
Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis

 , , , , , , , , , ,
, , , , , , , , , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
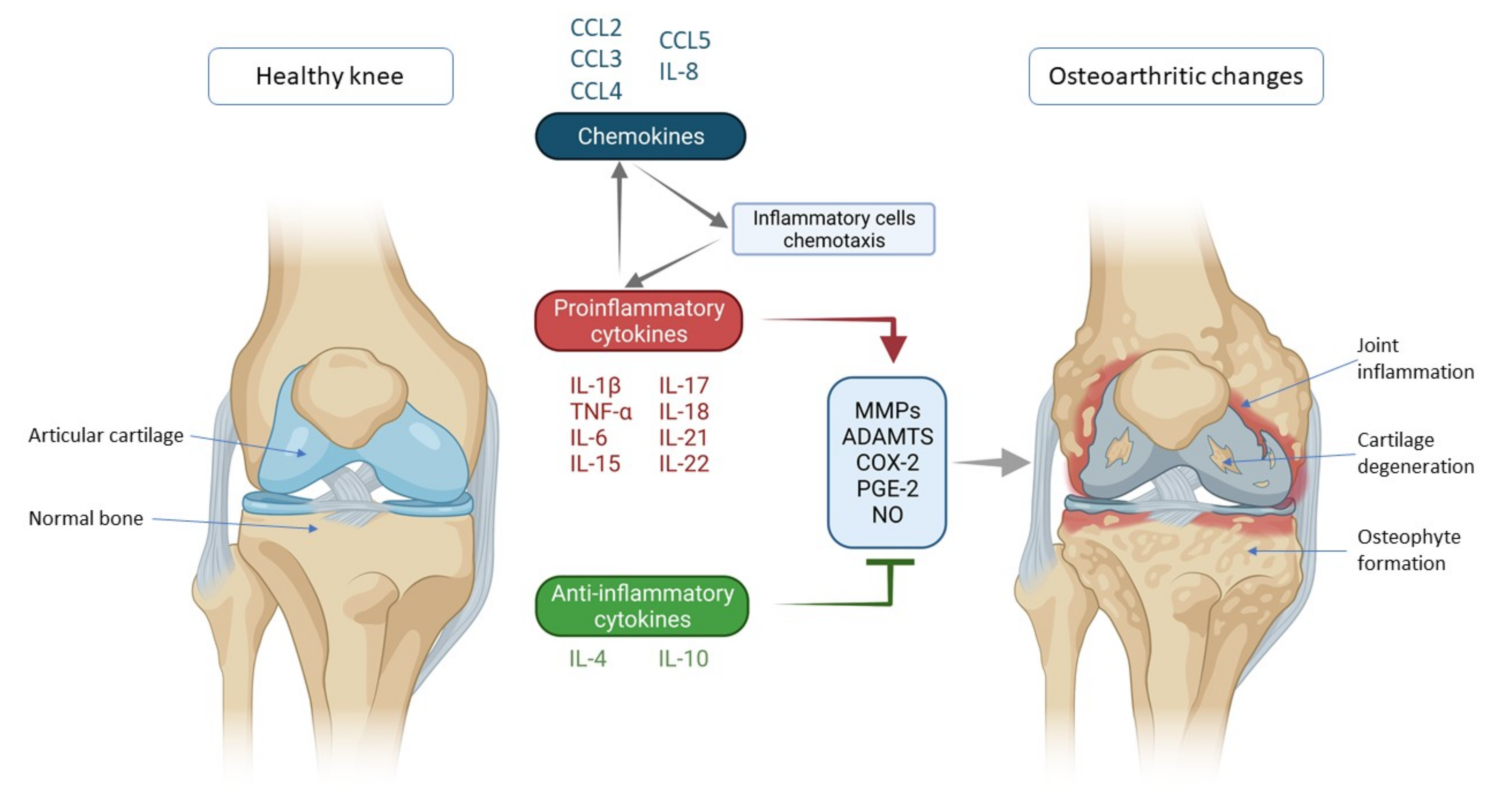
2. Cytokines and Chemokines Involved in Knee Osteoarthritis Pathogenesis
2.1. Proinflammatory Cytokines
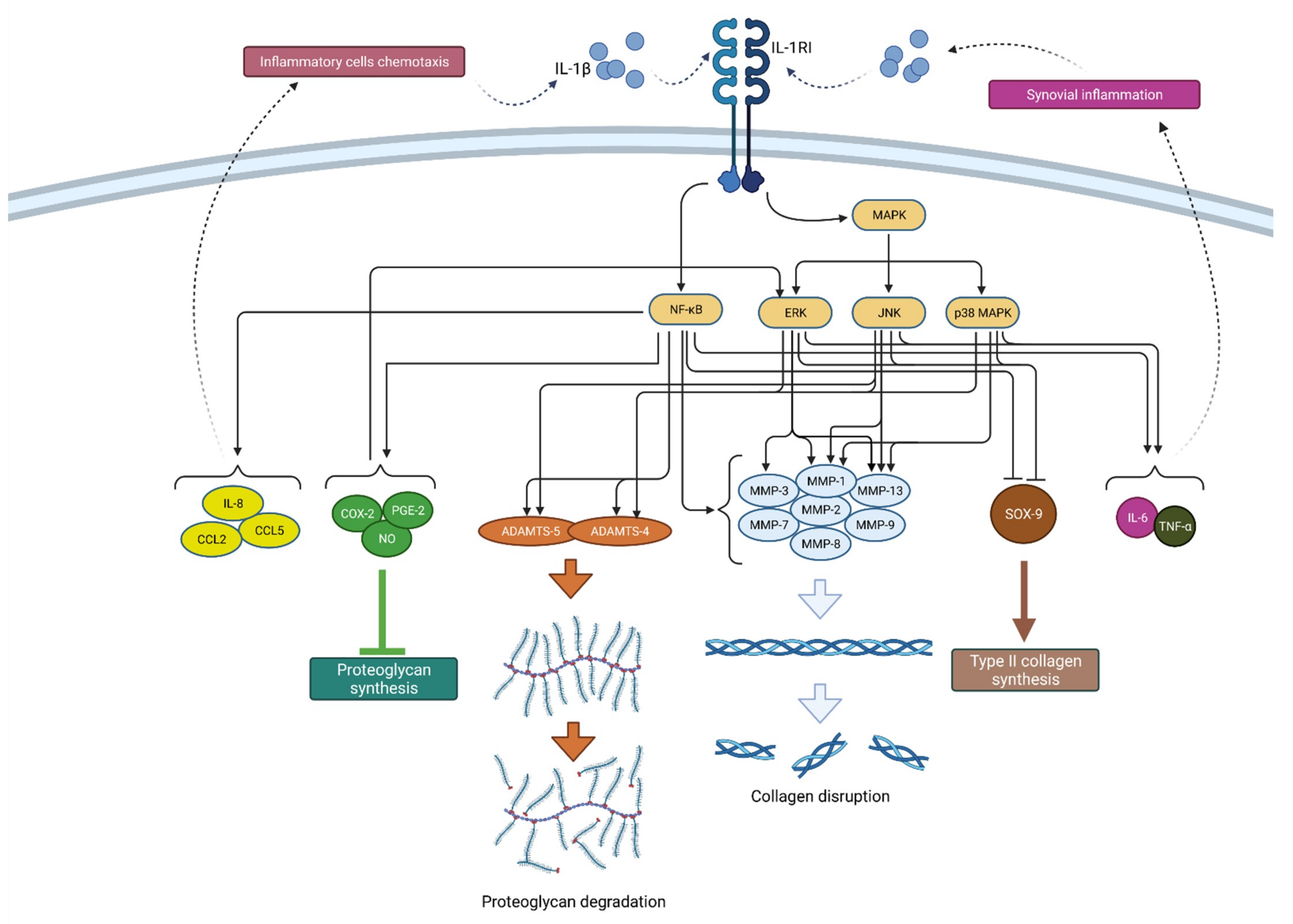
2.1.1. IL-1β
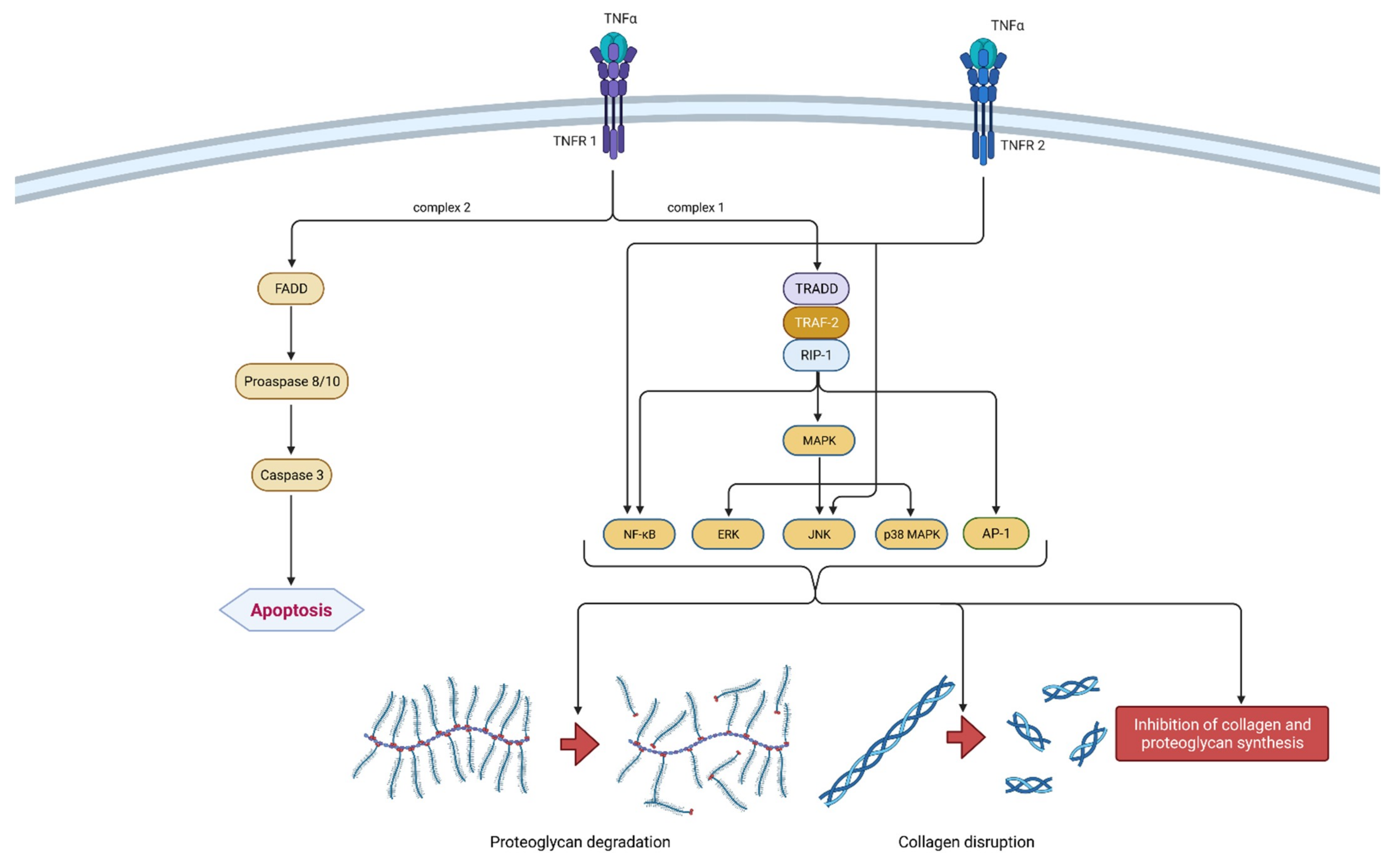
2.1.2. TNF-α
2.1.3. IL-6
2.1.4. IL-15
2.1.5. IL-17
2.1.6. IL-18
2.1.7. IL-21
2.1.8. IL-22
2.2. Anti-Inflammatory Cytokines
2.2.1. IL-4
2.2.2. IL-10
2.3. Chemokines
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Neogi, T. The epidemiology and impact of pain in osteoarthritis. Osteoarthr. Cartil. 2013, 21, 1145–1153. [Google Scholar] [CrossRef]
- Cui, A.; Li, H.; Wang, D.; Zhong, J.; Chen, Y.; Lu, H. Global, regional prevalence, incidence and risk factors of knee osteoarthritis in population-based studies. EClinicalMedicine 2020, 29–30, 100587. [Google Scholar] [CrossRef]
- Primorac, D.; Molnar, V.; Rod, E.; Jeleč, Ž.; Čukelj, F.; Matišić, V.; Vrdoljak, T.; Hudetz, D.; Hajsok, H.; Borić, I. Knee Osteoarthritis: A Review of Pathogenesis and State-Of-The-Art Non-Operative Therapeutic Considerations. Genes 2020, 11, 854. [Google Scholar] [CrossRef]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.-P.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Scanzello, C.R. Chemokines and inflammation in osteoarthritis: Insights from patients and animal models. J. Orthop. Res. 2017, 35, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural Basis of IL-1 Family Cytokine Signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Mccollum, R.; Dibattista, J.; Faure, M.-P.; Chin, J.A.; Fournier, S.; Sarfati, M.; Pelletier, J.-P. The interleukin-1 receptor in normal and osteoarthritic human articular chondrocytes. Identification as the type I receptor and analysis of binding kinetics and biologic function. Arthritis Rheum. 1992, 35, 530–540. [Google Scholar] [CrossRef]
