
COPD, Pulmonary Fibrosis and ILAs in Aging Smokers: The Paradox of Striking Different Responses to the Major Risk Factors



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Chronic Obstructive Pulmonary Disease
3. Interstitial Lung Abnormalities and Combined Pulmonary Fibrosis and Emphysema
4. Idiopathic Pulmonary Fibrosis
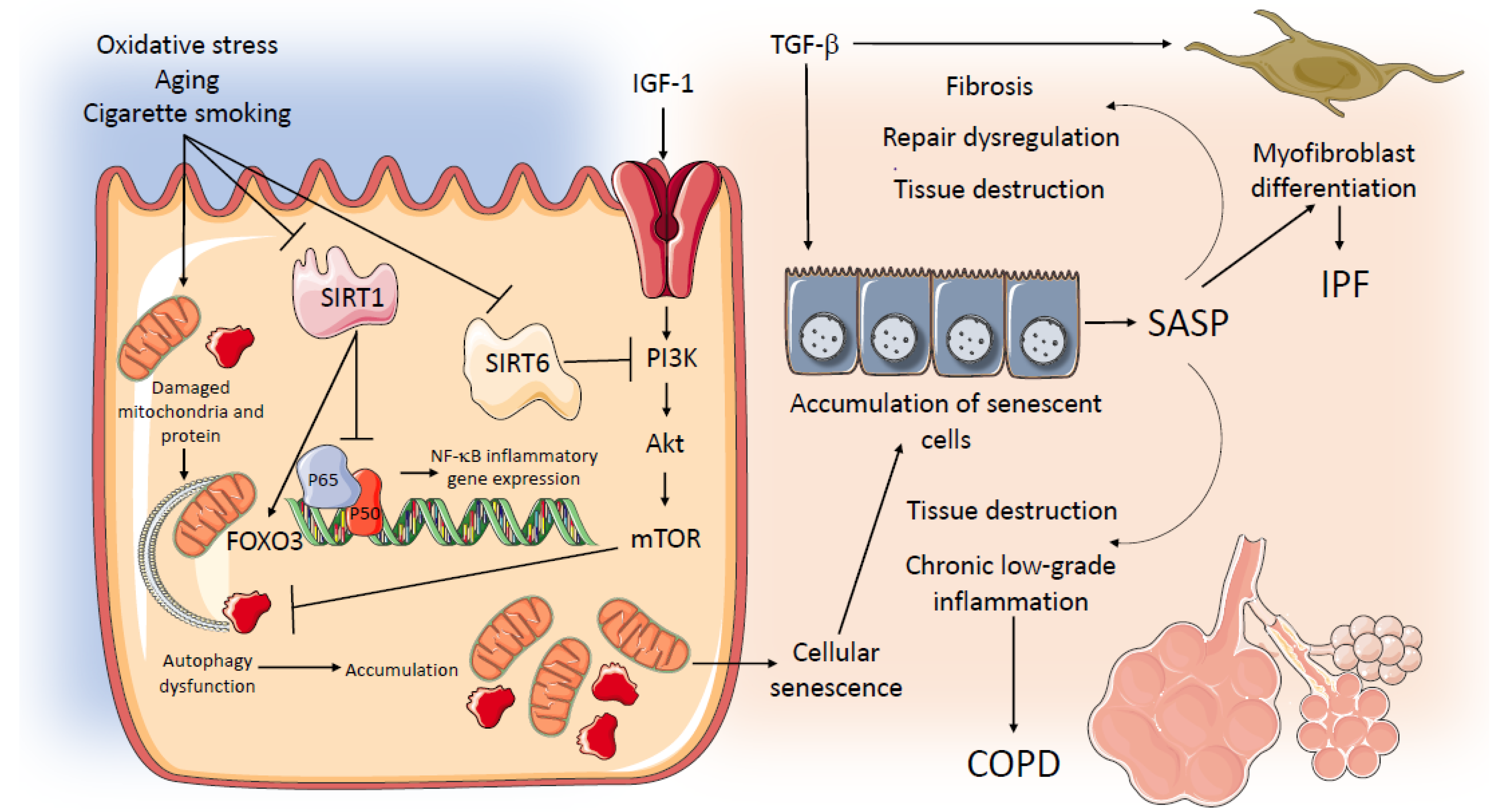
5. Aging-Related Pathways in COPD and IPF
6. Genetic Susceptibility to IPF and COPD
7. Severe Exacerbations of COPD, IPF, and Other Interstitial Lung Diseases
8. Severe Exacerbations of COPD
9. Severe Exacerbations of ILD
10. Exacerbations of CPFE
11. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Washko, G.R.; Hunninghake, G.M.; Fernandez, I.E.; Nishino, M.; Okajima, Y.; Yamashiro, T.; Ross, J.C.; Estepar, R.S.; Lynch, D.A.; Brehm, J.M.; et al. COPDGene Investigators. Lung volumes and emphysema in smokers with interstitial lung abnormalities. N. Engl. J. Med. 2011, 364, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabre, A.; Treacy, A.; Lavelle, L.P.; Narski, M.; Faheem, N.; Healy, D.; Dodd, J.D.; Keane, M.P.; Egan, J.J.; Jebrak, G.; et al. Smoking-Related Interstitial Fibrosis: Evidence of Radiologic Regression with Advancing Age and Smoking Cessation. COPD 2017, 14, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Putman, R.K.; Hatabu, H.; Araki, T.; Gudmundsson, G.; Gao, W.; Nishino, M.; Okajima, Y.; Dupuis, J.; Latourelle, J.C.; Cho, M.H.; et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators; COPDGene Investigators. Association between interstitial lung abnormalities and all-cause mortality. JAMA 2016, 315, 672–681. [Google Scholar] [CrossRef] [Green Version]
- Sauleda, J.; Nunez, B.; Sala, E.; Soriano, J.B. Idiopathic pulmonary fibrosis: Epidemiology, natural history, phenotypes. Med. Sci. 2018, 6, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, L.G.; Loughenbury, M.; Chaudhuri, N.; Spiteri, M.; Parfrey, H. Idiopathic pulmonary fibrosis in the UK: Analysis of the British Thoracic Society electronic registry between 2013 and 2019. ERJ Open Res. 2021, 7, 00187. [Google Scholar] [CrossRef]
- Hatabu, H.; Hunninghake, G.M.; Richeldi, L.; Brown, K.K.; Wells, A.U.; Remy-Jardin, M.; Verschakelen, J.; Nicholson, A.G.; Beasley, M.B.; Christiani, D.C.; et al. Interstitial lung abnormalities detected incidentally on CT: A Position Paper from the Fleischner Society. Lancet Respir. Med. 2020, 8, 726–737. [Google Scholar] [CrossRef]
- Janssens, J.P.; Pache, J.C.; Nicod, L.P. Physiological changes in respiratory function associated with ageing. Eur. Respir. J. 1999, 13, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Lange, P.; Celli, B.; Agusti, A.; Boje Jensen, G.; Divo, M.; Faner, R.; Guerra, S.; Marott, J.L.; Martinez, F.D.; Martinez-Camblor, P.; et al. Lung-function trajectories leading to chronic obstructive pulmonary disease. N. Engl. J. Med. 2015, 373, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Guerra, S.; Martinez, F.D. The complex beginnings of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2020, 201, 641–642. [Google Scholar] [CrossRef]
- Martinez, F.D. Early-life origins of chronic obstructive pulmonary disease. N. Engl. J. Med. 2016, 375, 871–878. [Google Scholar] [CrossRef] [Green Version]
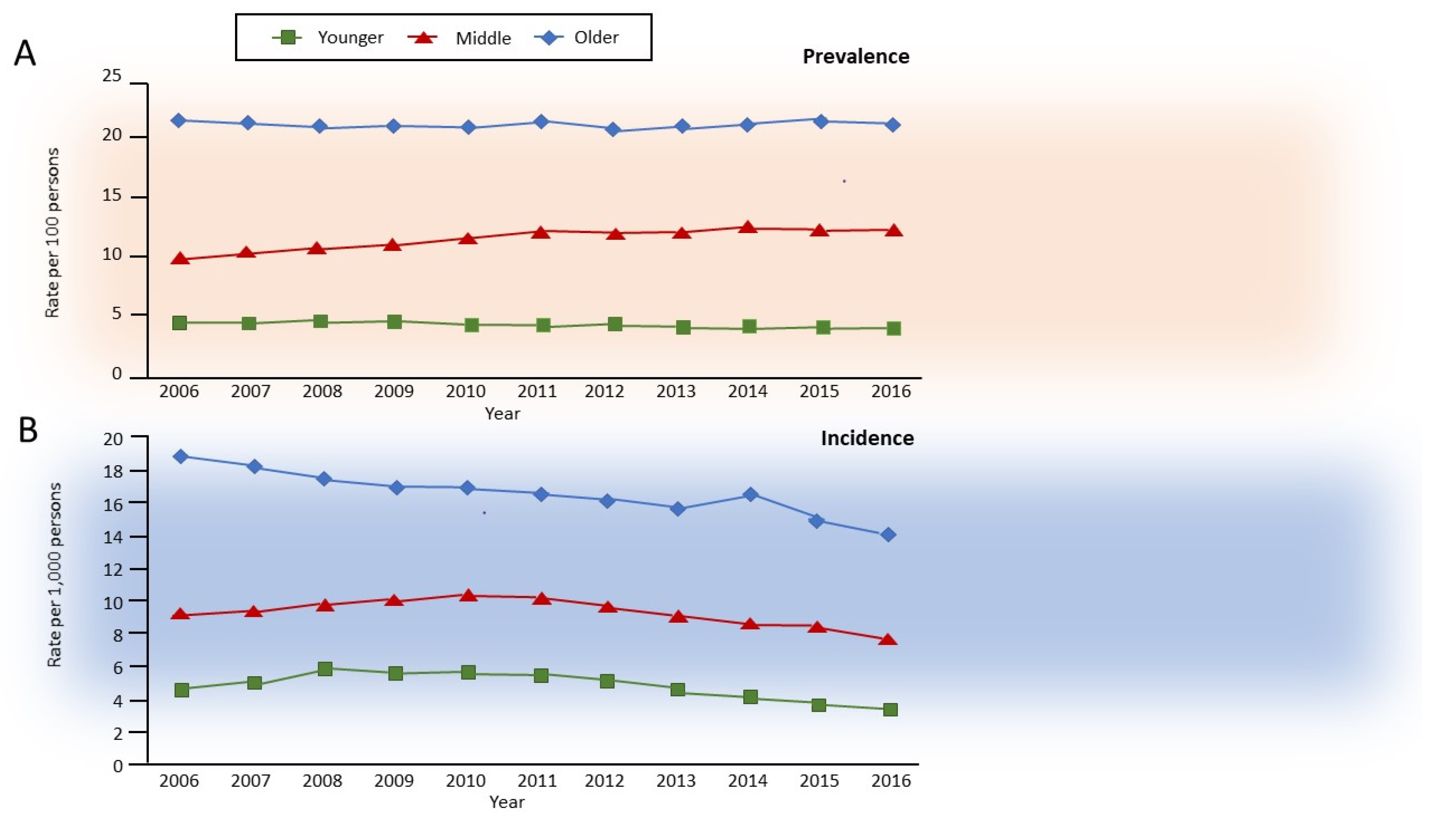
