
Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease

Abstract
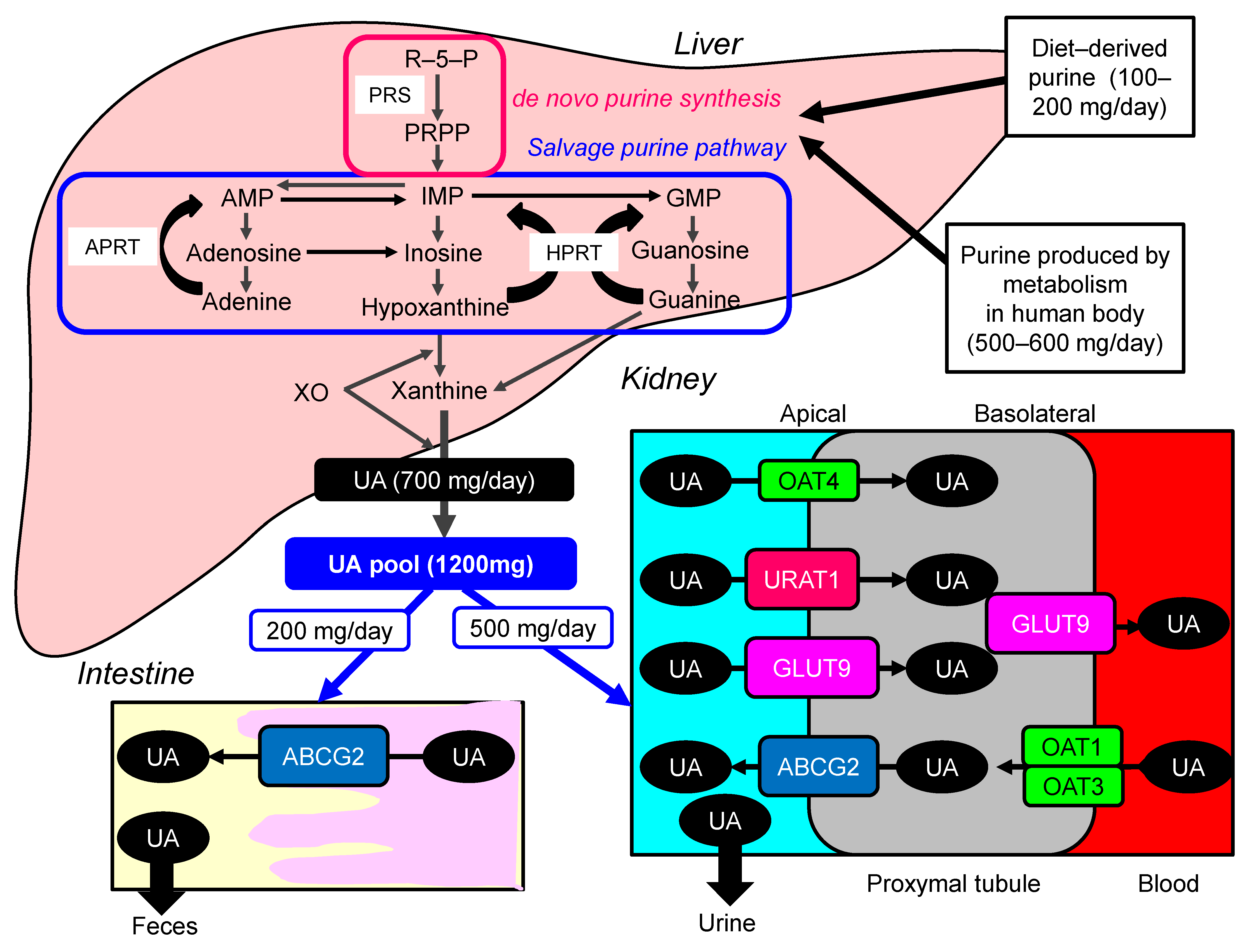
1. Introduction
2. Hyperuricemia and Metabolic Syndrome
2.1. Hyperuricemia and the Risk and Severity of Metabolic Syndrome
2.2. Hyperuricemia and the Components of Metabolic Syndrome
2.2.1. Hypertension
2.2.2. Diabetes
2.2.3. Dyslipidemia
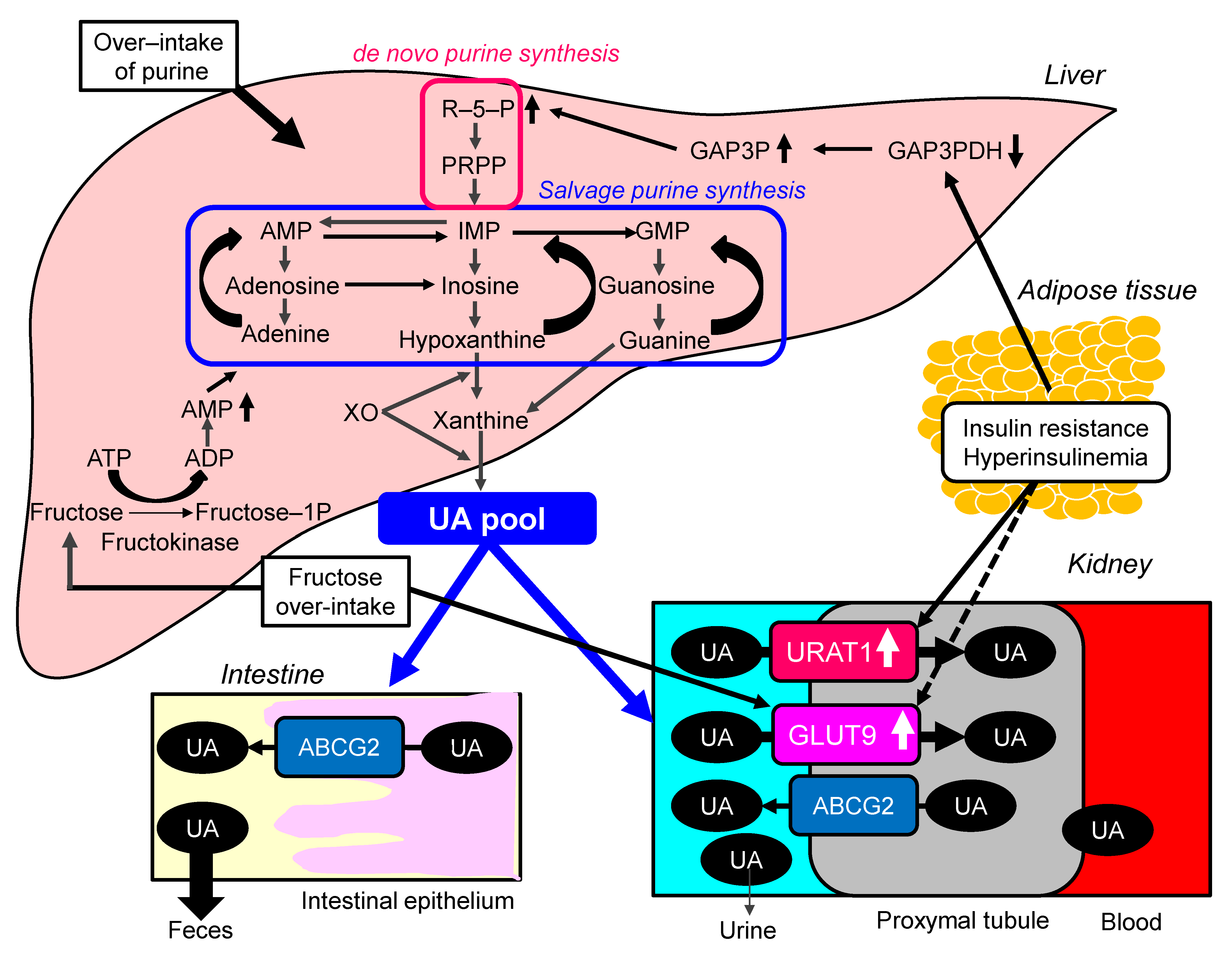
2.3. Possible Molecular Mechanisms for the Development of Hyperuricemia in Metabolic Syndrome
3. Hyperuricemia and CVD
3.1. Hyperuricemia and Atherosclerosis
3.2. Hyperuricemia and Coronary Heart Diseases (CHD)
3.3. Hyperuricemia and Stroke
3.4. Molecular Mechanisms of Hyperuricemia-Induced Atherogenesis and Thrombosis
4. Hyperuricemia and Renal Dysfunction
4.1. Hyperuricemia and CKD
4.2. Molecular Mechanisms of Hyperuricemia-Induced Renal Dysfunction
5. Effects of UA-Lowering Treatment (ULT) on CVD and CKD
5.1. ULT
5.2. Effects of ULT on Atherosclerosis and CVD
5.3. Effects of ULT on CKD
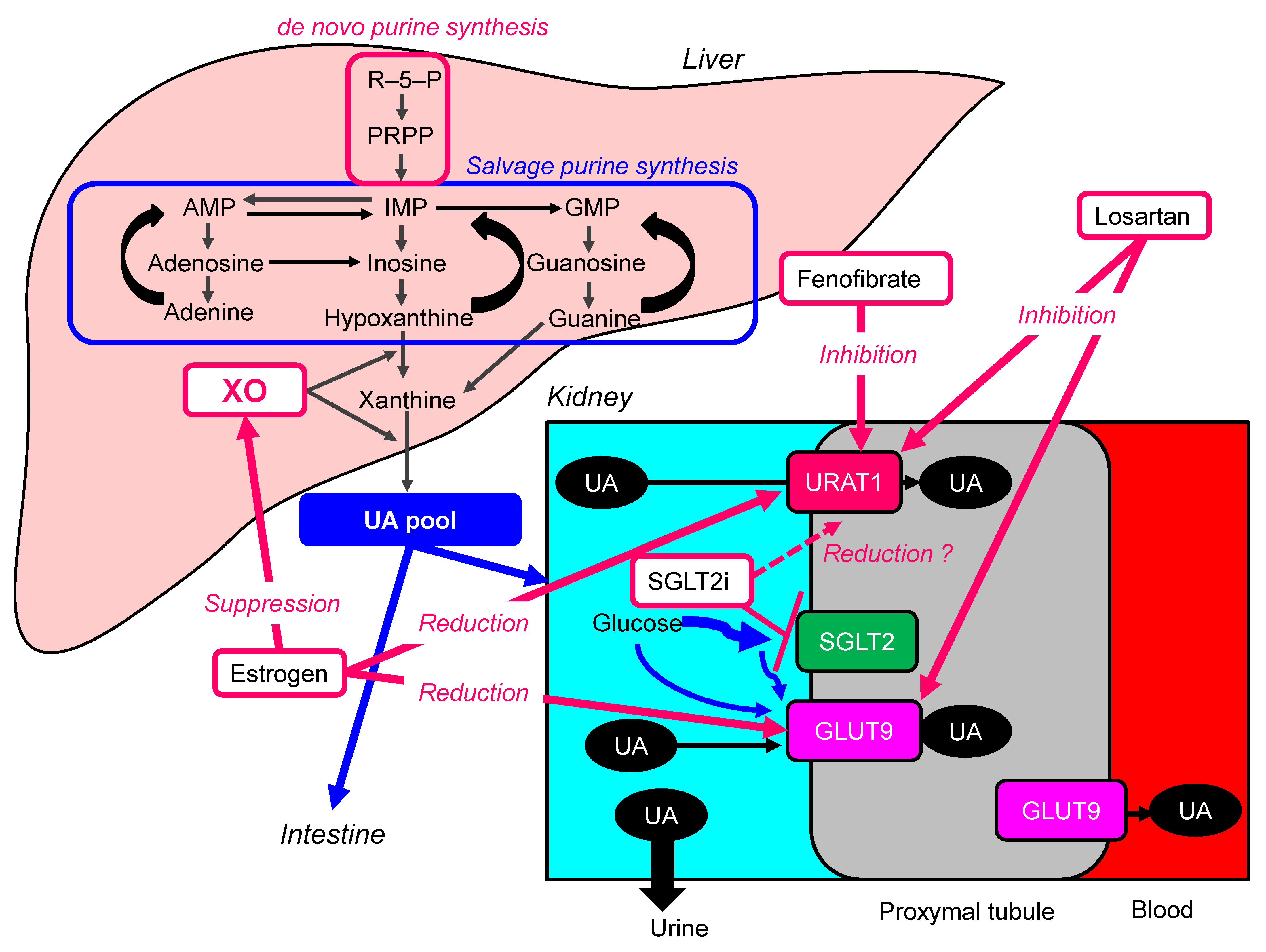
6. Other Drugs to Lower Serum UA
6.1. Estrogen
6.2. Losartan
6.3. Fenofibrate
6.4. Sodium-Glucose Cotransporter 2 Inhibitors (SGLT2i)
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABCG2 | AT P-binding cassette, subfamily G, 2 |
| ADP | adenosine diphosphate |
| AMP | adenosine monophosphate |
| APRT | adenine phosphoribosyltransferase |
| CABG | coronary artery bypass graft |
| CI | confidence intervals |
| CIMT | carotid intima-media thickness |
| CHD | coronary heart diseases |
| CKD | chronic kidney disease |
| CVD | cardiovascular diseases |
| ESRD | end-stage renal disease |
| eGFR | estimated glomerular filtration rate |
| Fructose-1-P | fructose-1-phosphate |
| FMD | flow-mediated dilation |
| GAP3P | glyceraldehyde-3-phosphate |
| GAP3PDH | glyceraldehyde-3-phosphate dehydrogenase |
| GLUT9 | glucose transporter 9 |
| GMP | guanine monophosphate |
| HPRT | hypoxanthine-guanine phosphor-ribosyl-transferase |
| IMP | inosine monophosphate |
| let 7-c | lethal 7-c (let 7-c) |
| MD | mean difference |
| MACE | major adverse cardiovascular events |
| OAT | organic anion transporter |
| OR | odds ratio |
| PDMPs | Platelet-derived microparticles |
| PPRP | phosphor-ribosyl-pyrophosphate |
| PRS | phosphor-ribosyl-pyrophosphate synthase |
| RCTs | randomized controlled trials |
| RD | risk difference |
| RR | relative risk |
| R-5-P | ribose-5-phosphate |
| SMD | standardized mean difference |
| SGLT2i | sodium-glucose cotransporter 2 inhibitors |
| UA | uric acid |
| URAT1 | urate transporter 1 |
| ULT | UA-lowering treatment |
| WMD | weighted mean difference |
| XO | xanthine oxidase |
References
- El Ridi, R.; Tallima, H. Physiological functions and pathogenic potential of uric acid: A review. J. Adv. Res. 2017, 8, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Maesaka, J.K.; Fishbane, S. Regulation of renal urate excretion: A critical review. Am. J. Kidney. Dis. 1998, 32, 917–933. [Google Scholar] [CrossRef]
- Sorensen, L.B. Role of the intestinal tract in the elimination of uric acid. Arthritis Rheum. 1965, 8, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Akaoka, I.; Kamatani, N. Abnormalities in urate metabolism: Concept and classification. Nihon Rinsho. Jpn. J. Clin. Med. 1996, 54, 3243–3247. [Google Scholar]
- Hisatome, I.; Ichida, K.; Mineo, I.; Ohtahara, A.; Ogino, K.; Kuwabara, M.; Ishizaka, N.; Uchida, S.; Kurajoh, M.; Kohagura, K.; et al. Japanese Society of Gout and Uric & Nucleic Acids. 2019 Guidelines for Management of Hyperuricemia and Gout 3rd Edition. Gout Uric Nucleic Acids 2020, 44 (Supplement), 1–40. [Google Scholar]
- Merriman, T.R.; Dalbeth, N. The genetic basis of hyperuricaemia and gout. Jt. Bone Spine 2011, 78, 35–40. [Google Scholar] [CrossRef]
- Gibson, T.; Waterworth, R.; Hatfield, P.; Robinson, G.; Bremner, K. Hyperuricaemia, gout and kidney function in New Zealand Maori men. Br. J. Rheumatol. 1984, 23, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, H.A.; McBride, M.B.; Hatfield, P.J.; Graham, R.; McCaskey, J.; Jackson, M. Polynesian women are also at risk for hyperuricaemia and gout because of a genetic defect in renal urate handling. Br. J. Rheumatol. 1994, 33, 932–937. [Google Scholar] [CrossRef]
- Dalbeth, N.; Merriman, T. Crystal ball gazing: New therapeutic targets for hyperuricaemia and gout. Rheumatology 2009, 48, 222–226. [Google Scholar] [CrossRef]
- Caulfield, M.J.; Munroe, P.B.; O’Neill, D.; Witkowska, K.; Charchar, F.J.; Doblado, M.; Evans, S.; Eyheramendy, S.; Onipinla, A.; Howard, P.; et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008, 5, e197. [Google Scholar] [CrossRef]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Köttgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Köttgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Shi, Y.; Zhuang, S.; Liu, N. Recent advances on uric acid transporters. Oncotarget 2017, 8, 100852–100862. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sanna, S.; Maschio, A.; Busonero, F.; Usala, G.; Mulas, A.; Lai, S.; Dei, M.; Orrù, M.; Albai, G. The GLUT9 gene is associated with serum uric acid levels in Sardinia and Chianti cohorts. PLoS Genet. 2007, 3, e194. [Google Scholar] [CrossRef]