- Attur, M.; Statnikov, A.; Samuels, J.; Li, Z.; Alekseyenko, A.V.; Greenberg, J.D.; Krasnokutsky, S.; Rybak, L.; Lu, Q.A.; Todd, J.; et al. Plasma levels of interleukin-1 receptor antagonist (IL1Ra) predict radiographic progression of symptomatic knee osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1915–1924. [Google Scholar] [CrossRef]
- Wang, X.; Li, F.; Fan, C.; Wang, C.; Ruan, H. Effects and relationship of ERK1 and ERK2 in interleukin-1β-induced alterations in MMP3, MMP13, type II collagen and aggrecan expression in human chondrocytes. Int. J. Mol. Med. 2011, 27, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-G.; Yu, S.-S.; Poo, H.; Chun, J.-S. c-Jun/Activator Protein-1 Mediates Interleukin-1β-induced Dedifferentiation but Not Cyclooxygenase-2 Expression in Articular Chondrocytes. J. Biol. Chem. 2005, 280, 29780–29787. [Google Scholar] [CrossRef]
- Jenei-Lanzl, Z.; Meurer, A.; Zaucke, F. Interleukin-1β signaling in osteoarthritis—chondrocytes in focus. Cell. Signal. 2019, 53, 212–223. [Google Scholar] [CrossRef]
- Hwang, H.; Kim, H. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef]
- Wang, X.; Li, F.; Fan, C.; Wang, C.; Ruan, H. Analysis of isoform specific ERK signaling on the effects of interleukin-1β on COX-2 expression and PGE2 production in human chondrocytes. Biochem. Biophys. Res. Commun. 2010, 402, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Chow, Y.Y.; Chin, K.-Y. The Role of Inflammation in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2020, 2020, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-B Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734. [Google Scholar] [CrossRef]
- Lepetsos, P.; Papavassiliou, K.A.; Papavassiliou, A.G. Redox and NF-κB signaling in osteoarthritis. Free Radic. Biol. Med. 2019, 132, 90–100. [Google Scholar] [CrossRef]
- Chevalier, X.; Eymard, F.; Richette, P. Biologic agents in osteoarthritis: Hopes and disappointments. Nat. Rev. Rheumatol. 2013, 9, 400–410. [Google Scholar] [CrossRef]
