
The PPARg System in Major Depression: Pathophysiologic and Therapeutic Implications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Organization of the Normal Stress System: Template for Depressive Illness
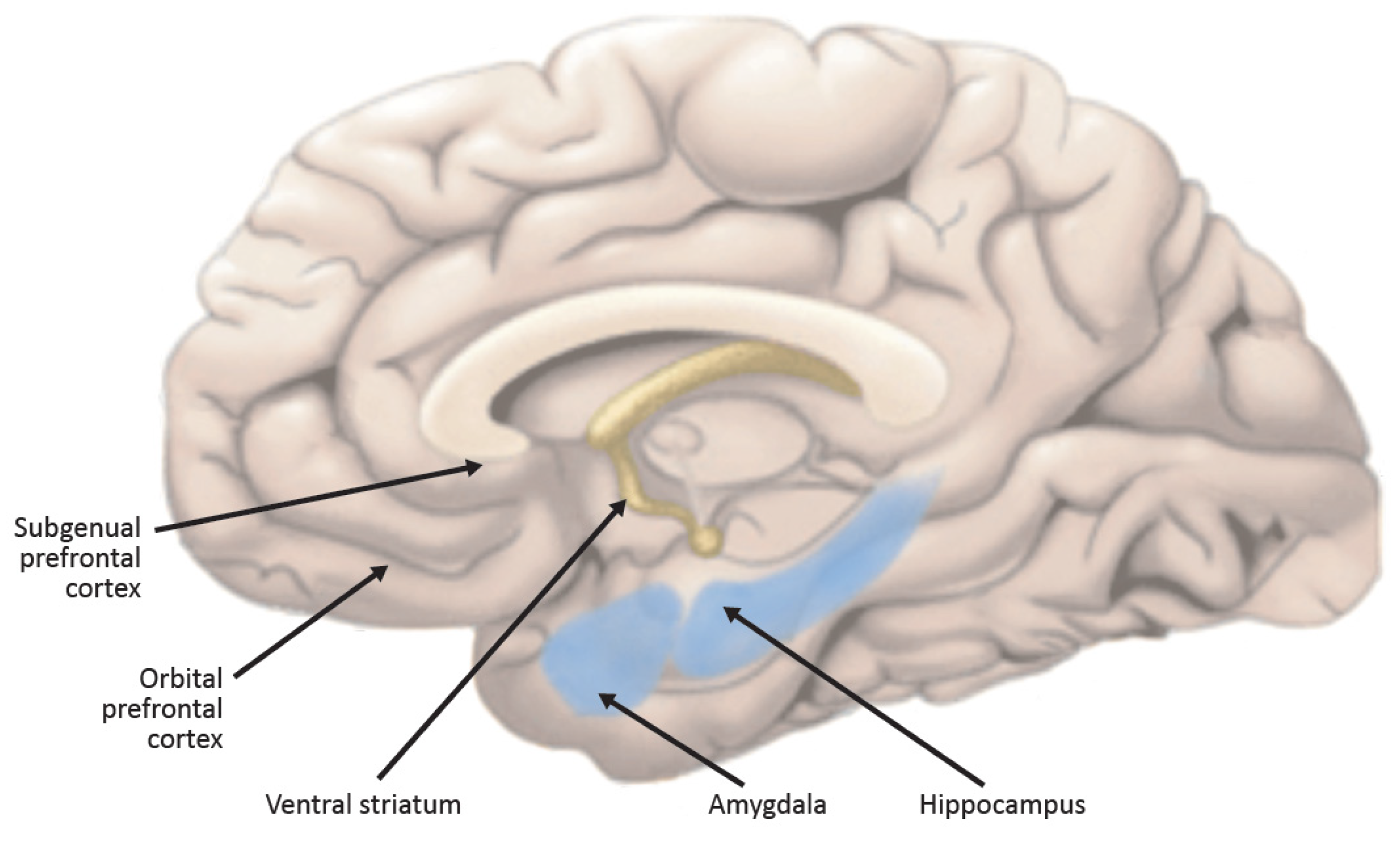
3. Behavioral and Cognitive Manifestations of Melancholic Depression
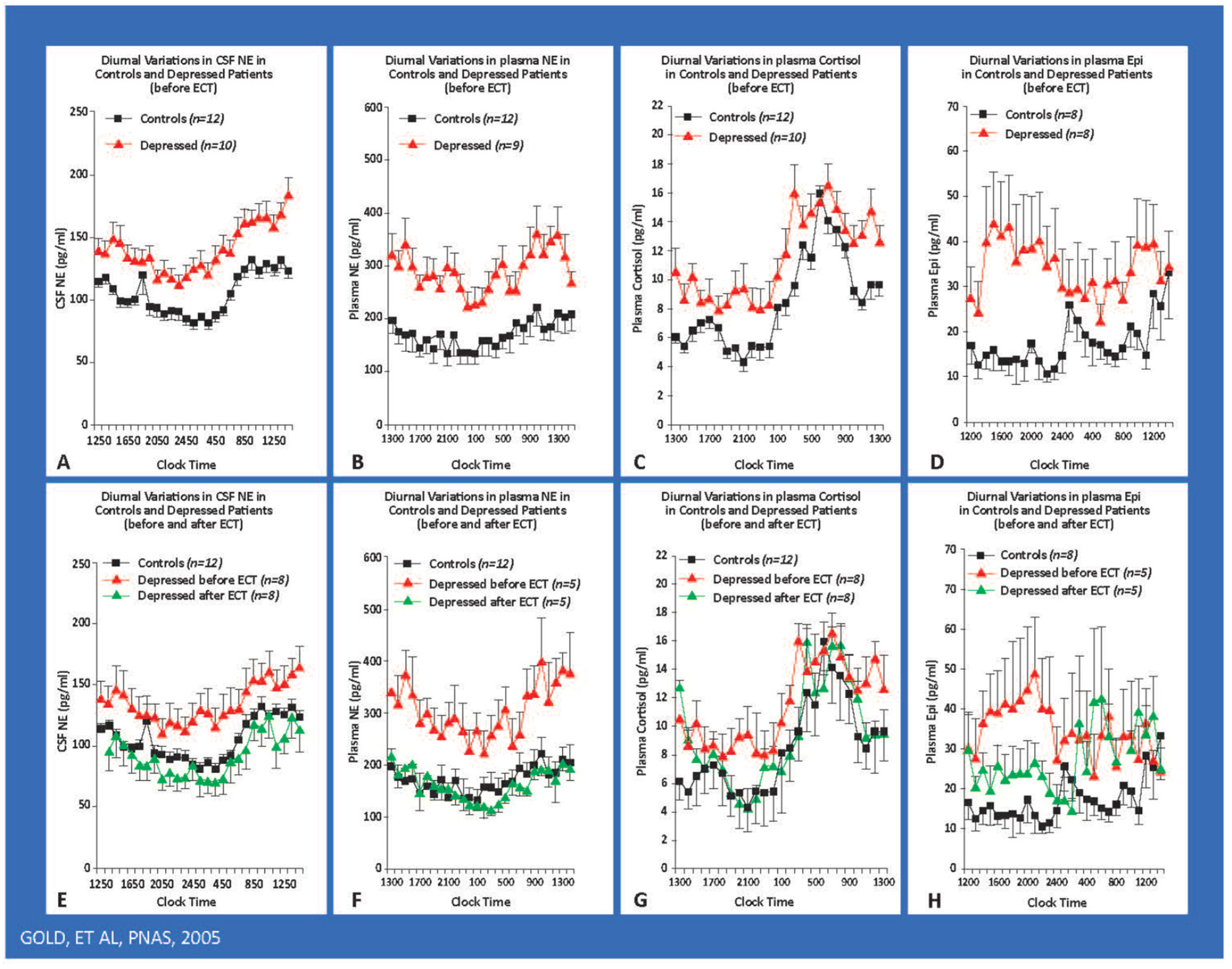
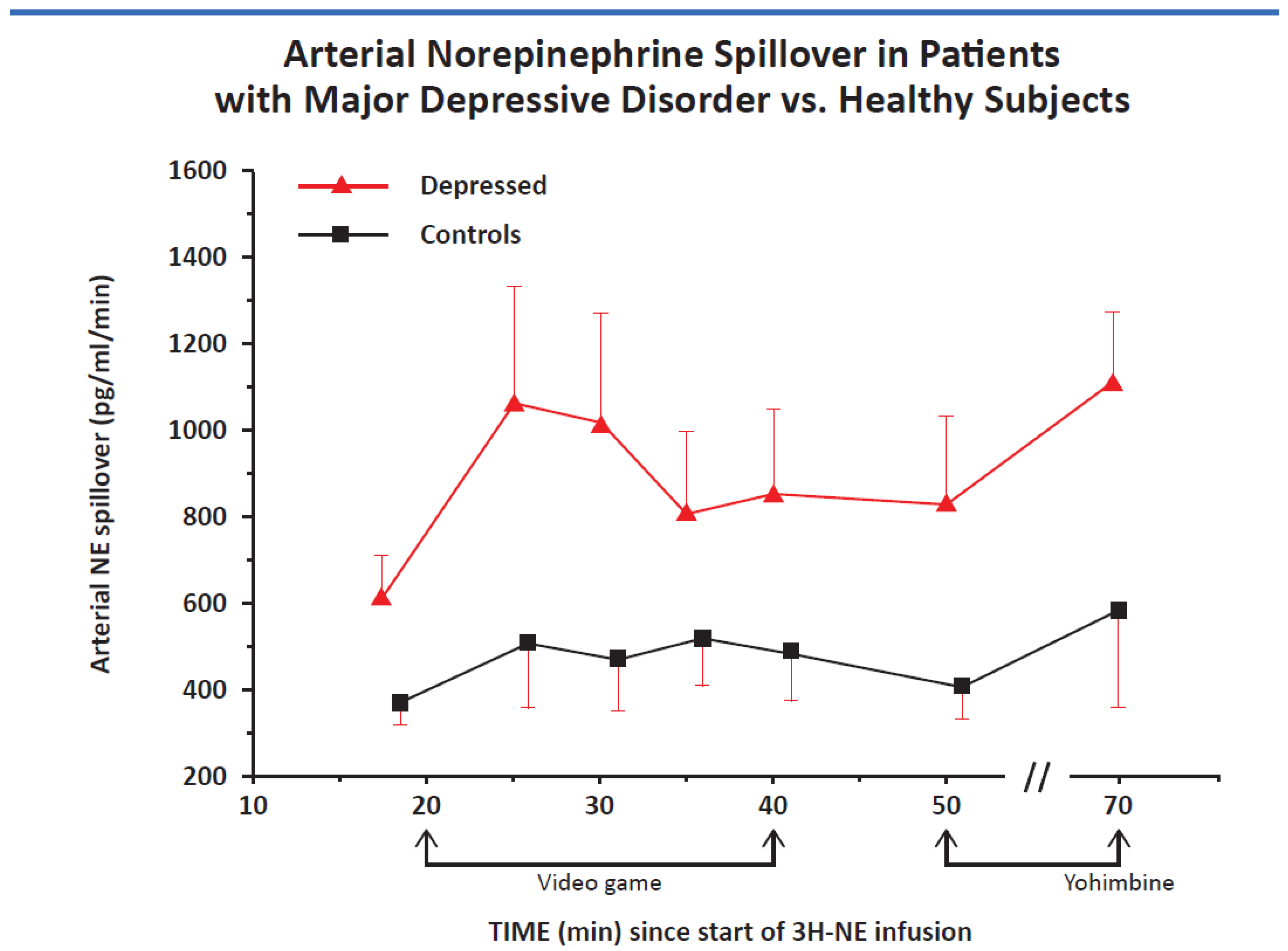
4. Dysregulation of the Stress System in Melancholic Depression: Evidence for a Pathological Activation of the Stress System
5. Inflammation in Depression
6. Manifestation of Inflammation in Patients with Depressive Illness
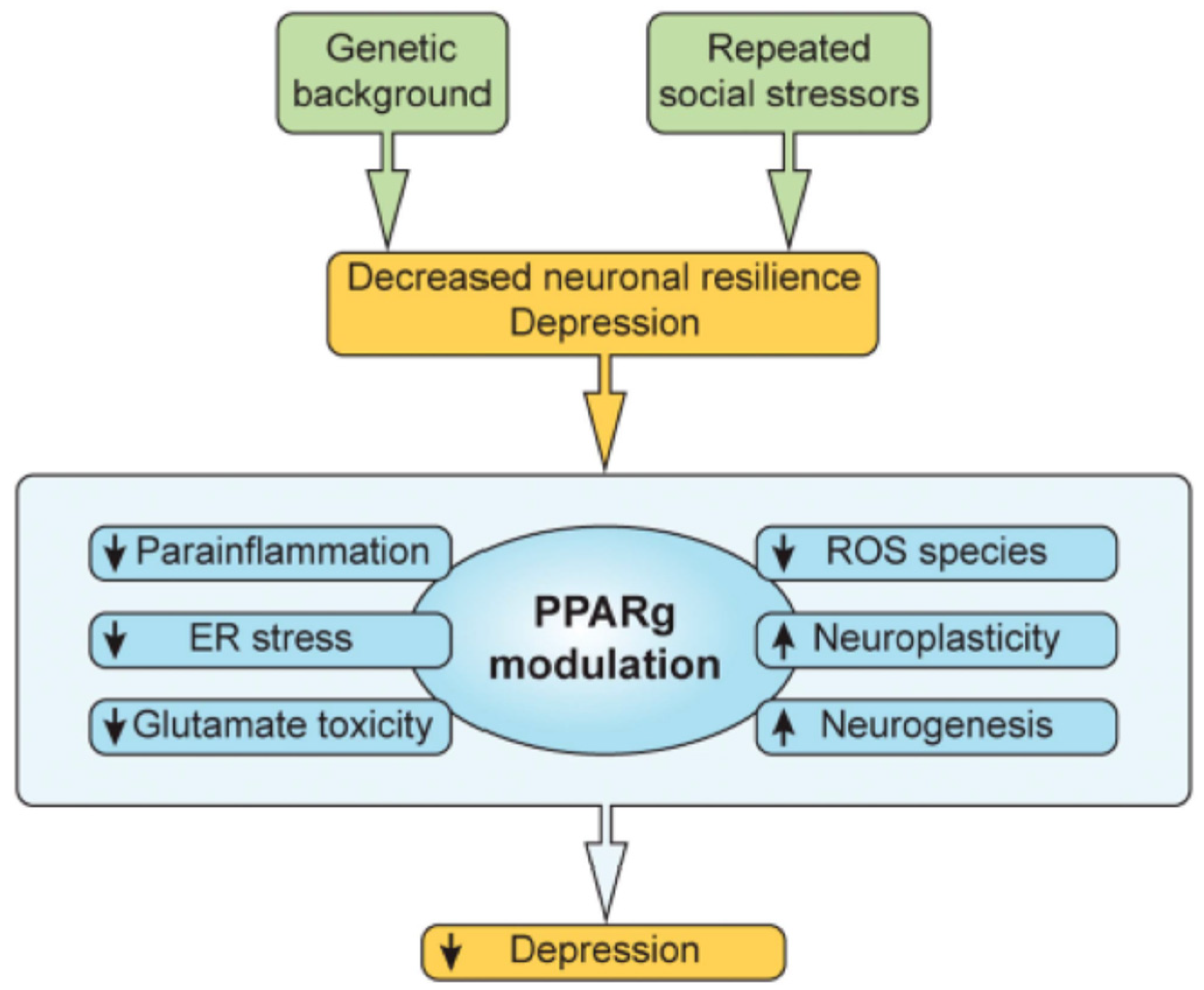
7. More on the PPARg System
Funding
Conflicts of Interest
References
- Barefoot, J.C.; Schroll, M. Symptoms of depression, acute myocardial infarction, and total mortality in a community sample. Circulation 1996, 93, 1976–1980. [Google Scholar] [CrossRef]
- Ford, C.D.; Gray, M.S.; Crowther, M.R.; Wadley, V.G.; Austin, A.L.; Crowe, M.G.; Pulley, L.; Unverzagt, F.; Kleindorfer, D.O.; Kissela, B.M.; et al. Depressive symptoms and risk of stroke in a national cohort of blacks and whites from RE-GARDS. Neurol. Clin. Pract. 2020, 6, 108–112. [Google Scholar]
- Silva, N.D.M.L.E.; Lam, M.P.; Soares, C.N.; Munoz, D.P.; Milev, R.; De Felice, F.G. Insulin Resistance as a Shared Pathogenic Mechanism Between Depression and Type 2 Diabetes. Front. Psychiatry 2019, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Knol, M.J.; Twisk, J.W.R.; Beekman, A.T.F.; Heine, R.J.; Snoek, F.J.; Pouwer, F. Depression as a risk factor for the onset of type II diabetes. Diabetologia 2005, 49, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Michelson, D.; Stratakis, C.; Hill, L.; Reynolds, J.; Galliven, E.; Chrousos, G.; Gold, P. Bone mineral density in women with depression. N. Engl. J. Med. 1996, 335, 1176–1181. [Google Scholar] [CrossRef] [PubMed]
- Whooley, M.A.; de Jonge, P.; Vittinghoff, E.; Otte, C.; Moos, R. Depressive symptoms, health behaviors, and risk of cardiovascular events in patients with coronary heart disease. JAMA 2008, 3, 2379–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, P.W.; Goodwin, F.K.; Chrousos, G.P. Clinical and biochemical manifestations of depression. Relation to the neurobiology of stress (2). N. Engl. J. Med. 1988, 319, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Gold, P.W.; Wong, M.-L. Re-assessing the catecholamine hypothesis of depression: The case of melancholic depression. Mol. Psychiatry 2021, 1–4. [Google Scholar] [CrossRef]