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.K.; Floyd, J.; Palmer, C.N.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442. [Google Scholar] [CrossRef]
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Ikebuchi, Y.; Ito, K.; Kusanagi, Y.; Chiba, T.; Tadokoro, S. Common defects of ABCG2, a high-capacity urate exporter, cause gout: A function-based genetic analysis in a Japanese population. Sci. Transl. Med. 2009, 1, 5ra11. [Google Scholar] [CrossRef]
- Perez-Ruiz, F.; Calabozo, M.; Erauskin, G.G.; Ruibal, A.; Herrero-Beites, A.M. Renal underexcretion of uric acid is present in patients with apparent high urinary uric acid output. Arthritis Rheum. 2002, 47, 610–613. [Google Scholar] [CrossRef]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef]
- Yuan, H.; Yu, C.; Li, X.; Sun, L.; Zhu, X.; Zhao, C.; Zhang, Z.; Yang, Z. Serum Uric Acid Levels and Risk of Metabolic Syndrome: A Dose-Response Meta-Analysis of Prospective Studies. J. Clin. Endocrinol. Metab. 2015, 100, 4198–4207. [Google Scholar] [CrossRef]
- Choi, H.K.; Ford, E.S. Prevalence of the metabolic syndrome in individuals with hyperuricemia. Am. J. Med. 2007, 120, 442–447. [Google Scholar] [CrossRef]
- Hjortnaes, J.; Algra, A.; Olijhoek, J.; Huisman, M.; Jacobs, J.; van der Graaf, Y.; Visseren, F. Serum uric acid levels and risk for vascular diseases in patients with metabolic syndrome. J. Rheumatol. 2007, 34, 1882–1887. [Google Scholar]
- Takahashi, S.; Yamamoto, T.; Tsutsumi, Z.; Moriwaki, Y.; Yamakita, J.; Higashino, K. Close correlation between visceral fat accumulation and uric acid metabolism in healthy men. Metabolism 1997, 46, 1162–1165. [Google Scholar] [CrossRef]
- Facchini, F.; Chen, Y.D.; Hollenbeck, C.B.; Reaven, G.M. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA 1991, 266, 3008–3011. [Google Scholar] [CrossRef]
- Wang, J.; Qin, T.; Chen, J.; Li, Y.; Wang, L.; Huang, H.; Li, J. Hyperuricemia and risk of incident hypertension: A systematic review and meta-analysis of observational studies. PLoS ONE 2014, 9, e114259. [Google Scholar] [CrossRef]
- Grayson, P.C.; Kim, S.Y.; LaValley, M.; Choi, H.K. Hyperuricemia and incident hypertension: A systematic review and meta-analysis. Arthritis Care. Res 2011, 63, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Zhang, X.; Kang, S.; Wu, Y. Serum uric acid levels and incidence of impaired fasting glucose and type 2 diabetes mellitus: A meta-analysis of cohort studies. Diabetes Res. Clin. Pract. 2013, 101, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Saito, K.; Yachi, Y.; Asumi, M.; Sugawara, S.; Totsuka, K.; Saito, A.; Sone, H. Association between serum uric acid and development of type 2 diabetes. Diabetes Care 2009, 32, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Meng, X.F.; He, F.F.; Chen, S.; Su, H.; Xiong, J.; Gao, P.; Tian, X.J.; Liu, J.S.; Zhu, Z.H. High serum uric acid and increased risk of type 2 diabetes: A systemic review and meta-analysis of prospective cohort studies. PLoS ONE 2013, 8, e56864. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, H.; Chen, Y.; Wang, J.; Xu, L.; Miao, M.; Xu, C. Association between serum uric acid levels and dyslipidemia in Chinese adults: A cross-sectional study and further meta-analysis. Medicine 2020, 99, e19088. [Google Scholar] [CrossRef] [PubMed]
- Goli, P.; Riahi, R.; Daniali, S.S.; Pourmirzaei, M.; Kelishadi, R. Association of serum uric acid concentration with components of pediatric metabolic syndrome: A systematic review and meta-analysis. J. Res. Med. Sci. 2020, 25, 43. [Google Scholar]
- Doshi, M.; Takiue, Y.; Saito, H.; Hosoyamada, M. The increased protein level of URAT1 was observed in obesity/metabolic syndrome model mice. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1290–1294. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Yan, S.; Wang, J.; Wang, B.; Li, Y.; Xing, X.; Yuan, Y.; Meng, D.; Wang, L.; Gu, J. Insulin resistance acts as an independent risk factor exacerbating high-purine diet induced renal injury and knee joint gouty lesions. Inflamm. Res. 2009, 58, 659–668. [Google Scholar] [CrossRef]
- Leyva, F.; Wingrove, C.S.; Godsland, I.F.; Stevenson, J.C. The glycolytic pathway to coronary heart disease: A hypothesis. Metabolism 1998, 47, 657–662. [Google Scholar] [CrossRef]
- Ter Horst, K.W.; Schene, M.R.; Holman, R.; Romijn, J.A.; Serlie, M.J. Effect of fructose consumption on insulin sensitivity in nondiabetic subjects: A systematic review and meta-analysis of diet-intervention trials. Am. J. Clin. Nutr. 2016, 104, 1562–1576. [Google Scholar] [CrossRef]
- Kelishadi, R.; Mansourian, M.; Heidari-Beni, M. Association of fructose consumption and components of metabolic syndrome in human studies: A systematic review and meta-analysis. Nutrition 2014, 30, 503–510. [Google Scholar] [CrossRef]
- Li, R.; Yu, K.; Li, C. Dietary factors and risk of gout and hyperuricemia: A meta-analysis and systematic review. Asia. Pac. J. Clin. Nutr. 2018, 27, 1344–1356. [Google Scholar] [PubMed]