- Chevalier, X.; Goupille, P.; Beaulieu, A.D.; Burch, F.X.; Bensen, W.G.; Conrozier, T.; Loeuille, D.; Kivitz, A.J.; Silver, D.; Appleton, B.E. Intraarticular injection of anakinra in osteoarthritis of the knee: A multicenter, randomized, double-blind, placebo-controlled study. Arthritis Care Res. 2009, 61, 344–352. [Google Scholar] [CrossRef]
- Chevalier, X.; Eymard, F. Anti-IL-1 for the treatment of OA: Dead or alive? Nat. Rev. Rheumatol. 2019, 15, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Theeuwes, W.F.; van den Bosch, M.H.J.; Thurlings, R.M.; Blom, A.B.; van Lent, P.L.E.M. The role of inflammation in mesenchymal stromal cell therapy in osteoarthritis, perspectives for post-traumatic osteoarthritis: A review. Rheumatology 2021, 60, 1042–1053. [Google Scholar] [CrossRef]
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11, 372–377. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef]
- Kriegler, M.; Perez, C.; DeFay, K.; Albert, I.; Lu, S.D. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: Ramifications for the complex physiology of TNF. Cell 1988, 53, 45–53. [Google Scholar] [CrossRef]
- Zhou, T.; Mountz, J.D.; Kimberly, R.P. Immunobiology of tumor necrosis factor receptor superfamily. Immunol. Res. 2002, 26, 323–336. [Google Scholar] [CrossRef]
- Appay, V.; Sauce, D. Immune activation and inflammation in HIV-1 infection: Causes and consequences. J. Pathol. 2008, 214, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Westacott, C.I.; Barakat, A.F.; Wood, L.; Perry, M.J.; Neison, P.; Bisbinas, I.; Armstrong, L.; Millar, A.B.; Elson, C.J. Tumor necrosis factor alpha can contribute to focal loss of cartilage in osteoarthritis. Osteoarthr. Cartil. 2000, 8, 213–221. [Google Scholar] [CrossRef]
- Wojdasiewicz, P.; Poniatowski, Ł.A.; Szukiewicz, D. The Role of Inflammatory and Anti-Inflammatory Cytokines in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2014, 2014, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef]
- Henderson, B.; Pettipher, E.R. Arthritogenic actions of recombinant IL-1 and tumour necrosis factor α in the rabbit: Evidence for synergistic interactions between cytokines in vivo. Clin. Exp. Immunol. 1989, 75, 306–310. [Google Scholar] [PubMed]