- Gold, P.W. The organization of the stress system and its dysregulation in depressive illness. Mol. Psychiatry 2015, 20, 32–47. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.L.; Kling, M.A.; Munson, P.J.; Listwak, S.; Licinio, J.; Karp, B.; McCutcheon, E.; Geracioti, T.; DeBellis, M.; Oldfield, E.; et al. Pronounced and sustained central hypernoradrenergic function in major depression with melancholic features: Relation to hypercortisolism and corticotropin-releasing hormone. Proc. Natl. Acad. Sci. USA 2000, 97, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Gold, P.W.; Loriaux, D.L.; Roy, A.; Kling, M.A.; Calabrese, J.; Kellner, C.; Post, R.M.; Gold, P.W. Responses to corticotropin-releasing hormone in the hypercortisolism of depression and Cushing’s disease. Pathophysiologic and diagnostic implications. N. Engl. J. Med. 1986, 314, 1329–1335. [Google Scholar] [CrossRef]
- Gold, P.W.; Licinio, J.; Pavlatou, M.G. Pathological parainflammation and endoplasmic reticulum stress in depression: Potential translational targets through the insulin, klotho and PPAR-gamma systems. Mol. Psychiatry 2013, 18, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, E.M.; Chrousos, G.P.; Wilder, R.L.; Gold, P.W. The Stress Response and the Regulation of Inflammatory Disease. Ann. Intern. Med. 1992, 117, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Spitz, R.A. Hospitalism: An inquiry into the genesis of psychiatric illness in early childhood. Psychoanal. Study Child 1945, 1, 53–74. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, L.; Yang, S.; Qin, D.; Wang, J.; Li, C.; Lv, L.; Ma, Y.; Hu, X. Maternal separation produces long-lasting changes in cortisol and behavior in rhesus monkeys. Proc. Natl. Acad. Med. USA 2010, 108, 14312–14317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villapol, S. Roles of Peroxisome Proliferator-Activated Receptor Gamma on Brain and Peripheral Inflammation. Cell Mol. Neurobiol. 2018, 38, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Domi, E.; Uhrig, S.; Soverchia, L.; Hansen, A.; Barbier, E.; Heilig, M.; Ubaldi, M. Genetic Deletion of Neuronal PPARgamma Enhances the Emotional Response to Acute Stress Exacerbates Anxiety: An Effect Reversed by Rescue of Amygdala PPARgamma Function. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 12611–12623. [Google Scholar] [CrossRef]