- Jamnik, J.; Rehman, S.; Blanco Mejia, S.; de Souza, R.J.; Khan, T.A.; Leiter, L.A.; Wolever, T.M.; Kendall, C.W.; Jenkins, D.J.; Sievenpiper, J.L. Fructose intake and risk of gout and hyperuricemia: A systematic review and meta-analysis of prospective cohort studies. BMJ Open 2016, 6, e013191. [Google Scholar] [CrossRef]
- Ng, H.Y.; Lee, Y.T.; Kuo, W.H.; Huang, P.C.; Lee, W.C.; Lee, C.T. Alterations of Renal Epithelial Glucose and Uric Acid Transporters in Fructose Induced Metabolic Syndrome. Kidney Blood Press. Res. 2018, 43, 1822–1831. [Google Scholar] [CrossRef]
- Dalbeth, N.; House, M.E.; Gamble, G.D.; Horne, A.; Pool, B.; Purvis, L.; Stewart, A.; Merriman, M.; Cadzow, M.; Phipps-Green, A.; et al. Population-specific influence of SLC2A9 genotype on the acute hyperuricaemic response to a fructose load. Ann. Rheum. Dis. 2013, 72, 1868–1873. [Google Scholar] [CrossRef]
- Hallfrisch, J. Metabolic effects of dietary fructose. FASEB J. 1990, 4, 2652–2660. [Google Scholar] [CrossRef]
- Van den Berghe, G. Fructose: Metabolism and shortterm effects on carbohydrate and purine metabolic pathways. In Metabolic Effects of Dietary Carbohydrates; Macdonald, I., Vrana, A., Eds.; Progress in Biochemical Pharmacology; Karger: Basel, Switzerland, 1986; Volume 21, pp. 1–32. [Google Scholar]
- Ma, M.; Wang, L.; Huang, W.; Zhong, X.; Li, L.; Wang, H.; Peng, B.; Mao, M. Meta-analysis of the correlation between serum uric acid level and carotid intima-media thickness. PLoS ONE 2021, 16, e0246416. [Google Scholar]
- Ji, X.; Leng, X.Y.; Dong, Y.; Ma, Y.H.; Xu, W.; Cao, X.P.; Hou, X.H.; Dong, Q.; Tan, L.; Yu, J.T. Modifiable risk factors for carotid atherosclerosis: A meta-analysis and systematic review. Ann. Transl. Med. 2019, 7, 632. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hu, X.; Fan, Y.; Li, K.; Zhang, X.; Hou, W.; Tang, Z. Hyperuricemia and the risk for coronary heart disease morbidity and mortality a systematic review and dose-response meta-analysis. Sci. Rep. 2016, 6, 19520. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Liu, X.; Jiang, L.; Mao, S.; Yin, X.; Guo, L. Hyperuricemia and coronary heart disease mortality: A meta-analysis of prospective cohort studies. BMC Cardiovasc. Disord. 2016, 16, 207. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Guevara, J.P.; Kim, K.M.; Choi, H.K.; Heitjan, D.F.; Albert, D.A. Hyperuricemia and coronary heart disease: A systematic review and meta-analysis. Arthritis Care Res. 2010, 62, 170–180. [Google Scholar] [CrossRef]
- Kim, S.Y.; Guevara, J.P.; Kim, K.M.; Choi, H.K.; Heitjan, D.F.; Albert, D.A. Hyperuricemia and risk of stroke: A systematic review and meta-analysis. Arthritis Rheum. 2009, 61, 885–892. [Google Scholar] [CrossRef]
- Li, M.; Hou, W.; Zhang, X.; Hu, L.; Tang, Z. Hyperuricemia and risk of stroke: A systematic review and meta-analysis of prospective studies. Atherosclerosis 2014, 232, 265–270. [Google Scholar] [CrossRef]
- Yu, W.; Cheng, J.D. Uric Acid and Cardiovascular Disease: An Update from Molecular Mechanism to Clinical Perspective. Front. Pharmacol. 2020, 11, 582680. [Google Scholar] [CrossRef]
- Men, P.; Punzo, G. Uric acid: Bystander or culprit in hypertension and progressive renal disease? J. Hypertens. 2008, 26, 2085–2092. [Google Scholar] [CrossRef]
- Peluso, I.; Morabito, G.; Urban, L.; Ioannone, F.; Serafini, M. Oxidative stress in atherosclerosis development: The central role of LDL and oxidative burst. Endocr. Metab. Immune Disord. Drug. Targets 2012, 12, 351–360. [Google Scholar] [CrossRef]
- Nomura, J.; Busso, N.; Ives, A.; Matsui, C.; Tsujimoto, S.; Shirakura, T.; Tamura, M.; Kobayashi, T.; So, A.; Yamanaka, Y. Xanthine oxidase inhibition by febuxostat attenuates experimental atherosclerosis in mice. Sci. Rep. 2014, 4, 4554. [Google Scholar] [CrossRef]
- Patetsios, P.; Song, M.; Shutze, W.P.; Pappas, C.; Rodino, W.; Ramirez, J.A.; Panetta, T.F. Identification of uric acid and xanthine oxidase in atherosclerotic plaque. Am. J. Cardiol. 2001, 88, 188–191. [Google Scholar] [CrossRef]
- Schröder, K.; Vecchione, C.; Jung, O.; Schreiber, J.G.; Shiri-Sverdlov, R.; van Gorp, P.J.; Busse, R.; Brandes, R.P. Xanthine oxidase inhibitor tungsten prevents the development of atherosclerosis in ApoE knockout mice fed a Western-type diet. Free Radic. Biol. Med. 2006, 41, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Guthikonda, S.; Sinkey, C.; Barenz, T.; Haynes, W.G. Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circulation 2003, 107, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Liu, T.; Ma, L.; Liu, Z.; Xin, Y.; Jia, Z.; Chen, Y.; Li, C.; Sun, R. Prothrombotic effects of high uric acid in mice via activation of MEF2C-dependent NF-κB pathway by upregulating let-7c. Aging 2020, 12, 17976–17989. [Google Scholar] [CrossRef] [PubMed]
- Yisireyili, M.; Hayashi, M.; Wu, H.; Uchida, Y.; Yamamoto, K.; Kikuchi, R.; Shoaib Hamrah, M.; Nakayama, T.; Wu Cheng, X.; Matsushita, T.; et al. Xanthine oxidase inhibition by febuxostat attenuates stress-induced hyperuricemia, glucose dysmetabolism, and prothrombotic state in mice. Sci. Rep. 2017, 7, 1266. [Google Scholar] [CrossRef]