- Séguin, C.A.; Bernier, S.M. TNFα Suppresses Link Protein and Type II Collagen Expression in Chondrocytes: Role of MEK1/2 and NF-κB Signaling Pathways. J. Cell. Physiol. 2003, 197, 356–369. [Google Scholar] [CrossRef]
- Xue, J.; Wang, J.; Liu, Q.; Luo, A. Tumor necrosis factor-α induces ADAMTS-4 expression in human osteoarthritis chondrocytes. Mol. Med. Rep. 2013, 8, 1755–1760. [Google Scholar] [CrossRef]
- Oo, W.M.; Yu, S.P.-C.; Daniel, M.S.; Hunter, D.J. Disease-modifying drugs in osteoarthritis: Current understanding and future therapeutics. Expert Opin. Emerg. Drugs 2018, 23, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Abul, K.; Abbas, M.; Lichtman, A.H.; Shiv Pillai, M. Cellular and Molecular Immunology; Elsevier: Philadeplphia, PA, USA, 2021. [Google Scholar]
- Murphy, K.M.; Weaver, C. Janeway’s Immunobiology: Ninth International Student Edition; Garland Science, Taylor & Francis Group, LLC: New York, NY, USA, 2017. [Google Scholar]
- Hirano, T.; Yasukawa, K.; Harada, H.; Taga, T.; Watanabe, Y.; Matsuda, T.; Kashiwamura, S.; Nakajima, K.; Koyama, K.; Iwamatsu, A.; et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 1986, 324, 73–76. [Google Scholar] [CrossRef]
- Hirano, T. Revisiting the 1986 Molecular Cloning of Interleukin 6. Front. Immunol. 2014, 5, 456. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028415. [Google Scholar] [CrossRef]
- Baran, P.; Hansen, S.; Waetzig, G.H.; Akbarzadeh, M.; Lamertz, L.; Huber, H.J.; Ahmadian, M.R.; Moll, J.M.; Scheller, J. The balance of interleukin (IL)-6, IL-6·soluble IL-6 receptor (sIL-6R), and IL-6·sIL-6R·sgp130 complexes allows simultaneous classic and trans-signaling. J. Biol. Chem. 2018, 293, 6762–6775. [Google Scholar] [CrossRef]
- Lutosławska, G. Interleukin-6 As An Adipokine And Myokine: The Regulatory Role Of Cytokine In Adipose Tissue And Skeletal Muscle Metabolism. Hum. Mov. 2012, 13, 372–379. [Google Scholar] [CrossRef]