- Warden, A.T.J.; Merriman, M.; Truitt, J.; Pomomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfild, R.D.; Harris, R.A. Localization of PPARg isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- García-Bueno, B.; Madrigal, J.L.; Pérez-Nievas, B.G.; Leza, J.C. Stress mediators regulate brain prostaglandin synthesis and peroxisome proliferator-activated receptor-γ activation after stress in rats. Endocrinology 2008, 149, 1969–1978. [Google Scholar] [CrossRef] [Green Version]
- Gold, P.W.; Wong, M.L.; Goldstein, D.S.; Gold, H.K.; Ronsaville, D.S.; Esler, M.; Alesci, S.; Masood, A.; Licinio, J.; Geracioti, T.D.; et al. Cardiac implications of increased arterial entry and reversible 24-h central and peripheral norepinephrine levels in melancholia. Proc. Natl. Acad. Sci. USA 2005, 102, 8303–8308. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, T.; Koeda, M.; Okubo, Y.; Kimura, M. Hypofunction of left dorsolateral prefrontal cortex in depression during verbal fluency task: A multi-channel near-infrared spectroscopy study. J. Affect. Disord. 2018, 231, 83–90. [Google Scholar] [CrossRef]
- Simpson, D.W.C., Jr.; Snyder, A.Z.; Gusnard, D.A.; Raichle, M.E. Emotion-induced changes in human medial prefrontal cortex: II. During anticipatory anxiety. Proc. Nat. Acad. Sci. USA 2001, 98, 688–693. [Google Scholar] [CrossRef] [Green Version]
- Roozendaal, B.; McEwen, B.S.; Chattarji, S. Stress, memory and the amygdala. Nat. Rev. Neurosci. 2009, 10, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Roozendaal, B.; Barsegyan, A.; Lee, S. Adrenal stress hormones, amygdala activation, and memory for emotionally arousing experiences. Prog. Brain Res. 2008, 167, 79–97. [Google Scholar]
- Brady, L.S.; Whitfield, H.J., Jr.; Fox, R.J.; Gold, P.W.; Herkenham, M. Long-term antidepressant administration alters corticotropin-releasing hormone, tyrosine hydroxylase, and mineralocorticoid receptor gene expression in rat brain. Therapeutic implications. J. Clin. Investig. 1991, 87, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Gold, P.W. Endocrine Factors in Key Structural and Intracellular Changes in Depression. Trends Endocrinol. Metab. 2021, 32, 212–223. [Google Scholar] [CrossRef]
