and Negative Symptoms and Jackson. Arch. Gen. Psychiatry 1985, 42, 95–97. [Google Scholar] [CrossRef]
- Fenton, W.S.; McGlashan, T.H. Natural history of schizophrenia subtypes. II. Positive and negative symptoms and long-term course. Arch. Gen. Psychiatry 1991, 48, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Davison, K. Schizophrenia-like psychoses associated with organic cerebral disorders: A review. Psychiatr. Dev. 1983, 1, 1–33. [Google Scholar] [PubMed]
- Liddle, P.F. Schizophrenic syndromes, cognitive performance and neurological dysfunction. Psychol. Med. 1987, 17, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Blumer, D. Personality changes with frontal and temporal lobe lesions. Psychiatr. Asp. Neurol. Dis. 1975, 1, 151–170. [Google Scholar]
- Heuvel, M.V.D.; Fornito, A. Brain Networks in Schizophrenia. Neuropsychol. Rev. 2014, 24, 32–48. [Google Scholar] [CrossRef]
- Johnstone, E.; Frith, C.; Crow, T.; Husband, J.; Kreel, L. Cerebral ventricular size and cognitive impairment in chronic schizophrenia. Lancet 1976, 308, 924–926. [Google Scholar] [CrossRef]
- Falkai, P.; Bogerts, B. Cell loss in the hippocampus of schizophrenics. Eur. Arch. Psychiatry Clin. Neurosci. 1986, 236, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, N.C.; Flashman, L.; Flaum, M.; Arndt, S.; Swayze, V.; O’Leary, D.S.; Ehrhardt, J.C.; Yuh, W.T. Regional brain abnormalities in schizophrenia measured with magnetic resonance imaging. JAMA 1994, 272, 1763–1769. [Google Scholar] [CrossRef]
- Lawrie, S.M.; Abukmeil, S.S. Brain abnormality in schizophrenia. A systematic and quantitative review of volumetric magnetic resonance imaging studies. Br. J. Psychiatry 1998, 172, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Mathalon, D.H.; Sullivan, E.; Lim, K.; Pfefferbaum, A. Progressive brain volume changes and the clinical course of schizophrenia in men: A longitudinal magnetic resonance imaging study. Arch. Gen. Psychiatry 2001, 58, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donovan, M.C.; Craddock, N.; Norton, N.; Williams, H.; Peirce, T.; Moskvina, V.; Nikolov, I.; Hamshere, M.; Carroll, L.; Georgieva, L.; et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 2008, 40, 1053–1055. [Google Scholar] [CrossRef]
- Potkin, S.G.; Turner, J.; Guffanti, G.; Lakatos, A.; Fallon, J.H.; Nguyen, D.D.; Mathalon, D.; Ford, J.; Lauriello, J.; Macciardi, F. A Genome-Wide Association Study of Schizophrenia Using Brain Activation as a Quantitative Phenotype. Schizophr. Bull. 2009, 35, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Ripke, S.; Sanders, A.; Kendler, K.; Levinson, D.; Sklar, P.; Holmans, P.; Lin, D.; Duan, J.; Ophoff, R.; Andreassen, O.; et al. Schizophrenia Psychiatric Genome-Wide Association Study (GWAS), Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011, 43, 969–976. [Google Scholar]
- Ripke, S.; O’Dushlaine, C.; Chambert, K.; Moran, J.; Kähler, A.K.; Akterin, S.; Bergen, S.; Collins, A.L.; Crowley, J.; Fromer, M.; et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013, 45, 1150–1159. [Google Scholar] [CrossRef]
- Ripke, S.; Neale, B.; Corvin, A.; Walters, J.; Farh, K.; Holmans, P.; Lee, P.; Sullivan, B.; Collier, D.; Huang, H.; et al. Schizophrenia Working Group of the Psychiatric Genomics, Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar]