- Pal, M.; Febbraio, M.A.; Whitham, M. From cytokine to myokine: The emerging role of interleukin-6 in metabolic regulation. Immunol. Cell Biol. 2014, 92, 331–339. [Google Scholar] [CrossRef]
- Akdis, M.; Burgler, S.; Crameri, R.; Eiwegger, T.; Fujita, H.; Gomez, E.; Klunker, S.; Meyer, N.; O’Mahony, L.; Palomares, O.; et al. Interleukins, from 1 to 37, and interferon-γ: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2011, 127, 701–721.e70. [Google Scholar] [CrossRef]
- Sanchez, C.; Gabay, O.; Salvat, C.; Henrotin, Y.E.; Berenbaum, F. Mechanical loading highly increases IL-6 production and decreases OPG expression by osteoblasts. Osteoarthr. Cartil. 2009, 17, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta- Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.-H.; Yang, S.; Shin, Y.; Rhee, J.; Chun, C.-H.; Chun, J.-S. Interleukin-6 plays an essential role in hypoxia-inducible factor 2α-induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum. 2011, 63, 2732–2743. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Scheller, J.; Elson, G.; Jones, S.A. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: Role in inflammation and cancer. J. Leukoc. Biol. 2006, 80, 227–236. [Google Scholar] [CrossRef]
- McGregor, N.E.; Murat, M.; Elango, J.; Poulton, I.J.; Walker, E.C.; Crimeen-Irwin, B.; Ho, P.W.M.; Gooi, J.H.; Martin, T.J.; Sims, N.A. IL-6 exhibits both cis- and trans-signaling in osteocytes and osteoblasts, but only trans-signaling promotes bone formation and osteoclastogenesis. J. Biol. Chem. 2019, 294, 7850–7863. [Google Scholar] [CrossRef]
- Reeh, H.; Rudolph, N.; Billing, U.; Christen, H.; Streif, S.; Bullinger, E.; Schliemann-Bullinger, M.; Findeisen, R.; Schaper, F.; Huber, H.J.; et al. Response to IL-6 trans- and IL-6 classic signalling is determined by the ratio of the IL-6 receptor α to gp130 expression: Fusing experimental insights and dynamic modelling. Cell Commun. Signal. 2019, 17, 46. [Google Scholar] [CrossRef]