- Berger, J.P.H.; Woods, J.; Hayes, N.S.; Parent, S.A.; Clemas, J.; Leibowitz, M.D.; Ebrecht, A.; Rachubinski, R.A.; Capone, J.P.; Moller, D.E. A PPARg mutant serves as a dominant negative inhibitor of PPARg signaling and is localized in the nucleus. Mol. Cell Endocrinol. 2000, 162, 57–67. [Google Scholar] [CrossRef]
- Pizzagalli, D.A.; Holmes, A.J.; Dillon, D.G.; Goetzz, E.L.; Birk, J.; Bogdan, R.; Dugherty, D.D.; Iosefescu, D.V.; Rouch, S.L.; Fava, M. Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am. J. Psychiatry 2009, 166, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauczyciel, C.; Robic, S.; Dondaine, T. The nucleus accumbens: A target for deep brain stimulation in resistant major depressive disorder. J. Mol. Psychiatry 2013, 1, 17. [Google Scholar] [CrossRef] [Green Version]
- Ulrich-Lai, Y.M.; Ryan, K.K. PPARgamma and stress: Implications for aging. Exp. Gerontol. 2013, 48, 671–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drevets, W.C.; Price, J.L.; Simpson, J.R., Jr.; Todd, R.D.; Reich, T.; Vannier, M.; Raichle, M.E. Subgenual prefrontal cortex abnormalities in mood disorders. Nature 1997, 386, 824–827. [Google Scholar] [CrossRef]
- Drevets, W.C.; Savitz, J.; Trimble, M.R.D.; Reich, T.; Vannier, M.; Raichle, M.E. The subgenual anterior cingulate cortex in mood disorders. CNS Spectr. 2008, 13, 663–681. [Google Scholar] [CrossRef] [PubMed]
- Bjorkholm, C.; Monteggia, L.M. BDNF–a key transducer of antidepressant effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.-H.; Kim, Y.-K. The Roles of BDNF in the Pathophysiology of Major Depression and in Antidepressant Treatment. Psychiatry Investig. 2010, 7, 231–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariharan, T.; Nanayakkara, G.; Parameshwaran, K.; Bagasrawala, I.; Ahuja, M.; Abdel-Rahman, E.; Amin, A.T.; Dhanasekaran, M.; Suppiramaniam, V.; Amin, R.H. Central activation of PPAR-gamma ameliorates diabetes induced cognitive dysfunction and improves BDNF expression. Neurobiol. Aging 2015, 36, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Aghajanian, G.K. Synaptic Dysfunction in Depression: Potential Therapeutic Targets. Science 2012, 338, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Holmes, S.E.; Scheinost, D.; Finnema, S.J.; Naganawa, M.; Davis, M.T.; Della Gioia, N.; Nabulsi, N.; Matuskey, D.; Angaria, G.A.; Pietrzak, R.H.; et al. Lower synaptic density is associated with depression severity and network alterations. Nat. Commun. 2019, 10, 1529. [Google Scholar] [CrossRef] [Green Version]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2011, 13, 22–37. [Google Scholar] [CrossRef] [Green Version]