- Rampino, A.; Marakhovskaia, A.; Soares-Silva, T.; Torretta, S.; Veneziani, F.; Beaulieu, J.M. Antipsychotic Drug Responsiveness and Dopamine Receptor Signaling; Old Players and New Prospects. Front. Psychiatry 2019, 9, 702. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Snyder, G.L.; Vanover, K.E. Dopamine Targeting Drugs for the Treatment of Schizophrenia: Past, Present and Future. Curr. Top. Med. Chem. 2016, 16, 3385–3403. [Google Scholar] [CrossRef] [Green Version]
- Gurung, R.; Prata, D. What is the impact of genome-wide supported risk variants for schizophrenia and bipolar disorder on brain structure and function? A systematic review. Psychol. Med. 2015, 45, 2461–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, L.; Bergen, S.; Akula, N.; Song, J.; Hultman, C.M.; Landén, M.; Adli, M.; Alda, M.; Ardau, R.; Arias, B.; et al. Genome-wide association study of 40,000 individuals identifies two novel loci associated with bipolar disorder. Hum. Mol. Genet. 2016, 25, 3383–3394. [Google Scholar] [CrossRef] [PubMed]
- Ruderfer, D.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Fanous, A.H.; Ripke, S.; McQuillin, A.; Amdur, R.L.; Gejman, P.V.; O’Donovan, M.; Andreassen, O.A.; Djurovic, S.; et al. Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol. Psychiatry 2014, 19, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusev, A.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Mancuso, N.; Won, H.; Kousi, M.; Finucane, H.K.; Reshef, Y.; Song, L.; Safi, A.; McCarroll, S.; et al. Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet. 2018, 50, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruderfer, D.; Ripke, S.; McQuillin, A.; Boocock, J.; Stahl, E.; Pavlides, J.; Mullins, N.; Charney, A.; Ori, P.; Loohuis, L.; et al. Genomic Dissection of Bipolar Disorder and Schizophrenia, Including 28 Subphenotypes. Cell 2018, 173, 1705–1715.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Cheng, W.; Zhang, X.; Wang, X.; Yue, W. Integration analysis of methylation quantitative trait loci and GWAS identify three schizophrenia risk variants. Neuropsychopharmacology 2020, 45, 1179–1187. [Google Scholar] [CrossRef]
- Mahmoudi, E.; Atkins, J.R.; Quidé, Y.; Reay, W.R.; Cairns, H.M.; Fitzsimmons, C.; Carr, V.J.; Green, M.J.; Cairns, M.J. The MIR137 VNTR rs58335419 Is Associated with Cognitive Impairment in Schizophrenia and Altered Cortical Morphology. Schizophr. Bull. 2021, 47, 495–504. [Google Scholar] [CrossRef]
- Graziadei, G.A.; Graziadei, P.P. Neurogenesis and neuron regeneration in the olfactory system of mammals. II. Degeneration and reconstitution of the olfactory sensory neurons after axotomy. J. Neurocytol. 1979, 8, 197–213. [Google Scholar] [CrossRef]
- Matigian, N.; Abrahamsen, G.; Sutharsan, R.; Cook, A.; Vitale, A.M.; Nouwens, A.; Bellette, B.; An, J.; Anderson, M.; Beckhouse, A.G.; et al. Disease-specific, neurosphere-derived cells as models for brain disorders. Dis. Model. Mech. 2010, 3, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Benítez-King, G.; Riquelme, A.; Ortíz-López, L.; Berlanga, C.; Rodríguez-Verdugo, M.; Romo, F.; Calixto, E.; Solís-Chagoyán, H.; Jímenez, M.; Montaño, L.; et al. A non-invasive method to isolate the neuronal linage from the nasal epithelium from schizophrenic and bipolar diseases. J. Neurosci. Methods 2011, 201, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.H.; Su, Y.; Wen, Z.; Yoritomo, N.; Ross, C.A.; Margolis, R.L.; Song, H.; Ming, G.I. Integration-free induced pluripotent stem cells derived from schizophrenia patients with a DISC1 mutation. Mol. Psychiatry 2011, 16, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Brennand, K.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef]