- Nishizawa, T.; Taniura, T.; Nomura, S. Effects of febuxostat on platelet-derived microparticles and adiponectin in patients with hyperuricemia. Blood Coagul. Fibrinolysis 2015, 26, 887–892. [Google Scholar] [CrossRef]
- Zhu, P.; Liu, Y.; Han, L.; Xu, G.; Ran, J.M. Serum uric acid is associated with incident chronic kidney disease in middle-aged populations: A meta-analysis of 15 cohort studies. PLoS ONE 2014, 9, e100801. [Google Scholar] [CrossRef]
- Li, L.; Yang, C.; Zhao, Y.; Zeng, X.; Liu, F.; Fu, P. Is hyperuricemia an independent risk factor for new-onset chronic kidney disease?: A systematic review and meta-analysis based on observational cohort studies. BMC Nephrol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Johnson, R.J.; Kang, D.H.; Feig, D.; Kivlighn, S.; Kanellis, J.; Watanabe, S.; Tuttle, K.R.; Rodriguez-Iturbe, B.; Herrera-Acosta, J.; Mazzali, M. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 2003, 41, 1183–1190. [Google Scholar] [CrossRef]
- Berger, L.; Yü, T.F. Renal function in gout. IV. An analysis of 524 gouty subjects including long-term follow-up studies. Am. J. Med. 1975, 59, 605–613. [Google Scholar] [CrossRef]
- Feig, D.I.; Madero, M.; Jalal, D.I.; Sanchez-Lozada, L.G.; Johnson, R.J. Uric acid and the origins of hypertension. J. Pediatr. 2013, 162, 896–902. [Google Scholar] [CrossRef]
- Mazzali, M.; Hughes, J.; Kim, Y.G.; Jefferson, J.A.; Kang, D.H.; Gordon, K.L.; Lan, H.Y.; Kivlighn, S.; Johnson, R.J. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001, 38, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Uedono, H.; Tsuda, A.; Ishimura, E.; Yasumoto, M.; Ichii, M.; Ochi, A.; Ohno, Y.; Nakatani, S.; Mori, K.; Uchida, J.; et al. Relationship between serum uric acid levels and intrarenal hemodynamic parameters. Kidney Blood Press. Res. 2015, 40, 315–322. [Google Scholar] [CrossRef]
- Mallat, S.G.; Al Kattar, S.; Tanios, B.Y.; Jurjus, A. Hyperuricemia, Hypertension, and Chronic Kidney Disease: An Emerging Association. Curr. Hypertens. Rep. 2016, 18, 74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Doherty, M.; Bardin, T.; Pascual, E.; Barskova, V.; Conaghan, P.; Gerster, J.; Jacobs, J.; Leeb, B.; Lioté, F. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann. Rheum. Dis. 2006, 65, 1312–1324. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Fitzgerald, J.D.; Khanna, P.P.; Bae, S.; Singh, M.K.; Neogi, T.; Pillinger, M.H.; Merill, J.; Lee, S.; Prakash, S. 2012 American College of Rheumatology guidelines for management of gout. Part 1: Systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012, 64, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Sattui, S.E.; Gaffo, A.L. Treatment of hyperuricemia in gout: Current therapeutic options, latest developments and clinical implications. Ther. Adv. Musculoskelet. Dis. 2016, 8, 145–159. [Google Scholar] [CrossRef]
- Reinders, M.K.; van Roon, E.N.; Houtman, P.M.; Brouwers, J.R.; Jansen, T.L. Biochemical effectiveness of allopurinol and allopurinol-probenecid in previously benzbromarone-treated gout patients. Clin. Rheumatol. 2007, 26, 1459–1465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taniguchi, T.; Ashizawa, N.; Matsumoto, K.; Saito, R.; Motoki, K.; Sakai, M.; Chikamatsu, N.; Hagihara, C.; Hashiba, M.; Iwanaga, T. Pharmacological Evaluation of Dotinurad, a Selective Urate Reabsorption Inhibitor. J. Pharmacol. Exp. Ther. 2019, 371, 162–170. [Google Scholar] [CrossRef]
- Hosoya, T.; Furuno, K.; Kanda, S. A non-inferiority study of the novel selective urate reabsorption inhibitor dotinurad versus febuxostat in hyperuricemic patients with or without gout. Clin. Exp. Nephrol. 2020, 24 (Suppl. 1), 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Siriopol, D.; Nistor, I.; Elcioglu, O.C.; Telci, O.; Takir, M.; Johnson, R.J.; Covic, A. Effects of allopurinol on endothelial dysfunction: A meta-analysis. Am. J. Nephrol. 2014, 39, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Mi, S.; Lin, Z. Allopurinol therapy improves vascular endothelial function in subjects at risk for cardiovascular diseases: A meta-analysis of randomized controlled trials. Cardiovasc. Ther. 2016, 34, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Pirro, M.; Watts, G.F.; Mikhailidis, D.P.; Banach, M.; Sahebkar, A. Effects of Allopurinol on Endothelial Function: A Systematic Review and Meta-Analysis of Randomized Placebo-Controlled Trials. Drugs 2018, 78, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Alem, M.M. Allopurinol and endothelial function: A systematic review with meta-analysis of randomized controlled trials. Cardiovasc. Ther. 2018, 36, e12432. [Google Scholar] [CrossRef]
- Zhang, T.; Pope, J.E. Cardiovascular effects of urate-lowering therapies in patients with chronic gout: A systematic review and meta-analysis. Rheumatology 2017, 56, 1144–1153. [Google Scholar] [CrossRef]
- Bredemeier, M.; Lopes, L.M.; Eisenreich, M.A.; Hickmann, S.; Bongiorno, G.K.; d’Avila, R.; Morsch, A.L.B.; da Silva Stein, F.; Campos, G.G.D. Xanthine oxidase inhibitors for prevention of cardiovascular events: A systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2018, 18, 24. [Google Scholar] [CrossRef]