- Rose-John, S. The soluble interleukin-6 receptor and related proteins. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 787–797. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Chanalaris, A.; Troeberg, L. ADAMTS and ADAM metalloproteinases in osteoarthritis—Looking beyond the ‘usual suspects’. Osteoarthr. Cartil. 2017, 25, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Chalaris, A.; Garbers, C.; Rabe, B.; Rose-John, S.; Scheller, J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur. J. Cell Biol. 2011, 90, 484–494. [Google Scholar] [CrossRef]
- Garbers, C.; Thaiss, W.; Jones, G.W.; Waetzig, G.H.; Lorenzen, I.; Guilhot, F.; Lissilaa, R.; Ferlin, W.G.; Grötzinger, J.; Jones, S.A.; et al. Inhibition of Classic Signaling Is a Novel Function of Soluble Glycoprotein 130 (sgp130), Which Is Controlled by the Ratio of Interleukin 6 and Soluble Interleukin 6 Receptor. J. Biol. Chem. 2011, 286, 42959–42970. [Google Scholar] [CrossRef]
- Narazaki, M.; Yasukawa, K.; Saito, T.; Ohsugi, Y.; Fukui, H.; Koishihara, Y.; Yancopoulos, G.D.; Taga, T.; Kishimoto, T. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 1993, 82, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Jostock, T.; Müllberg, J.; Özbek, S.; Atreya, R.; Blinn, G.; Voltz, N.; Fischer, M.; Neurath, M.F.; Rose-John, S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 2001, 268, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Waetzig, G.H.; Chalaris, A.; Reinheimer, T.M.; Wege, H.; Rose-John, S.; Garbers, C. Different Soluble Forms of the Interleukin-6 Family Signal Transducer gp130 Fine-tune the Blockade of Interleukin-6 Trans-signaling. J. Biol. Chem. 2016, 291, 16186–16196. [Google Scholar] [CrossRef] [PubMed]
- Silacci, P.; Dayer, J.-M.; Desgeorges, A.; Peter, R.; Manueddu, C.; Guerne, P.-A. Interleukin (IL)-6 and Its Soluble Receptor Induce TIMP-1 Expression in Synoviocytes and Chondrocytes, and Block IL-1-induced Collagenolytic Activity. J. Biol. Chem. 1998, 273, 13625–13629. [Google Scholar] [CrossRef]