- Hook, V.; Brennand, K.; Kim, Y.; Toneff, T.; Funkelstein, L.; Lee, K.C.; Ziegler, M.; Gage, F.H. Human iPSC Neurons Display Activity-Dependent Neurotransmitter Secretion: Aberrant Catecholamine Levels in Schizophrenia Neurons. Stem Cell Rep. 2014, 3, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Hammond, W. A Treatise on Diseases of the Nervous System. Med. Chir. Rev. J. Med. Sci. Anal. Ser. 1822, 3, 277–306. [Google Scholar]
- Cade, J.F. Lithium salts in the treatment of psychotic excitement. 1949. Bull. World Health Organ. 2000, 78, 518–520. [Google Scholar]
- Gershon, S.; Yuwiler, A. Lithium ion: A specific psychopharmacological approach to the treatment of mania. J. Neuropsychiatry 1960, 1, 229–241. [Google Scholar]
- Mitchell, P.B.; Hadzi-Pavlovic, D. Lithium treatment for bipolar disorder. Bull. World Health Organ. 2000, 78, 515–517. [Google Scholar]
- Carmassi, C.; Del Grande, C.; Gesi, C.; Musetti, L.; Dell’Osso, L. A new look at an old drug: Neuroprotective effects and therapeutic potentials of lithium salts. Neuropsychiatr. Dis. Treat. 2016, 12, 1687–1703. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, S.; Hough, C.J.; Chuang, D.-M. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proc. Natl. Acad. Sci. USA 1998, 95, 2642–2647. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.C.; Lucas, F.R.; Salinas, P.C. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell 2000, 100, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.S.B.; Cheng, L.; Mudge, A.W.; Harwood, A. A common mechanism of action for three mood-stabilizing drugs. Nature 2002, 417, 292–295. [Google Scholar] [CrossRef]
- Burgoyne, R.D.; Weiss, J.L. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem. J. 2001, 353 Pt 1, 1–12. [Google Scholar] [CrossRef]
- Schlecker, C.; Boehmerle, W.; Jeromin, A.; DeGray, B.; Varshney, A.; Sharma, Y.; Szigeti-Buck, K.; Ehrlich, B.E. Neuronal calcium sensor-1 enhancement of InsP3 receptor activity is inhibited by therapeutic levels of lithium. J. Clin. Investig. 2006, 116, 1668–1674. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Zeng, W.Z.; Yuan, P.X.; Huang, L.D.; Jiang, Y.M.; Zhao, Z.H.; Manji, H. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J. Neurochem. 1999, 72, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Klein, P.S. Molecular targets of lithium action. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 789–813. [Google Scholar] [CrossRef]
- Gurvich, N.; Klein, P.S. Lithium and valproic acid: Parallels and contrasts in diverse signaling contexts. Pharmacol. Ther. 2002, 96, 45–66. [Google Scholar] [CrossRef]
- Berridge, M.J.; Downes, C.P.; Hanley, M.R. Neural and developmental actions of lithium: A unifying hypothesis. Cell 1989, 59, 411–419. [Google Scholar] [CrossRef]
- Lyoo, I.K.; Dager, S.R.; E Kim, J.; Yoon, S.J.; Friedman, S.; Dunner, D.L.; Renshaw, P.F. Lithium-Induced Gray Matter Volume Increase as a Neural Correlate of Treatment Response in Bipolar Disorder: A Longitudinal Brain Imaging Study. Neuropsychopharmacology 2010, 35, 1743–1750. [Google Scholar] [CrossRef]