- Singh, T.P.; Skalina, T.; Nour, D.; Murali, A.; Morrison, S.; Moxon, J.V.; Golledge, J. A meta-analysis of the efficacy of allopurinol in reducing the incidence of myocardial infarction following coronary artery bypass grafting. BMC Cardiovasc. Disord. 2018, 18, 143. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, L.; Zhao, T.Y.; Yang, X.; Zhu, X.X.; Zou, H.J.; Wan, W.G.; Xue, Y. Cardiovascular events in hyperuricemia population and a cardiovascular benefit-risk assessment of urate-lowering therapies: A systematic review and meta-analysis. Chin. Med. J. 2020, 133, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Ullah, W.; Khanal, S.; Khan, R.; Basyal, B.; Munir, S.; Minalyan, A.; Alraies, M.C.; Fischman, D.L. Efficacy of Allopurinol in Cardiovascular Diseases: A Systematic Review and Meta-Analysis. Cardiol. Res. 2020, 11, 226–232. [Google Scholar] [CrossRef] [PubMed]
- White, W.B.; Saag, K.G.; Becker, M.A.; Borer, J.S.; Gorelick, P.B.; Whelton, A.; Hunt, B.; Castillo, M.; Gunawardhana, L.; CARES Investigators. Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout. N. Engl. J. Med. 2018, 378, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, J.A.; Balda, J.; Palacio, A.; Young, L.; Pillinger, M.H.; Tamariz, L. Febuxostat and Cardiovascular Events: A Systematic Review and Meta-Analysis. Int. J. Rheumatol. 2019, 2019, 1076189. [Google Scholar] [CrossRef]
- Barrientos-Regala, M.; Macabeo, R.A.; Ramirez-Ragasa, R.; Pestaño, N.S.; Punzalan, F.E.R.; Tumanan-Mendoza, B.; Castillo, R.R. The Association of Febuxostat Compared with Allopurinol on Blood Pressure and Major Adverse Cardiac Events Among Adult Patients With Hyperuricemia: A Meta-analysis. J. Cardiovasc. Pharmacol. 2020, 76, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Al-Abdouh, A.; Khan, S.U.; Barbarawi, M.; Upadhrasta, S.; Munira, S.; Bizanti, A.; Elias, H.; Jat, A.; Zhao, D.; Michos, E.D. Effects of Febuxostat on Mortality and Cardiovascular Outcomes: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Mayo Clin. Proc. Innov. Qual. Outcomes 2020, 4, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, B.; Pan, Y.; Lu, Y.; Cheng, R. Cardiovascular safety of febuxostat compared to allopurinol for the treatment of gout: A systematic and meta-analysis. Clin. Cardiol. 2021, 44, 907–916. [Google Scholar] [CrossRef]
- Kanji, T.; Gandhi, M.; Clase, C.M.; Yang, R. Urate lowering therapy to improve renal outcomes in patients with chronic kidney disease: Systematic review and meta-analysis. BMC Nephrol. 2015, 16, 58. [Google Scholar] [CrossRef]
- Fleeman, N.; Pilkington, G.; Dundar, Y.; Dwan, K.; Boland, A.; Dickson, R.; Anijeet, H.; Kennedy, T.; Pyatt, J. Allopurinol for the treatment of chronic kidney disease: A systematic review. Health. Technol. Assess. 2014, 18, 1–77. [Google Scholar] [CrossRef] [PubMed]
- Bose, B.; Badve, S.V.; Hiremath, S.S.; Boudville, N.; Brown, F.G.; Cass, A.; de Zoysa, J.R.; Fassett, R.G.; Faull, R.; Harris, D.C.; et al. Effects of uric acid-lowering therapy on renal outcomes: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2014, 29, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Pisano, A.; Cernaro, V.; Gembillo, G.; D’Arrigo, G.; Buemi, M.; Bolignano, D. Xanthine Oxidase Inhibitors for Improving Renal Function in Chronic Kidney Disease Patients: An Updated Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2017, 18, 2283. [Google Scholar] [CrossRef]
- Siu, Y.P.; Leung, K.T.; Tong, M.K.H.; Kwan, T.H. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am. J. Kidney Dis. 2006, 47, 51–59. [Google Scholar] [CrossRef]
- Goicoechea, M.; de Vinuesa, S.G.; Verdalles, U.; Ruiz-Caro, C.; Ampuero, J.; Rincón, A.; Arroyo, D.; Luño, J. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin. J. Am. Soc. Nephrol. 2010, 5, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea, M.; Garcia de Vinuesa, S.; Verdalles, U.; Verde, E.; Macias, N.; Santos, A.; Pérez de Jose, A.; Cedeño, S.; Linares, T.; Luño, J. Allopurinol and progression of CKD and cardiovascular events: Long-term follow-up of a randomized clinical trial. Am. J. Kidney Dis. 2015, 65, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Whelton, A.; Macdonald, P.A.; Zhao, L.; Hunt, B.; Gunawardhana, L. Renal function in gout: Long-term treatment effects of febuxostat. J. Clin. Rheumatol. 2011, 17, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Shibagaki, Y.; Ohno, I.; Hosoya, T.; Kimura, K. Safety, efficacy and renal effect of febuxostat in patients with moderate-to-severe kidney dysfunction. Hypertens. Res. 2014, 37, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Hosoya, T.; Uchida, S.; Inaba, M.; Makino, H.; Maruyama, S.; Ito, S.; Yamamoto, T.; Tomino, Y.; Ohno, I.; et al. FEATHER Study Investigators. Febuxostat Therapy for Patients with Stage 3 CKD and Asymptomatic Hyperuricemia: A Randomized Trial. Am. J. Kidney Dis. 2018, 72, 798–810. [Google Scholar] [CrossRef]
- Tsuruta, Y.; Mochizuki, T.; Moriyama, T.; Itabashi, M.; Takei, T.; Tsuchiya, K.; Nitta, K. Switching from allopurinol to febuxostat for the treatment of hyperuricemia and renal function in patients with chronic kidney disease. Clin. Rheumatol. 2014, 33, 1643–1648. [Google Scholar] [CrossRef]