- Gershon, A.S.; McGihon, R.E.; Luo, J.; Blazer, A.J.; Kendzerska, T.; To, T.; Aaron, S.D. Trends in chronic obstructive pulmonary disease prevalence, incidence, and health services use in younger adults in Ontario, Canada, 2006–2016. Am. J. Respir. Crit. Care Med. 2021, 203, 1196–1199. [Google Scholar] [CrossRef]
- 2021 Global Strategy for Prevention, Diagnosis and Management of COPD. Global Initiative of Chronic Obstructive Pulmonary Disease. Available online: www.goldcopd.org (accessed on 28 May 2021).
- MacLeod, M.; Papi, A.; Contoli, M.; Beghe, B.; Celli, B.R.; Wedzicha, J.A.; Fabbri, L.M. Chronic obstructive pulmonary disease exacerbation fundamentals: Diagnosis, treatment, prevention and disease impact. Respirology 2021, 26, 532–551. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.E.; Yuan, R.; Suzuki, M.; Seyednejad, N.; Elliott, W.M.; Sanchez, P.G.; Wright, A.C.; Gefter, W.B.; Litzky, L.; Coxson, H.O.; et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N. Engl. J. Med. 2011, 365, 1567–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Divo, M.; Celli, B.R. Multimorbidity in patients with chronic obstructive pulmonary disease. Clin. Chest Med. 2020, 41, 405–419. [Google Scholar] [CrossRef]
- Cottin, V.; Cordier, J.F. Combined pulmonary fibrosis and emphysema in connective tissue disease. Curr. Opin. Pulm. Med. 2012, 18, 418–427. [Google Scholar] [CrossRef]
- Cottin, V.; Nunes, H.; Brillet, P.Y.; Delaval, P.; Devouassoux, G.; Tillie-Leblond, I.; Israel-Biet, D.; Court-Fortune, I.; Valeyre, D.; Cordier, J.F. Groupe d’Etude et de Recherche sur les Maladies Orphelines, Pulmonaires. Combined pulmonary fibrosis and emphysema: A distinct underrecognised entity. Eur. Respir. J. 2005, 26, 586–593. [Google Scholar] [CrossRef]
- Hage, R.; Gautschi, F.; Steinack, C.; Schuurmans, M.M. Combined Pulmonary Fibrosis and Emphysema (CPFE) Clinical Features and Management. Int. J. Chron. Obstruct. Pulmon. Dis. 2021, 16, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, J.; Strickland, B.; Turner-Warwick, M. Combined cryptogenic fibrosing alveolitis and emphysema: The value of high resolution computed tomography in assessment. Respir. Med. 1990, 84, 365–369. [Google Scholar] [CrossRef]
- Ryerson, C.J.; Hartman, T.; Elicker, B.M.; Ley, B.; Lee, J.S.; Abbritti, M.; Jones, K.D.; King, T.E., Jr.; Ryu, J.; Collard, H.R. Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest 2013, 144, 234–240. [Google Scholar] [CrossRef]
- Shih, A.R.; Nitiwarangkul, C.; Little, B.P.; Roop, B.W.; Nandy, S.; Szabari, M.V.; Mercaldo, N.; Mercaldo, S.; Montesi, S.B.; Muniappan, A.; et al. Practical application and validation of the 2018 ATS/ERS/JRS/ALAT and Fleischner Society guidelines for the diagnosis of idiopathic pulmonary fibrosis. Respir. Res. 2021, 22, 124. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Selman, M.; Martinez, F.J.; Pardo, A. Why does an aging smoker’s lung develop idiopathic pulmonary fibrosis and not chronic obstructive pulmonary disease? Am. J. Respir. Crit. Care Med. 2019, 199, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Luppi, F.; Kalluri, M.; Faverio, P.; Kreuter, M.; Ferrara, G. Idiopathic pulmonary fibrosis beyond the lung: Understanding disease mechanisms to improve diagnosis and management. Respir. Res. 2021, 22, 109. [Google Scholar] [CrossRef] [PubMed]