- Benedetti, F.; Radaelli, D.; Poletti, S.; Locatelli, C.; Falini, A.; Colombo, C.; Smeraldi, E. Opposite effects of suicidality and lithium on gray matter volumes in bipolar depression. J. Affect. Disord. 2011, 135, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Hallahan, B.; Newell, J.; Soares, J.; Brambilla, P.; Strakowski, S.; Fleck, D.; Kieseppa, T.; Altshuler, L.; Fornito, A.; Malhi, G.; et al. Structural magnetic resonance imaging in bipolar disorder: An international collaborative mega-analysis of individual adult patient data. Biol. Psychiatry 2011, 69, 326–335. [Google Scholar] [CrossRef]
- Bowley, M.P.; Drevets, W.C.; Öngür, D.; Price, J.L. Low glial numbers in the amygdala in major depressive disorder. Biol. Psychiatry 2002, 52, 404–412. [Google Scholar] [CrossRef]
- Hibar, D.P.; the Costa Rica/Colombia Consortium for Genetic Investigation of Bipolar Endophenotypes; Westlye, L.T.; Van Erp, T.G.M.; Rasmussen, J.; Leonardo, C.D.; Faskowitz, J.; Haukvik, U.K.; Hartberg, C.B.; Doan, N.T.; et al. Subcortical volumetric abnormalities in bipolar disorder. Mol. Psychiatry 2016, 21, 1710–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajek, T.; Bauer, M.; Simhandl, C.; Rybakowski, J.; O’Donovan, C.; Pfennig, A.; König, B.; Suwalska, A.; Yucel, K.; Uher, R.; et al. Neuroprotective effect of lithium on hippocampal volumes in bipolar disorder independent of long-term treatment response. Psychol. Med. 2013, 44, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Gildengers, A.G.; Butters, M.A.; Aizenstein, H.; Marron, M.; Emanuel, J.; Anderson, S.; Weissfeld, L.A.; Becker, J.T.; Lopez, O.L.; Mulsant, B.H.; et al. Longer lithium exposure is associated with better white matter integrity in older adults with bipolar disorder. Bipolar Disord. 2015, 17, 248–256. [Google Scholar] [CrossRef] [Green Version]
- de Sousa, R.T.; Zarate, C., Jr.; Zanetti, M.; Costa, A.; Talib, L.; Gattaz, W.; Machado-Vieira, R. Oxidative stress in early stage bipolar disorder and the association with response to lithium. J. Psychiatr. Res. 2014, 50, 36–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Findling, R.L.; Robb, A.; McNamara, N.K.; Pavuluri, M.N.; Kafantaris, V.; Scheffer, R.; Frazier, J.A.; Rynn, M.; DelBello, M.P.; Kowatch, R.A.; et al. Lithium in the Acute Treatment of Bipolar I Disorder: A Double-Blind, Placebo-Controlled Study. Pediatrics 2015, 136, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Findling, R.L.; McNamara, N.; Pavuluri, M.; Frazier, J.; Rynn, M.; Scheffer, R.; Kafantaris, V.; Robb, A.; DelBello, M.; Kowatch, R.; et al. Lithium for the maintenance treatment of bipolar I disorder: A double-blind, placebo-controlled discontinuation study. J. Am. Acad. Child Adolesc. Psychiatry 2019, 58, 287–296.e4. [Google Scholar] [CrossRef]
- Velosa, J.; Delgado, A.; Finger, E.; Berk, M.; Kapczinski, F.; Cardoso, T.D.A. Risk of dementia in bipolar disorder and the interplay of lithium: A systematic review and meta-analyses. Acta Psychiatr. Scand. 2020, 141, 510–521. [Google Scholar] [CrossRef]
- Mertens, J.; Wang, Q.W.; Kim, Y.; Yu, D.; Pham, S.; Yang, B.; Zheng, Y.; Diffenderfer, K.; Zhang, J.; Soltani, S.; et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 2015, 527, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Tobe, B.T.D.; Crain, A.M.; Winquist, A.M.; Calabrese, B.; Makihara, H.; Zhao, W.; Lalonde, J.; Nakamura, H.; Konopaske, G.; Sidor, M.; et al. Probing the lithium-response pathway in hiPSCs implicates the phosphoregulatory set-point for a cytoskeletal modulator in bipolar pathogenesis. Proc. Natl. Acad. Sci. USA 2017, 114, E4462–E4471. [Google Scholar] [CrossRef] [Green Version]