- Hosoya, T.; Ohno, I.; Nomura, S.; Hisatome, I.; Uchida, S.; Fujimori, S.; Yamamoto, T.; Hara, S. Effects of topiroxostat on the serum urate levels and urinary albumin excretion in hyperuricemic stage 3 chronic kidney disease patients with or without gout. Clin. Exp. Nephrol. 2014, 18, 876–884. [Google Scholar] [CrossRef]
- Horino, T.; Hatakeyama, Y.; Ichii, O.; Matsumoto, T.; Shimamura, Y.; Inoue, K.; Terada, Y.; Okuhara, Y. Effects of topiroxostat in hyperuricemic patients with chronic kidney disease. Clin. Exp. Nephrol. 2018, 22, 337–345. [Google Scholar] [CrossRef]
- Katsuyama, H.; Yanai, H.; Hakoshima, M. Renoprotective Effect of Xanthine Oxidase Inhibitor, Topiroxostat. J. Clin. Med. Res. 2019, 11, 614–616. [Google Scholar] [CrossRef][Green Version]
- Fujimori, S.; Ooyama, K.; Ooyama, H.; Moromizato, H. Efficacy of benzbromarone in hyperuricemic patients associated with chronic kidney disease. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Liu, X.; Song, Y.; Cheng, J.; Bao, H.; Qin, L.; Zhou, X.; Wang, L.; Peng, A. Safety and Efficacy of Benzbromarone and Febuxostat in Hyperuricemia Patients with Chronic Kidney Disease: A Prospective Pilot Study. Clin. Exp. Nephrol. 2018, 22, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.W.; Chiu, H.T.; Tsai, C.W.; Ting, I.W.; Yeh, H.C.; Huang, H.C.; Kuo, C.C.; CMUH Kidney Research Group. Comparative effectiveness of allopurinol, febuxostat and benzbromarone on renal function in chronic kidney disease patients with hyperuricemia: A 13-year inception cohort study. Nephrol. Dial. Transplant. 2018, 33, 1620–1627. [Google Scholar] [CrossRef]
- Sumino, H.; Ichikawa, S.; Kanda, T.; Nakamura, T.; Sakamaki, T. Reduction of serum uric acid by hormone replacement therapy in postmenopausal women with hyperuricaemia. Lancet 1999, 354, 650. [Google Scholar] [CrossRef]
- Fan, Y.; Wei, F.; Lang, Y.; Wang, S. Losartan treatment for hypertensive patients with hyperuricaemia in Chinese population: A meta-analysis. J. Hypertens. 2015, 33, 681–688. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, X.; Dong, Z.; Lu, J.; Zhao, Y.; Li, R.; Li, C.; Chen, Y. Impact of fenofibrate therapy on serum uric acid concentrations: A review and meta-analysis. Endocr. J. 2021, 68, 829–837. [Google Scholar] [CrossRef]
- Derosa, G.; Maffioli, P.; Sahebkar, A. Plasma uric acid concentrations are reduced by fenofibrate: A systematic review and meta-analysis of randomized placebo-controlled trials. Pharmacol. Res. 2015, 102, 63–70. [Google Scholar] [CrossRef]
- Bailey, C.J. Uric acid and the cardio-renal effects of SGLT2 inhibitors Diabetes. Obes. Metab. 2019, 21, 1291–1298. [Google Scholar] [CrossRef]
- Xin, Y.; Guo, Y.; Li, Y.; Ma, Y.; Li, L.; Jiang, H. Effects of sodium glucose cotransporter-2 inhibitors on serum uric acid in type 2 diabetes mellitus: A systematic review with an indirect comparison meta-analysis. Saudi J. Biol. Sci. 2019, 26, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, L.; Tian, D.; Xia, P.; Zheng, H.; Wang, L.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2018, 20, 458–462. [Google Scholar] [CrossRef]
- Takiue, Y.; Hosoyamada, M.; Kimura, M.; Saito, H. The effect of female hormones upon urate transport systems in the mouse kidney. Nucleosides Nucleotides Nucleic Acids 2011, 30, 113–119. [Google Scholar] [CrossRef]
- Huh, K.; Shin, U.S.; Choi, J.W.; Lee, S.I. Effect of sex hormones on lipid peroxidation in rat liver. Arch. Pharm. Res. 1994, 17, 109–114. [Google Scholar] [CrossRef]
- Bach, M.H.; Simkin, P.A. Uricosuric drugs: The once and future therapy for hyperuricemia? Curr. Opin. Rheumatol. 2014, 26, 169–175. [Google Scholar] [CrossRef]
- Uetake, D.; Ohno, I.; Ichida, K.; Yamaguchi, Y.; Saikawa, H.; Endou, H.; Hosoya, T. Effect of fenofibrate on uric acid metabolism and urate transporter 1. Intern. Med. 2010, 49, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Kawaguchi, A.; Waragai, Y.; Harigae, T.; Masui, Y.; Kakuta, K.; Hamasaki, H.; Katsuyama, H.; et al. Effects of Six Kinds of Sodium-Glucose Cotransporter 2 Inhibitors on Metabolic Parameters, and Summarized Effect and Its Correlations with Baseline Data. J. Clin. Med. Res. 2017, 9, 605–612. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Katsuyama, H. Multi-Organ Protective Effects of Sodium Glucose Cotransporter 2 Inhibitors. Int. J. Mol. Sci. 2021, 22, 4416. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Xanthine Oxidase Inhibitors (The First-Line Treatment for Hyperuricemia) | |||
|---|---|---|---|
| Author | Study Design | Subjects Studied | Renal Outcomes |
| Allopurinol (The most widely used drug approved for the treatment of hyperuricemia) | |||