- Alhamad, E.H.; Cal, J.G.; Alrajhi, N.N.; AlBoukai, A.A. Acute exacerbation in interstitial lung disease. Ann. Thorac. Med. 2021, 16, 178–187. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of idiopathic pulmonary fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Celli, B.R.; Anderson, J.A.; Cowans, N.J.; Crim, C.; Hartley, B.F.; Martinez, F.J.; Morris, A.N.; Quasny, H.; Yates, J.; Vestbo, J.; et al. Pharmacotherapy and lung function decline in patients with chronic obstructive pulmonary disease. A systematic review. Am. J. Respir. Crit. Care Med. 2021, 203, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Lange, P.; Ahmed, E.; Lahmar, Z.M.; Martinez, F.J.; Bourdin, A. Natural history and mechanisms of COPD. Respirology 2021, 26, 298–321. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Lipson, D.A.; Barnhart, F.; Brealey, N.; Brooks, J.; Criner, G.J.; Day, N.C.; Dransfield, M.T.; Halpin, D.M.G.; Han, M.K.; Jones, C.E.; et al. IMPACT Investigators. Once-daily single-inhaler triple versus dual therapy in patients with COPD. N. Engl. J. Med. 2018, 378, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F.; Martinez, F.J.; Ferguson, G.T.; Wang, C.; Singh, D.; Wedzicha, J.A.; Trivedi, R.; St Rose, E.; Ballal, S.; McLaren, J.; et al. Triple inhaled therapy at two glucocorticoid doses in moderate-to-very-severe COPD. N. Engl. J. Med. 2020, 383, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Kardatzke, D.R.; Daigl, M.; Kirchgaessler, K.U.; Lancaster, L.H.; et al. Effect of pirfenidone on mortality: Pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir. Med. 2017, 5, 33–41. [Google Scholar] [CrossRef]
- World Health Organization. Noncommunicable Diseases. Available online: https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases (accessed on 25 August 2021).
- Potter, A.L.; Bajaj, S.S.; Yang, C.J. The 2021 USPSTF lung cancer screening guidelines: A new frontier. Lancet Respir. Med. 2021, 9, 689–691. [Google Scholar] [CrossRef]
- Schneider, J.L.; Rowe, J.H.; Garcia-de-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The aging lung: Physiology, disease, and immunity. Cell 2021, 184, 1990–2019. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Kuwano, K.; Araya, J.; Hara, H.; Minagawa, S.; Takasaka, N.; Ito, S.; Kobayashi, K.; Nakayama, K. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir. Investig. 2016, 54, 397–406. [Google Scholar] [CrossRef]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Romero, Y.; Bueno, M.; Ramirez, R.; Alvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Araya, J.; Takasaka, N.; Fujii, S.; Kojima, J.; Yumino, Y.; Shimizu, K.; Ishikawa, T.; Numata, T.; Kawaishi, M.; et al. Involvement of creatine kinase B in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Respir. Cell Mol. Biol. 2012, 46, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.C.; Welker, L.; Paasch, K.; Feindt, B.; Erpenbeck, V.J.; Hohlfeld, J.M.; Krug, N.; Nakashima, M.; Branscheid, D.; Magnussen, H.; et al. Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respir. Res. 2006, 7, 32. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Alveolar cell senescence in patients with pulmonary emphysema. Am. J. Respir. Crit. Care Med. 2006, 174, 886–893. [Google Scholar] [CrossRef]
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 177, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Bitto, A.; Lerner, C.; Torres, C.; Roell, M.; Malaguti, M.; Perez, V.; Lorenzini, A.; Hrelia, S.; Ikeno, Y.; Matzko, M.E.; et al. Long-term IGF-I exposure decreases autophagy and cell viability. PLoS ONE 2010, 5, e12592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Yoshii, Y.; Yumino, Y.; Fujii, S.; et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Cigarette smoke induces senescence in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, E.H.; Greider, C.W.; Szostak, J.W. Telomeres and telomerase: The path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 2006, 12, 1133–1138. [Google Scholar] [CrossRef]