- Latourte, A.; Cherifi, C.; Maillet, J.; Ea, H.-K.; Bouaziz, W.; Funck-Brentano, T.; Cohen-Solal, M.; Hay, E.; Richette, P. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann. Rheum. Dis. 2017, 76, 748–755. [Google Scholar] [CrossRef]
- Porée, B.; Kypriotou, M.; Chadjichristos, C.; Beauchef, G.; Renard, E.; Legendre, F.; Melin, M.; Gueret, S.; Hartmann, D.-J.J.; Malléin-Gerin, F.; et al. Interleukin-6 (IL-6) and/or soluble IL-6 receptor down-regulation of human type II collagen gene expression in articular chondrocytes requires a decrease of Sp1·Sp3 ratio and of the binding activity of both factors to the COL2A1 promoter. J. Biol. Chem. 2008, 283, 4850–4865. [Google Scholar] [CrossRef]
- De Hooge, A.S.K.; van de Loo, F.A.J.; Bennink, M.B.; Arntz, O.J.; de Hooge, P.; van den Berg, W.B. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthr. Cartil. 2005, 13, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Beekhuizen, M.; Gierman, L.M.M.; van Spil, W.E.E.; Van Osch, G.J.V.M.J.V.M.; Huizinga, T.W.J.W.J.; Saris, D.B.F.B.F.; Creemers, L.B.B.; Zuurmond, A.-M.M. An explorative study comparing levels of soluble mediators in control and osteoarthritic synovial fluid. Osteoarthr. Cartil. 2013, 21, 918–922. [Google Scholar] [CrossRef]
- Li, L.; Li, Z.; Li, Y.; Hu, X.; Zhang, Y.; Fan, P. Profiling of inflammatory mediators in the synovial fluid related to pain in knee osteoarthritis. BMC Musculoskelet. Disord. 2020, 21, 99. [Google Scholar] [CrossRef]
- Stannus, O.; Jones, G.; Cicuttini, F.; Parameswaran, V.; Quinn, S.; Burgess, J.; Ding, C. Circulating levels of IL-6 and TNF-α are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthr. Cartil. 2010, 18, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Goekoop, R.J.; Kloppenburg, M.; Kroon, H.M.; Frölich, M.; Huizinga, T.W.J.; Westendorp, R.G.J.; Gussekloo, J. Low innate production of interleukin-1β and interleukin-6 is associated with the absence of osteoarthritis in old age. Osteoarthr. Cartil. 2010, 18, 942–947. [Google Scholar] [CrossRef]