| Siu, Y.P., et al. [91] | Patients were randomly assigned to treatment with allopurinol, 100 to 300 mg/day, or to continue the usual therapy for 12 months. Study end points included stable kidney function with less than 40% increase in serum creatinine level; impaired renal function with creatinine level increase greater than 40% of baseline value; initiation of dialysis therapy; and death. | 54 hyperuricemic patients with CKD | There was a trend toward a lower serum creatinine level in the treatment group compared with controls after 12 months, although it did not reach statistical significance (p = 0.08). Less patients (16%) in the allopurinol group reached the combined end points of significant deterioration in renal function and dialysis dependence compared with patients (46.1%) in the control group (p = 0.015). |
| Goicoechea, M., et al. [92] | Patients were randomly assigned to treatment with allopurinol 100 mg/day or to continue the usual therapy. Study end points included renal disease progression; cardiovascular events; and hospitalizations of any causes. | 113 patients with eGFR < 60 mL/min. | In the control group, eGFR decreased by 3.3 ± 1.2 mL/min/1.73 m2, and in the allopurinol group, eGFR increased by 1.3 ± 1.3 mL/min/1.73 m2 after 24 months. Allopurinol treatment reduced risk of CV events in 71% compared with standard therapy. |
| Goicoechea, M., et al. [93] | Post hoc analysis of a long-term follow-up after completion of the 2-year trial. Intervention is continuation of allopurinol treatment, 100 mg/day, or standard treatment. Study end points included renal event (starting dialysis therapy and/or doubling serum creatinine and/or decrease in eGFR by over 50%) and CV events (myocardial infarction, coronary revascularization or angina pectoris, congestive heart failure, cerebrovascular disease, and peripheral artery disease). | 113 participants (57 in the allopurinol group and 56 in the control group) initially followed up for 2 years and 107 participants followed up to 5 additional years. | During the initial and long-term follow-up (median, 84 months), 9 patients in the allopurinol group had a renal event compared with 24 patients in the control group (adjested HR, 0.32; 95%CI, 0.15–0.69; p = 0.004). Overall, 16 patients treated with allopurinol experienced CV events compared with 23 in the control group (adjusted HR, 0.43; 95% CI, 0.21–0.88; p = 0.02). |
| Febuxostat (The widely used drug approved for the treatment of hyperuricemia) | |||
| Whelton, A., et al. [94] | A post hoc analysis of the Febuxostat Open-label Clinical trial of Urate-lowering efficacy and Safety study, during which subjects received daily doses of febuxostat (40, 80, or 120 mg) for up to 5 years. | 116 hyperuricemic gout subjects | Maintenance or improvement in eGFR was inversely correlated with the reduction in serum UA from baseline. For every 1 mg/dL decrease in serum UA, the model projected an expected improvement in eGFR of 1 mL/min from the untreated value. |
| Shibagaki, Y., et al. [95] | The safety and efficacy of escalating doses of febuxostat over a 24-week period in patients with CKD stages 3b, 4 and 5 were studied. | 70 patients with CKD stages 3b, 4 and 5 | Multivariate analysis showed that a greater reduction in serum UA with febuxostat was associated with an increase in eGFR and a tendency toward decreased proteinuria. |
| Kimura, K., et al. [96] | Participants were randomly assigned in a 1:1 ratio to receive febuxostat or placebo for 108 weeks. The primary end point was the slope of eGFR | 467 patients with stage 3 CKD and asymptomatic hyperuricemia at 55 medical institutions in Japan | There was no significant difference in mean eGFR slope between the febuxostat and placebo groups. |
| Tsuruta, Y. et al. [97] | A 1-year cohort study. In 51 patients, treatment was changed from allopurinol to febuxostat, and the other 22 patients were continued on allopurinol. | 73 hyperuricemic patients who had an eGFR below 45 mL/min and were being treated with urate-lowering therapy | The eGFR decreased 27.3 to 25.7 mL/min in the febuxostat group and from 26.1 to 19.9 mL/min in the allopurinol group. The switch from allopurinol to febuxostat was significantly associated with the changes in eGFR according to a multiple regression analysis (β = −0.22145, p < 0.05). |
| Topiroxostat (The drug approved for the treatment of hyperuricemia) | |||
| Hosoya, T., et al. [98] | A 22-week, randomized, multicenter, double-blind study. The enrolled patients were randomly assigned to treatment with topiroxostat 160 mg/day or to the placebo. The endpoints included change in the eGFR, the urinary albumin-to-creatinine ratio. | 123 hyperuricemic stage 3 CKD patients with or without gout | After 22 weeks, although the changes in the eGFR was not significant, the percent change in urinary albumin-to-creatinine ratio (−33.0 vs. −6.0 %, p = 0.0092) was found to have decreased in the topiroxostat as compared with the placebo. |
| Horino, T., et al. [99] | Patients were administered 40 mg/day of topiroxostat twice daily. All patients were followed for a year. | 30 hyperuricemic patients with CKD (20 male, 10 female) | Topiroxostat treatment resulted in significant reduction in urinary protein excretion (−795.5 mg/gCr) compared with baseline values. However, serum creatinine level, and eGFR did not change significantly. |