- Levy, M.Z.; Allsopp, R.C.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere end-replication problem and cell aging. J. Mol. Biol. 1992, 225, 951–960. [Google Scholar] [CrossRef]
- Verdun, R.E.; Karlseder, J. Replication and protection of telomeres. Nature 2007, 447, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Anderson, R.K.; Correia-Melo, C.; Jurk, D.; Hewitt, G.; Marques, F.M.; Green, N.J.; Moisey, E.; Birrell, M.A.; Belvisi, M.G.; et al. DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1124–L1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duckworth, A.; Gibbons, M.A.; Allen, R.J.; Almond, H.; Beaumont, R.N.; Wood, A.R.; Lunnon, K.; Lindsay, M.A.; Wain, L.V.; Tyrrell, J.; et al. Telomere length and risk of idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease: A mendelian randomisation study. Lancet Respir. Med. 2021, 9, 285–294. [Google Scholar] [CrossRef]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [Green Version]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [Green Version]
- Snetselaar, R.; van Batenburg, A.A.; van Oosterhout, M.F.M.; Kazemier, K.M.; Roothaan, S.M.; Peeters, T.; van der Vis, J.J.; Goldschmeding, R.; Grutters, J.C.; van Moorsel, C.H.M. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS ONE 2017, 12, e0189467. [Google Scholar] [CrossRef] [Green Version]
- Houben, J.M.; Mercken, E.M.; Ketelslegers, H.B.; Bast, A.; Wouters, E.F.; Hageman, G.J.; Schols, A.M. Telomere shortening in chronic obstructive pulmonary disease. Respir. Med. 2009, 103, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Savale, L.; Chaouat, A.; Bastuji-Garin, S.; Marcos, E.; Boyer, L.; Maitre, B.; Sarni, M.; Housset, B.; Weitzenblum, E.; Matrat, M.; et al. Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2009, 179, 566–571. [Google Scholar] [CrossRef]
- Cordoba-Lanus, E.; Cazorla-Rivero, S.; Garcia-Bello, M.A.; Mayato, D.; Gonzalvo, F.; Ayra-Plasencia, J.; Celli, B.; Casanova, C. Telomere length dynamics over 10-years and related outcomes in patients with COPD. Respir. Res. 2021, 22, 56. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Sandford, A.J.; Connett, J.E.; Yan, J.; Mui, T.; Li, Y.; Daley, D.; Anthonisen, N.R.; Brooks-Wilson, A.; Man, S.F.; et al. The relationship between telomere length and mortality in chronic obstructive pulmonary disease (COPD). PLoS ONE 2012, 7, e35567. [Google Scholar] [CrossRef]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Fingerlin, T.E.; Zhang, W.; Yang, I.V.; Ainsworth, H.C.; Russell, P.H.; Blumhagen, R.Z.; Schwarz, M.I.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; et al. Genome-wide imputation study identifies novel HLA locus for pulmonary fibrosis and potential role for auto-immunity in fibrotic idiopathic interstitial pneumonia. BMC Genet. 2016, 17, 74. [Google Scholar] [CrossRef] [Green Version]
- Mushiroda, T.; Wattanapokayakit, S.; Takahashi, A.; Nukiwa, T.; Kudoh, S.; Ogura, T.; Taniguchi, H.; Kubo, M.; Kamatani, N.; Nakamura, Y. Pirfenidone Clinical Study Group. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J. Med. Genet. 2008, 45, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.F.; Garcia, J.G.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.E.; et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.J.W.; Renzoni, E.A. Telomeres in Interstitial Lung Disease. J. Clin. Med. 2021, 10, 1384. [Google Scholar] [CrossRef]