- Fales, C.L.; Barch, D.M.; Rundle, M.M.; Mintun, M.A.; Mathews, J.; Snyder, A.Z.; Sheline, Y.I. Antidepressant treatment normalizes hypoactivity in dorsolateral prefrontal cortex during emotional interference processing in major depression. J. Affect. Disord. 2009, 112, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Duman, R.S. Neuronal damage and protection in the pathophysiology and treatment of psychiatric illness: Stress and depression. Dialogues Clin. Neurosci. 2009, 11, 239–255. [Google Scholar]
- Pittenger, C.D.R. Stress, deprression, and neuroplasticity: A comvergence of mechanisms. Neuropsychopharmacology 2008, 33, 88–109. [Google Scholar] [CrossRef] [PubMed]
- Laje, G.; Lally, N.; Mathews, D.; Brutche, N.; Chemerinski, A.; Akula, N.; Kelmendi, B.; Simen, A.; McMahon, F.J.; Sanacora, G.S.; et al. Brain-derived neurotrophic factor Val66Met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biol. Psychiatry 2012, 72, e27–e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banasr, M.; Dwyer, J.M.; Duman, R.S. Cell atrophy and loss in depression: Reversal by antidepressant treatment. Curr. Opin. Cell Biol. 2011, 23, 730–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Sa, C.; Duman, R.S. Antidepressants and neuroplasticity. Bipolar Disord. 2002, 4, 183–194. [Google Scholar] [CrossRef]
- Chang, C.H.; Chen, M.C.; Lu, J. Effect of antidepressant drugs on the vmPFC-limbic circuitry. Neuropharmacology 2015, 92, 116–124. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S.; Nasca, C.; Gray, J.D. Stress Effects on Neuronal Structure: Hippocampus, Amygdala, and Prefrontal Cortex. Neuropsychopharmacology 2016, 41, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Warner-Schmidt, J.L.; Duman, R.S. Hippocampal neurogenesis: Opposing effects of stress and antidepressant treatment. Hippocampus 2006, 16, 239–249. [Google Scholar] [CrossRef]
- Gold, P.W.; Chrousos, G.P. The endocrinology of melancholic and atypical depression: Relation to neurocircuitry and somatic consequences. Proc. Assoc. Am. Physicians 1999, 111, 22–34. [Google Scholar] [CrossRef]
- Gold, P.W.; Wong, M.L.; Chrousos, G.P.; Licinio, J. Stress system abnormalities in melancholic and atypical depression: Molecular, pathophysiological, and therapeutic implications. Mol. Psychiatry 1996, 1, 257–264. [Google Scholar]