| Katsuyama, H., et al. [100] | Patients who had been continuously prescribed topiroxostat for 3 months or more were retrospectively picked up, and compared serum UA, eGFR and urinary protein before the topiroxostat treatment with the data at 3 and 6 months after the topiroxostat treatment started. | 27 hyperuricemic patients | eGFR did not change 3 months after, however, eGFR showed a trend to increase 6 months after. The number of patients who showed positivity for urinary protein significantly decreased at 3 and 6 months after the start of topiroxostat as compared with baseline. |
| Uricosuric agent (The second-line treatment for hyperuricemia) | |||
| Benzbromarone (The widely used drug approved for the treatment of hyperuricemia) | |||
| Fujimori, S., et al. [101] | Renal function changes over a period of up to 7 years were retrospectively evaluated in patients with CKD associated with hyperuricemia and were receiving monotherapy with benzbromarone | 35 patients with CKD (stage 3, 32 patients; stage 4, 2 patients; stage 5, 1 patient) associated with hyperuricemia | No significant changes in eGFR from the baseline value of 46.2 ± 11.5 mL/min/1.73 m2 were found after benzbromarone therapy. |
| Yu, H., et al. [102] | A single-centered, parallel-grouped, RCT. Patients were randomly assigned into benzbromarone and febuxostat treatment group. | 66 hyperuricemia participants with eGFR 20–60 mL/min/1.73 m2 | After 12-month treatment, eGFR did not have significant changes in both groups. |
| Chou, H.W., et al. [103] | A pharmacoepidemiology cohort study by including patients from Taiwan’s long-term integrated CKD care program to compare the effectiveness among allopurinol, febuxostat and benzbromarone in reducing the risk of progression to dialysis. Patients with hyperuricemia who were newly treated with allopurinol, febuxostat or benzbromarone were included. | 874 CKD patients with hyperuricemia | Compared with allopurinol, benzbromarone therapy was associated with a reduced risk of progression to dialysis, the adjusted HR was 0.50 (95%CI, 0.25–0.99). Among patients who reached the therapeutic target, those with febuxostat and benzbromarone initiation had a significantly lower risk of end-stage renal disease. |
| Drugs | Main Pharmacological Action | Possible Mechanisms to Reduce Serum UA | Studies Which Showed Its UA-Lowering Effects |
|---|---|---|---|
| Estrogen | Female sex hormone | Suppression of the protein levels of URAT1 and GLUT9, and reduction of XO activity | Serum UA in 61 postmenopausal women before and during HRT were measured [104]. HRT significantly reduced serum UA concentrations throughout 3–12 months in postmenopausal women with hyperuricemia. On the other hand, serum UA concentrations in the control group showed no significant changes for 12 months. |
| Losartan | Antihypertensive drugs, angiotensin II receptor antagonist | Inhibition of URAT1 and GLUT9 | In the meta-analysis including 31 RCTs, losartan reduced serum UA levels (−1.57 mg/dL; 95%CI, −1.83 to −1.30) as compared with other antihypertensive agents [105]. |
| Fenofibrate | Anti-lipidemic drug | Inhibition of URAT1 | The meta-analysis including 9 studies demonstrated that fenofibrate significantly reduced serum UA levels (−1.32 mg/dL; 95%CI, −1.61 to −1.03; p < 0.001) [106]. Another meta-analysis showed a significant reduction in plasma UA concentrations following fenofibrate therapy [107]. |
| Sodium-Glucose Cotransporter 2 Inhibitors (SGLT2i) | Oral anti-diabetic drugs | SGLT2i may reduce over-expressed URAT1 due to insulin resistance in patients with type 2 diabetes, by improving insulin resistance. SGLT2i increase renal UA elimination by another mechanism. SGLT2i increase the concentration of glucose in the proximal tubules, and glucose may compete with UA for apical GLUT9, reducing UA reabsorption [108]. | In the meta-analysis including 31 studies, SGLT2i significantly decreased serum UA levels compared with placebo, canagliflozin (−37.02 μmol/L; 95%CI, −38.41 to −35.63), dapagliflozin (−38.05 μmol/L; 95%CI, −44.47 to −31.62), empagliflozin (−42.07 μmol/L; 95%CI, −46.27 to −37.86) [109]. Another meta-analysis also demonstrated that any of the SGLT2i (empagliflozin, canagliflozin, dapagliflozin, tofogliflozin, luseogliflozin or ipragliflozin) significantly decreased serum UA levels compared with control (−37.73 μmol/L; 95%CI, −40.51 to −34.95]) [110]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 9221. https://doi.org/10.3390/ijms22179221
Yanai H, Adachi H, Hakoshima M, Katsuyama H. Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease. International Journal of Molecular Sciences. 2021; 22(17):9221. https://doi.org/10.3390/ijms22179221
Chicago/Turabian StyleYanai, Hidekatsu, Hiroki Adachi, Mariko Hakoshima, and Hisayuki Katsuyama. 2021. "Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease" International Journal of Molecular Sciences 22, no. 17: 9221. https://doi.org/10.3390/ijms22179221
APA StyleYanai, H., Adachi, H., Hakoshima, M., & Katsuyama, H. (2021). Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease. International Journal of Molecular Sciences, 22(17), 9221. https://doi.org/10.3390/ijms22179221