- Allen, R.J.; Guillen-Guio, B.; Oldham, J.M.; Ma, S.F.; Dressen, A.; Paynton, M.L.; Kraven, L.M.; Obeidat, M.; Li, X.; Ng, M.; et al. Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Das, F.; Bera, A.; Ghosh-Choudhury, N.; Abboud, H.E.; Kasinath, B.S.; Choudhury, G.G. TGFbeta-induced deptor suppression recruits mTORC1 and not mTORC2 to enhance collagen I (alpha2) gene expression. PLoS ONE 2014, 9, e109608. [Google Scholar] [CrossRef] [PubMed]
- Coe, B.P.; Lee, E.H.; Chi, B.; Girard, L.; Minna, J.D.; Gazdar, A.F.; Lam, S.; MacAulay, C.; Lam, W.L. Gain of a region on 7p22.3, containing MAD1L1, is the most frequent event in small-cell lung cancer cell lines. Genes Chromosomes Cancer 2006, 45, 11–19. [Google Scholar] [CrossRef]
- Hawkins, G.A.; Mora, A.L. FAM13A, A fatty acid oxidation switch in mitochondria. Friend or foe in chronic obstructive pulmonary disease pathogenesis? Am. J. Respir. Cell Mol. Biol. 2017, 56, 689–691. [Google Scholar] [CrossRef]
- Silverman, E.K. Genetics of COPD. Annu. Rev. Physiol. 2020, 82, 413–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakornsakolpat, P.; Prokopenko, D.; Lamontagne, M.; Reeve, N.F.; Guyatt, A.L.; Jackson, V.E.; Shrine, N.; Qiao, D.; Bartz, T.M.; Kim, D.K.; et al. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat. Genet. 2019, 51, 494–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, B.D.; de Jong, K.; Lamontagne, M.; Bosse, Y.; Shrine, N.; Artigas, M.S.; Wain, L.V.; Hall, I.P.; Jackson, V.E.; Wyss, A.B.; et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat. Genet. 2017, 49, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, A.I.; Wedzicha, J.A. Definition, Causes, Pathogenesis, and Consequences of Chronic Obstructive Pulmonary Disease Exacerbations. Clin. Chest Med. 2020, 41, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Mathioudakis, A.G.; Janssens, W.; Sivapalan, P.; Singanayagam, A.; Dransfield, M.T.; Jensen, J.S.; Vestbo, J. Acute exacerbations of chronic obstructive pulmonary disease: In search of diagnostic biomarkers and treatable traits. Thorax 2020, 75, 520–527. [Google Scholar] [CrossRef] [Green Version]
- Sivapalan, P.; Lapperre, T.S.; Janner, J.; Laub, R.R.; Moberg, M.; Bech, C.S.; Eklof, J.; Holm, F.S.; Armbruster, K.; Sivapalan, P.; et al. Eosinophil-guided corticosteroid therapy in patients admitted to hospital with COPD exacerbation (CORTICO-COP): A multicentre, randomised, controlled, open-label, non-inferiority trial. Lancet Respir. Med. 2019, 7, 699–709. [Google Scholar] [CrossRef]
- Vedel-Krogh, S.; Nielsen, S.F.; Lange, P.; Vestbo, J.; Nordestgaard, B.G. Blood eosinophils and exacerbations in chronic obstructive pulmonary disease. The Copenhagen general population study. Am. J. Respir. Crit. Care Med. 2016, 193, 965–974. [Google Scholar] [CrossRef]
- Keir, H.R.; Dicker, A.; Lonergan, M.; Crichton, M.; Miller, B.E.; Tal-Singer, R.; Chalmers, J.D. Clinical endotypes of exacerbation are associated with differences in microbial composition and diversity in COPD. Eur. Respir. J. 2020, 56, 2000391. [Google Scholar] [CrossRef] [PubMed]