- Stern, S.; Segal, M.; Moses, E. Involvement of Potassium and Cation Channels in Hippocampal Abnormalities of Embryonic Ts65Dn and Tc1 Trisomic Mice. EBioMedicine 2015, 2, 1048–1062. [Google Scholar] [CrossRef] [Green Version]
- Boris Brant, T.S.; Shekhidem, H.A.; Mizrahi, L.; Rosh, I.; Stern, Y.; Ofer, P.; Asleh, A.; Umanah, G.K.E.; Jada, R.; Levy, N.S.; et al. IQSEC2 Mutation Associated with Epilepsy, Intellectual Disability and Autism Results in Hyperexcitability of Patient Derived Neurons and Deficient Synaptic transmission. Mol. Psychiatry 2021, in press. [Google Scholar]
- Quraishi, I.H.; Stern, S.; Mangan, K.; Zhang, Y.; Ali, S.; Mercier, M.; Marchetto, M.; McLachlan, M.; Jones, E.; Gage, F.; et al. An Epilepsy-Associated KCNT1 Mutation Enhances Excitability of Human iPSC-Derived Neurons by Increasing Slack KNa Currents. J. Neurosci. 2019, 39, 7438–7449. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, G.; Rodriguez, M.J.; Pomata, P.; Rela, L.; Murer, M.G. Reduction of an Afterhyperpolarization Current Increases Excitability in Striatal Cholinergic Interneurons in Rat Parkinsonism. J. Neurosci. 2011, 31, 6553–6564. [Google Scholar] [CrossRef] [Green Version]
- Stern, S.; Santos, R.; Marchetto, M.C.; Mendes, A.P.D.; Rouleau, G.A.; Biesmans, S.; Wang, Q.-W.; Yao, J.; Charnay, P.; Bang, A.G.; et al. Neurons derived from patients with bipolar disorder divide into intrinsically different sub-populations of neurons, predicting the patients’ responsiveness to lithium. Mol. Psychiatry 2018, 23, 1453–1465. [Google Scholar] [CrossRef]
- Stern, S.; Sarkar, A.; Stern, T.; Mei, A.; Mendes, A.P.; Stern, Y.; Goldberg, G.; Galor, D.; Nguyen, T.; Randolph-Moore, L.; et al. Mechanisms Underlying the Hyperexcitability of CA3 and Dentate Gyrus Hippocampal Neurons Derived From Patients With Bipolar Disorder. Biol. Psychiatry 2020, 88, 139–149. [Google Scholar] [CrossRef]
- Stern, S.; Sarkar, A.; Galor, D.; Stern, T.; Mei, A.; Stern, Y.; Mendes, A.P.; Randolph-Moore, L.; Rouleau, G.; Bang, A.G.; et al. A Physiological Instability Displayed in Hippocampal Neurons Derived From Lithium-Nonresponsive Bipolar Disorder Patients. Biol. Psychiatry 2020, 88, 150–158. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.J.; Wei, H.; Nievergelt, C.M.; Stautland, A.; Maihofer, A.X.; Welsh, D.K.; Shilling, P.; Alda, M.; Alliey-Rodriguez, N.; Anand, A.; et al. Chronotype and cellular circadian rhythms predict the clinical response to lithium maintenance treatment in patients with bipolar disorder. Neuropsychopharmacology 2019, 44, 620–628. [Google Scholar] [CrossRef]
- Mishra, H.K.; Ying, N.; Luis, A.; Wei, H.; Nguyen, M.; Nakhla, T.; Vandenburgh, S.; Alda, M.; Berrettini, W.; Brennand, K.; et al. Circadian rhythms in bipolar disorder patient-derived neurons predict lithium response: Preliminary studies. Mol. Psychiatry 2021, in press. [Google Scholar]
- Lerner, Y.; Mintzer, Y.; Schestatzky, M. Lithium Combined with Haloperidol in Schizophrenic Patients. Br. J. Psychiatry 1988, 153, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Terao, T.; Oga, T.; Nozaki, S.; Ohta, A.; Ohtsubo, Y.; Yamamoto, S.; Zamami, M.; Okada, M. Lithium addition to neuroleptic treatment in chronic schizophrenia: A randomized, double-blind, placebo-controlled, cross-over study. Acta Psychiatr. Scand. 1995, 92, 220–224. [Google Scholar] [CrossRef]