- Disner, S.G.; Beevers, C.G.; Haigh, E.A.; Beck, A.T. Neural mechanisms of the cognitive model of depression. Nat. Rev. Rev. Neurosci. 2011, 12, 467–477. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Franklin, T.; Iwata, M.; Duman, R.S. Integrating neuroimmune systems in the neurobiology of depression. Nat. Rev. Neurosci. 2016, 17, 497–511. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Kritas, S.K.; Saggini, A.; Cerulli, G. Corticotropin-releasing hormone, microglia and mental disorders. Int. J. Immunopathol. Pharmacol. 2014, 27, 163–167. [Google Scholar] [CrossRef]
- Brady, L.S.; Gold, P.W.; Herkenham, M.; Lynn, A.B.; Whitfield, H.J., Jr. The antidepressants fluoxetine, idazoxan and phenelzine alter corticotropin-releasing hormone and tyrosine hydroxylase mRNA levels in rat brain: Therapeutic implications. Brain Res. 1992, 572, 117–125. [Google Scholar] [CrossRef]
- Ayyadurai, S.; Gibson, A.J.; D’Costa, S.; Overman, E.; Somerville, L.J.; Poopal, A.; MacKey, E.; Yihang, L.; Moeser, A.J. Frontline Science: Corticotropin-releasing factor receptor subtype 1 is a critical modulator of mast cell degranulation and stress-induced pathophysiology. J. Leukoc. Biol. 2017, 102, 1299–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, P.; Chandler, N.; Kandere, K.; Basli, S.; Jacobson, S.; Connolly, R.; Tutor, D.; Theoharides, T.C. Corticotropin-releasing hormone and brain mast cells regulate blood-brain-barrier permeability induced by acute stress. J. Pharmacol. Exp. Ther. 2002, 303, 1061–1066. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Singh, L.K.; Boucher, W.; Pang, X.; Leterneau, R.; Webster, E.; Chrousos, G.P. Corticotropin-releasing hormone induces skin mast cell degranulation and increased vascular permeability, a possible explanation for its proinflammatory effects. Endocrinology 1998, 139, 403–413. [Google Scholar] [CrossRef]
- Aoun, P.; Watson, D.G.; Simpkins, J.W. Neuroprotective effects of PPARgamma agonists against oxidative insults in HT-22 cells. Eur. J. Pharmacol. 2003, 472, 65–71. [Google Scholar] [CrossRef]