- Vogelmeier, C.F.; Criner, G.J.; Martinez, F.J.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Chen, R.; Decramer, M.; Fabbri, L.M.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report. GOLD Executive Summary. Am. J. Respir. Crit. Care Med. 2017, 195, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Kurai, D.; Saraya, T.; Ishii, H.; Takizawa, H. Virus-induced exacerbations in asthma and COPD. Front. Microbiol. 2013, 4, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, A.C.; Parsons, K.; Moheimani, F.; Knight, D.A.; Hansbro, P.M.; Fujita, T.; Wark, P.A. Impaired antiviral stress granule and IFN-beta enhanceosome formation enhances susceptibility to influenza infection in chronic obstructive pulmonary disease epithelium. Am. J. Respir. Cell Mol. Biol. 2016, 55, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Berenson, C.S.; Kruzel, R.L.; Eberhardt, E.; Dolnick, R.; Minderman, H.; Wallace, P.K.; Sethi, S. Impaired innate immune alveolar macrophage response and the predilection for COPD exacerbations. Thorax 2014, 69, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.; Wang, K.; Zhang, W.; Xiao, K.; Yan, P.; Li, Y.; He, W.; Zhang, Y.; Xie, L. Immunodeficiency in Patients with Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Inflammation 2018, 41, 1582–1589. [Google Scholar] [CrossRef]
- Abe, Y.; Murphy, T.F.; Sethi, S.; Faden, H.S.; Dmochowski, J.; Harabuchi, Y.; Thanavala, Y.M. Lymphocyte proliferative response to P6 of Haemophilus influenzae is associated with relative protection from exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2002, 165, 967–971. [Google Scholar] [CrossRef]
- Sharma, A.; Rudra, D. Emerging Functions of Regulatory T Cells in Tissue Homeostasis. Front. Immunol. 2018, 9, 883. [Google Scholar] [CrossRef] [PubMed]
- Kalathil, S.G.; Lugade, A.A.; Pradhan, V.; Miller, A.; Parameswaran, G.I.; Sethi, S.; Thanavala, Y. T-regulatory cells and programmed death 1+ T cells contribute to effector T-cell dysfunction in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2014, 190, 40–50. [Google Scholar] [CrossRef] [Green Version]
- McKendry, R.T.; Spalluto, C.M.; Burke, H.; Nicholas, B.; Cellura, D.; Al-Shamkhani, A.; Staples, K.J.; Wilkinson, T.M. Dysregulation of antiviral function of CD8(+) T cells in the chronic obstructive pulmonary disease lung. Role of the PD-1-PD-L1 axis. Am. J. Respir. Crit. Care Med. 2016, 193, 642–651. [Google Scholar] [CrossRef] [Green Version]
- Agusti, A.; Edwards, L.D.; Rennard, S.I.; MacNee, W.; Tal-Singer, R.; Miller, B.E.; Vestbo, J.; Lomas, D.A.; Calverley, P.M.; Wouters, E.; et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: A novel phenotype. PLoS ONE 2012, 7, e37483. [Google Scholar] [CrossRef]
- Kunisaki, K.M.; Dransfield, M.T.; Anderson, J.A.; Brook, R.D.; Calverley, P.M.A.; Celli, B.R.; Crim, C.; Hartley, B.F.; Martinez, F.J.; Newby, D.E.; et al. Exacerbations of chronic obstructive pulmonary disease and cardiac events. A Post Hoc Cohort Analysis from the SUMMIT Randomized Clinical Trial. Am. J. Respir. Crit. Care Med. 2018, 198, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Hillas, G.; Perlikos, F.; Tzanakis, N. Acute exacerbation of COPD: Is it the “stroke of the lungs”? Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 1579–1586. [Google Scholar] [PubMed] [Green Version]