- Bender, S.; Linka, T.; Wolstein, J.; Gehendges, S.; Paulus, H.-J.; Schall, U.; Gastpar, M. Safety and efficacy of combined clozapine–lithium pharmacotherapy. Int. J. Neuropsychopharmacol. 2004, 7, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.L.; Conley, R.R.; Feldman, S.; Yü, Y.; McMahon, R.P.; Richardson, C.M. Adjunct Divalproex or Lithium to Clozapine in Treatment-Resistant Schizophrenia. Psychiatr. Q. 2006, 77, 81–95. [Google Scholar] [CrossRef]
- Löscher, W. The discovery of valproate. In Valproate; Springer: Berlin/Heidelberg, Germany, 1999; pp. 1–3. [Google Scholar]
- Lemperiere, T. Brief history of the development of valproate in bipolar disorders. Encephale 2001, 27, 365–372. [Google Scholar] [PubMed]
- Bowden, C.L.; Brugger, A.M.; Swann, A.C.; Calabrese, J.R.; Janicak, P.G.; Petty, F.; Dilsaver, S.C.; Davis, J.M.; Rush, A.J.; Small, J.G.; et al. Efficacy of divalproex vs lithium and placebo in the treatment of mania. The Depakote Mania Study Group. JAMA 1994, 271, 918–924. [Google Scholar] [CrossRef]
- Plitman, E.; Nakajima, S.; de la Fuente-Sandoval, C.; Gerretsen, P.; Chakravarty, M.M.; Kobylianskii, J.; Chung, J.K.; Caravaggio, F.; Iwata, Y.; Remington, G.; et al. Glutamate-mediated excitotoxicity in schizophrenia: A review. Eur. Neuropsychopharmacol. 2014, 24, 1591–1605. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, R.; Hough, C.; Nakazawa, T.; Yamamoto, T.; Chuang, D.-M. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: Involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J. Neurochem. 2002, 80, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Berg, R.J.V.D.; Kok, P.; Voskuyl, R.A. Valproate and sodium currents in cultured hippocampal neurons. Exp. Brain Res. 1993, 93, 279–287. [Google Scholar] [CrossRef]
- Löscher, W.; Schmidt, D. Increase of Human Plasma GABA by Sodium Valproate. Epilepsia 1980, 21, 611–615. [Google Scholar] [CrossRef]
- Treiman, D.M. GABAergic Mechanisms in Epilepsy. Epilepsia 2001, 42, 8–12. [Google Scholar] [CrossRef]
- Frey, H.H.; Loscher, W. Di-n-propylacetic acid--profile of anticonvulsant activity in mice. Arzneimittelforschung 1976, 26, 299–301. [Google Scholar]
- Wang, L.; Liu, Y.; Li, S.; Long, Z.-Y.; Wu, Y.-M. Wnt signaling pathway participates in valproic acid-induced neuronal differentiation of neural stem cells. Int. J. Clin. Exp. Pathol. 2015, 8, 578–585. [Google Scholar]
- de Ruijter, A.J.; Gennip, A.H.V.; Caron, H.N.; Kemp, S.; Kuilenburg, A.B.P.V. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370 Pt 3, 737–749. [Google Scholar] [CrossRef]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [Green Version]
- Dunn, R.T.; Frye, M.S.; Kimbrell, T.A.; Denicoff, K.D.; Leverich, G.S.; Post, R.M. The efficacy and use of anticonvulsants in mood disorders. Clin. Neuropharmacol. 1998, 21, 215–235. [Google Scholar]
- McElroy, S.L.; Kech, P.E., Jr.; Pope, H.G., Jr.; Hudson, J.I. Valproate in the treatment of rapid-cycling bipolar disorder. J. Clin. Psychopharmacol. 1988, 8, 275–279. [Google Scholar] [CrossRef]
- Calabrese, J.R.; Markovitz, P.J.; Kimmel, S.E.; Wagner, S.C. Spectrum of Efficacy of Valproate in 78 Rapid-Cycling Bipolar Patients. J. Clin. Psychopharmacol. 1992, 12 (Suppl. 1), 53S–56S. [Google Scholar] [CrossRef]