- Boden, G. Endoplasmic reticulum stress: Another link between obesity and insulin resistance/inflammation? Diabetes 2009, 58, 518–519. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Zhao, X.; Strong, R.; Zhang, J.; Sun, G.; Tsien, J.Z.; Cui, Z.; Grotta, J.G.; Aronowski, J. Neuronal PPARgamma deficiency increases susceptibility to brain damage after cerebral ischemia. J. Neurosci. 2009, 29, 6186–6195. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zheng, F. PPAR-gamma and aging: One link through klotho? Kidney Int. 2008, 74, 702–704. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuro-O, M. Klotho as a regulator of oxidative stress and senescence. Biol. Chem. 2008, 389, 233–241. [Google Scholar] [CrossRef]
- Nagai, T.; Yamada, K.; Kim, H.C.; Kim, Y.S.; Yukihiro, N.; Imura, A.; Nabachima, N.; Nebaschima, T. Cognition impairment in the genetic model of aging klotho gene mutant mice: A role of oxidative stress. FASEB J. 2003, 17, 50–52. [Google Scholar] [CrossRef]
- Luna-Medina, R.; Cortes-Canteli, M.; Sanchez-Galiano, S. NP031112, a thiadiazolidinone compound, prevents inflammation and neurodegeneration under excitotoxic conditions: Potential therapeutic role in brain disorders. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 5766–5776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoun, P.; Simpkins, J.W.; Agarwal, N. Role of PPAR-gamma ligands in neuroprotection against glutamate-induced cytotoxicity in retinal ganglion cells. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2999–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitao, Y.; Ozawa, K.; Miyazaki, M. Expression of the endoplasmic reticulum molecular chaperone (ORP150) rescues hippocampal neurons from glutamate toxicity. J. Clin. Investig. 2001, 108, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Cimini, A.; Ceru, M.P. Emerging roles of peroxisome proliferator-activated receptors (PPARs) in the regulation of neural stem cells proliferation and differentiation. Stem Cell Rev. 2008, 4, 293–303. [Google Scholar] [CrossRef]
- Imayama, I.; Ichiki, T.; Inanaga, K.; Ohtsubo, H.; Fukayama, K.; Ono, H.; Hashaguchi, Y.; Sunagawa, K. Telmisartan downregulates angiotensin II type 1 receptor through activation of peroxisome proliferator-activated receptor gamma. Cardiovasc. Res. 2006, 72, 184–190. [Google Scholar] [CrossRef]
- Festuccia, W.T.; Oztezcan, S.; Laplante, M.; Berthiaume Chantal, M.; Doghu, S.; Denis, R.G.; Brito, M.N.; Brito, N.A.; Miller, D.S. Peroxisome proliferator-activated receptor-gamma-mediated positive energy balance in the rat is associated with reduced sympathetic drive to adipose tissues and thyroid status. Endocrinology 2008, 149, 2121–2130. [Google Scholar] [CrossRef] [PubMed]