- MacIntyre, N.; Huang, Y.C. Acute exacerbations and respiratory failure in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2008, 5, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Marchioni, A.; Tonelli, R.; Fantini, R.; Tabbi, L.; Castaniere, I.; Livrieri, F.; Bedogni, S.; Ruggieri, V.; Pisani, L.; Nava, S.; et al. Respiratory mechanics and diaphragmatic dysfunction in COPD patients who failed non-invasive mechanical ventilation. Int. J. Chron. Obstruct. Pulmon. Dis. 2019, 14, 2575–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watz, H.; Waschki, B.; Meyer, T.; Kretschmar, G.; Kirsten, A.; Claussen, M.; Magnussen, H. Decreasing cardiac chamber sizes and associated heart dysfunction in COPD: Role of hyperinflation. Chest 2010, 138, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Wedzicha, J.A.E.C.-C.; Miravitlles, M.; Hurst, J.R.; Calverley, P.M.; Albert, R.K.; Anzueto, A.; Criner, G.J.; Papi, A.; Rabe, K.F.; Rigau, D.; et al. Management of COPD exacerbations: A European Respiratory Society/American Thoracic Society guideline. Eur. Respir. J. 2017, 49, 1600791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochwerg, B.; Brochard, L.; Elliott, M.W.; Hess, D.; Hill, N.S.; Nava, S.; Navalesi, P.M.O.T.S.C.; Antonelli, M.; Brozek, J.; Conti, G.; et al. Official ERS/ATS clinical practice guidelines: Noninvasive ventilation for acute respiratory failure. Eur. Respir. J. 2017, 50, 1602426. [Google Scholar] [CrossRef] [PubMed]
- Conti, G.; Antonelli, M.; Navalesi, P.; Rocco, M.; Bufi, M.; Spadetta, G.; Meduri, G.U. Noninvasive vs. conventional mechanical ventilation in patients with chronic obstructive pulmonary disease after failure of medical treatment in the ward: A randomized trial. Intensive Care Med. 2002, 28, 1701–1707. [Google Scholar] [CrossRef]
- Kolb, M.; Bondue, B.; Pesci, A.; Miyazaki, Y.; Song, J.W.; Bhatt, N.Y.; Huggins, J.T.; Oldham, J.M.; Padilla, M.L.; Roman, J.; et al. Acute exacerbations of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180071. [Google Scholar] [CrossRef]
- Invernizzi, R.; Molyneaux, P.L. The contribution of infection and the respiratory microbiome in acute exacerbations of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2019, 28, 190045. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.J.; Wells, A.U.; Kim, H.C.; Ji, W.; Cookson, W.O.; Moffatt, M.F.; Kim, D.S.; Maher, T.M. Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am. J. Respir. Crit. Care Med 2016, 194, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Marchioni, A.; Tonelli, R.; Ball, L.; Fantini, R.; Castaniere, I.; Cerri, S.; Luppi, F.; Malerba, M.; Pelosi, P.; Clini, E. Acute exacerbation of idiopathic pulmonary fibrosis: Lessons learned from acute respiratory distress syndrome? Crit. Care 2018, 22, 80. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.S.; Park, J.H.; Park, B.K.; Lee, J.S.; Nicholson, A.G.; Colby, T. Acute exacerbation of idiopathic pulmonary fibrosis: Frequency and clinical features. Eur. Respir. J. 2006, 27, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moye, S.; Bormann, T.; Maus, R.; Sparwasser, T.; Sandrock, I.; Prinz, I.; Warnecke, G.; Welte, T.; Gauldie, J.; Kolb, M.; et al. Regulatory T cells limit pneumococcus-induced exacerbation of lung fibrosis in mice. J. Immunol. 2020, 204, 2429–2438. [Google Scholar] [CrossRef] [PubMed]
- Marchioni, A.; Tonelli, R.; Rossi, G.; Spagnolo, P.; Luppi, F.; Cerri, S.; Cocconcelli, E.; Pellegrino, M.R.; Fantini, R.; Tabbi, L.; et al. Ventilatory support and mechanical properties of the fibrotic lung acting as a “squishy ball”. Ann. Intensive Care 2020, 10, 13. [Google Scholar] [CrossRef]