- Denicoff, K.D.; E Smith-Jackson, E.; Bryan, A.L.; O Ali, S.; Post, R.M. Valproate prophylaxis in a prospective clinical trial of refractory bipolar disorder. Am. J. Psychiatry 1997, 154, 1456–1458. [Google Scholar]
- Schneider, A.L.; Wilcox, C.S. Divalproate augmentation in lithium-resistant rapid cycling mania in four geriatric patients. J. Affect. Disord. 1998, 47, 201–205. [Google Scholar] [CrossRef]
- Bowden, C.L.; Calabrese, J.R.; McElroy, S.L.; Gyulai, L.; Wassef, A.; Petty, F.; Pope, H.G., Jr.; Chou, J.C.; Keck, P.E., Jr.; Rhodes, L.J.; et al. A randomized, placebo-controlled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Divalproex Maintenance Study Group. Arch. Gen. Psychiatry 2000, 57, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Salloum, I.M.; Cornelius, J.R.; Daley, D.C.; Kirisci, L.; Himmelhoch, J.M.; Thase, M.E. Efficacy of valproate maintenance in patients with bipolar disorder and alcoholism: A double-blind placebo-controlled study. Arch. Gen. Psychiatry 2005, 62, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geddes, J.R.; Goodwin, G.M.; Rendell, J.; Azorin, J.M.; Cipriani, A.; Ostacher, J.M.; Morriss, R.; Alder, N.; Juszczak, E. Lithium plus valproate combination therapy versus monotherapy for relapse prevention in bipolar I disorder (BALANCE): A randomised open-label trial. Lancet 2010, 375, 385–395. [Google Scholar]
- Smith, L.; Cornelius, V.; Azorín, J.; Perugi, G.; Vieta, E.; Young, A.; Bowden, C. Valproate for the treatment of acute bipolar depression: Systematic review and meta-analysis. J. Affect. Disord. 2010, 122, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; Bonaccorso, S.; Bobo, W.V.; Chen, Y.; Jayathilake, K. A 12-month randomized, open-label study of the metabolic effects of olanzapine and risperidone in psychotic patients: Influence of valproic acid augmentation. J. Clin. Psychiatry 2011, 72, 1602–1610. [Google Scholar] [CrossRef]
- Chen, S.-L.; Lee, S.-Y.; Chang, Y.-H.; Chen, P.-S.; Lee, I.-H.; Wang, T.-Y.; Chen, K.-C.; Yang, Y.-K.; Hong, J.-S.; Lu, R.-B. Therapeutic effects of add-on low-dose dextromethorphan plus valproic acid in bipolar disorder. Eur. Neuropsychopharmacol. 2014, 24, 1753–1759. [Google Scholar] [CrossRef]
- Chen, D.T.; Jiang, X.; Akula, N.; Shugart, Y.Y.; Wendland, J.R.; Steele, C.J.M.; Kassem, L.; Park, J.H.; Chatterjee, N.; Jamain, S.; et al. Genome-wide association study meta-analysis of European and Asian-ancestry samples identifies three novel loci associated with bipolar disorder. Mol. Psychiatry 2013, 18, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Mühleisen, T.W.; Leber, M.; Schulze, T.G.; Strohmaier, J.; Degenhardt, F.; Treutlein, J.; Mattheisen, M.; Forstner, A.J.; Schumacher, J.; Breuer, R.; et al. Genome-wide association study reveals two new risk loci for bipolar disorder. Nat. Commun. 2014, 5, 3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Detera-Wadleigh, S.D.; Akula, N.; Mallon, B.S.; Hou, L.; Xiao, T.; Felsenfeld, G.; Gu, X.; McMahon, F.J. Sodium valproate rescues expression of TRANK1 in iPSC-derived neural cells that carry a genetic variant associated with serious mental illness. Mol. Psychiatry 2019, 24, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Linker, S.B.; Stern, S.; Mendes, A.P.D.; Shokhirev, M.N.; Erikson, G.; Randolph-Moore, L.; Racha, V.; Kim, Y.; Kelsoe, J.R.; et al. Deficient LEF1 expression is associated with lithium resistance and hyperexcitability in neurons derived from bipolar disorder patients. Mol. Psychiatry 2021, in press. [Google Scholar]