- Craft, S. Insulin resistance syndrome and Alzheimer disease: Pathophysiologic mechanisms and therapeutic implications. Alzheimer Dis. Assoc. Disord. 2006, 20, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Role of PPAR in the brain: Promising target in therapy of Alzheimer’s Disease and other neurodegenerative disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahmida, A.A.A.; Mafauzy, M. Efficacy and safety of pioglitizone monotherapy an type II diabetes:A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2019, 9, 5839. [Google Scholar]
- Klutzky, J.K. PPARs as Therapeutic Agents in Reverse Cardiology and Endocrinology. Science 2010, 2010. [Google Scholar]
- Chiu, S.L.; Chen, C.M.; Cline, H.T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Ko, J.; Racz, B.; Burette, A.; Lee, J.; Kim, S.; Na, M.; Lee, H.W.; Kim, S.; Weinberg, R.J.; et al. Regulation of dendritic spine morphogenesis by insulin receptor substrate 53, a downstream effector of Rac1 and Cdc42 small GTPases. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Nelson, T.J.; Sun, M.K.; Hongpaisan, J.; Alkon, D.L. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur. J. Pharmacol. 2008, 585, 76–87. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Porte, D., Jr. Diabetes, obesity, and the brain. Science 2005, 307, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, D.E.; Schinagle, M.; Gao, K.; Calabrese, J. PPAR-gamma agonism as a modulator of mood: Proof-of-concept for pioglitazone in bipolar depression. CNS Drugs 2014, 28, 571–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gold, P.W. The PPARg System in Major Depression: Pathophysiologic and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 9248. https://doi.org/10.3390/ijms22179248
Gold PW. The PPARg System in Major Depression: Pathophysiologic and Therapeutic Implications. International Journal of Molecular Sciences. 2021; 22(17):9248. https://doi.org/10.3390/ijms22179248
Chicago/Turabian StyleGold, Philip W. 2021. "The PPARg System in Major Depression: Pathophysiologic and Therapeutic Implications" International Journal of Molecular Sciences 22, no. 17: 9248. https://doi.org/10.3390/ijms22179248
APA StyleGold, P. W. (2021). The PPARg System in Major Depression: Pathophysiologic and Therapeutic Implications. International Journal of Molecular Sciences, 22(17), 9248. https://doi.org/10.3390/ijms22179248