- Farrand, E.; Vittinghoff, E.; Ley, B.; Butte, A.J.; Collard, H.R. Corticosteroid use is not associated with improved outcomes in acute exacerbation of IPF. Respirology 2020, 25, 629–635. [Google Scholar] [CrossRef]
- Woodworth, T.G.; Suliman, Y.A.; Li, W.; Furst, D.E.; Clements, P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat. Rev. Nephrol. 2016, 12, 678–691. [Google Scholar] [CrossRef]
- Cottin, V.; Crestani, B.; Valeyre, D.; Wallaert, B.; Cadranel, J.; Dalphin, J.C.; Delaval, P.; Israel-Biet, D.; Kessler, R.; Reynaud-Gaubert, M.; et al. French National Reference Centre; Network of Competence Centres for Rare Lung Diseases. Diagnosis and management of idiopathic pulmonary fibrosis: French practical guidelines. Eur. Respir. Rev. 2014, 23, 193–214. [Google Scholar] [CrossRef] [Green Version]
- Tamm, A.M.; Kremens, K. Rituximab for Salvage Therapy of Refractory Hypersensitivity Pneumonitis. WMJ 2019, 118, 95–97. [Google Scholar] [PubMed]
- Mathai, S.C.; Danoff, S.K. Management of interstitial lung disease associated with connective tissue disease. BMJ 2016, 352, h6819. [Google Scholar] [CrossRef] [PubMed]
- Vianello, A.; Arcaro, G.; Molena, B.; Turato, C.; Braccioni, F.; Paladini, L.; Vio, S.; Ferrarese, S.; Peditto, P.; Gallan, F.; et al. High-flow nasal cannula oxygen therapy to treat acute respiratory failure in patients with acute exacerbation of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2019, 13, 1753466619847130. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Perez, E.R.; Yilmaz, M.; Jenad, H.; Daniels, C.E.; Ryu, J.H.; Hubmayr, R.D.; Gajic, O. Ventilator settings and outcome of respiratory failure in chronic interstitial lung disease. Chest 2008, 133, 1113–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zantah, M.; Dotan, Y.; Dass, C.; Zhao, H.; Marchetti, N.; Criner, G.J. Acute exacerbations of COPD versus IPF in patients with combined pulmonary fibrosis and emphysema. Respir. Res. 2020, 21, 164. [Google Scholar] [CrossRef] [PubMed]
- Ikuyama, Y.; Ushiki, A.; Kosaka, M.; Akahane, J.; Mukai, Y.; Araki, T.; Kitaguchi, Y.; Tateishi, K.; Urushihata, K.; Yasuo, M.; et al. Prognosis of patients with acute exacerbation of combined pulmonary fibrosis and emphysema: A retrospective single-centre study. BMC Pulm. Med. 2020, 20, 144. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beghé, B.; Cerri, S.; Fabbri, L.M.; Marchioni, A. COPD, Pulmonary Fibrosis and ILAs in Aging Smokers: The Paradox of Striking Different Responses to the Major Risk Factors. Int. J. Mol. Sci. 2021, 22, 9292. https://doi.org/10.3390/ijms22179292
Beghé B, Cerri S, Fabbri LM, Marchioni A. COPD, Pulmonary Fibrosis and ILAs in Aging Smokers: The Paradox of Striking Different Responses to the Major Risk Factors. International Journal of Molecular Sciences. 2021; 22(17):9292. https://doi.org/10.3390/ijms22179292
Chicago/Turabian StyleBeghé, Bianca, Stefania Cerri, Leonardo M. Fabbri, and Alessandro Marchioni. 2021. "COPD, Pulmonary Fibrosis and ILAs in Aging Smokers: The Paradox of Striking Different Responses to the Major Risk Factors" International Journal of Molecular Sciences 22, no. 17: 9292. https://doi.org/10.3390/ijms22179292
APA StyleBeghé, B., Cerri, S., Fabbri, L. M., & Marchioni, A. (2021). COPD, Pulmonary Fibrosis and ILAs in Aging Smokers: The Paradox of Striking Different Responses to the Major Risk Factors. International Journal of Molecular Sciences, 22(17), 9292. https://doi.org/10.3390/ijms22179292