- Facciolà, G.; Avenoso, A.; Scordo, M.G.; Madia, A.G.; Ventimiglia, A.; Perucca, E.; Spina, E. Small effects of valproic acid on the plasma concentrations of clozapine and its major metabolites in patients with schizophrenic or affective disorders. Ther. Drug Monit. 1999, 21, 341–345. [Google Scholar] [CrossRef]
- Wassef, A.A.; Dott, S.G.; Harris, A.; Brown, A.; O’Boyle, M.; Meyer, W.J.; Rose, R.M. Randomized, Placebo-Controlled Pilot Study of Divalproex Sodium in the Treatment of Acute Exacerbations of Chronic Schizophrenia. J. Clin. Psychopharmacol. 2000, 20, 357–361. [Google Scholar] [CrossRef]
- Wassef, A.A.; Hafiz, N.G.; Hampton, D.; Molloy, M. Divalproex Sodium Augmentation of Haloperidol in Hospitalized Patients with Schizophrenia: Clinical and Economic Implications. J. Clin. Psychopharmacol. 2001, 21, 21–26. [Google Scholar] [CrossRef]
- Casey, D.E.; Daniel, D.G.; Wassef, A.A.; Tracy, K.A.; Wozniak, P.; Sommerville, K.W. Effect of divalproex combined with olanzapine or risperidone in patients with an acute exacerbation of schizophrenia. Neuropsychopharmacology 2003, 28, 182–192. [Google Scholar] [CrossRef]
- Sajatovic, M.; Coconcea, N.; Ignacio, R.V.; Blow, F.C.; Hays, R.W.; Cassidy, K.A.; Meyer, W.J. Adjunct extended-release valproate semisodium in late life schizophrenia. Int. J. Geriatr. Psychiatry 2008, 23, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Vadodaria, K.C.; Amatya, D.N.; Marchetto, M.C.; Gage, F.H. Modeling psychiatric disorders using patient stem cell-derived neurons: A way forward. Genome Med. 2018, 10, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolmetsch, R.; Geschwind, D.H. The human brain in a dish: The promise of iPSC-derived neurons. Cell 2011, 145, 831–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofer, M.; Lutolf, M.P. Engineering organoids. Nat. Rev. Mater. 2021, 6, 402–420. [Google Scholar] [CrossRef]
- Sheridan, M.A.; Fernando, R.C.; Gardner, L.; Hollinshead, M.S.; Burton, G.J.; Moffett, A.; Turco, M.Y. Establishment and differentiation of long-term trophoblast organoid cultures from the human placenta. Nat. Protoc. 2020, 15, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nayak, R.; Rosh, I.; Kustanovich, I.; Stern, S. Mood Stabilizers in Psychiatric Disorders and Mechanisms Learnt from In Vitro Model Systems. Int. J. Mol. Sci. 2021, 22, 9315. https://doi.org/10.3390/ijms22179315
Nayak R, Rosh I, Kustanovich I, Stern S. Mood Stabilizers in Psychiatric Disorders and Mechanisms Learnt from In Vitro Model Systems. International Journal of Molecular Sciences. 2021; 22(17):9315. https://doi.org/10.3390/ijms22179315
Chicago/Turabian StyleNayak, Ritu, Idan Rosh, Irina Kustanovich, and Shani Stern. 2021. "Mood Stabilizers in Psychiatric Disorders and Mechanisms Learnt from In Vitro Model Systems" International Journal of Molecular Sciences 22, no. 17: 9315. https://doi.org/10.3390/ijms22179315
APA StyleNayak, R., Rosh, I., Kustanovich, I., & Stern, S. (2021). Mood Stabilizers in Psychiatric Disorders and Mechanisms Learnt from In Vitro Model Systems. International Journal of Molecular Sciences, 22(17), 9315. https://doi.org/10.3390/ijms22179315