- Eymard, F.; Pigenet, A.; Citadelle, D.; Flouzat-Lachaniette, C.-H.; Poignard, A.; Benelli, C.; Berenbaum, F.; Chevalier, X.; Houard, X. Induction of an Inflammatory and Prodegradative Phenotype in Autologous Fibroblast-like Synoviocytes by the Infrapatellar Fat Pad From Patients With Knee Osteoarthritis. Arthritis Rheumatol. 2014, 66, 2165–2174. [Google Scholar] [CrossRef]
- Pearson, M.J.; Herndler-Brandstetter, D.; Tariq, M.A.; Nicholson, T.A.; Philp, A.M.; Smith, H.L.; Davis, E.T.; Jones, S.W.; Lord, J.M. IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci. Rep. 2017, 7, 3451. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. IL-15 in the life and death of lymphocytes: Immunotherapeutic implications. Trends Mol. Med. 2003, 9, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Allard-Chamard, H.; Mishra, H.K.; Nandi, M.; Mayhue, M.; Menendez, A.; Ilangumaran, S.; Ramanathan, S. Interleukin-15 in autoimmunity. Cytokine 2020, 136, 155258. [Google Scholar] [CrossRef] [PubMed]
- Dell’Isola, A.; Steultjens, M. Classification of patients with knee osteoarthritis in clinical phenotypes: Data from the osteoarthritis initiative. PLoS ONE 2018, 13, e0191045. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Al-Mughales, J.; Field, M.; Leung, B.P.; Huang, F.; Dixon, R.; Sturrock, R.D.; Wilkinson, P.C.; Liew, F.Y. The role of interleukin–15 in T–cell migration and activation in rheumatoid arthritis. Nat. Med. 1996, 2, 175–182. [Google Scholar] [CrossRef]
- Savio, A.S.; Diaz, A.C.M.; Capote, A.C.; Navarro, J.M.; Alvarez, Y.R.; Pérez, R.B.; del Toro, M.E.; Nieto, G.E.G. Differential expression of pro-inflammatory cytokines IL-15Ralpha, IL-15, IL-6 and TNFalpha in synovial fluid from Rheumatoid arthritis patients. BMC Musculoskelet. Disord. 2015, 16, 51. [Google Scholar] [CrossRef]
- Scanzello, C.R.R.; Umoh, E.; Pessler, F.; Diaz-Torne, C.; Miles, T.; DiCarlo, E.; Potter, H.G.G.; Mandl, L.; Marx, R.; Rodeo, S.; et al. Local cytokine profiles in knee osteoarthritis: Elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthr. Cartil. 2009, 17, 1040–1048. [Google Scholar] [CrossRef]
- Sun, J.-M.; Sun, L.-Z.; Liu, J.; Su, B.; Shi, L. Serum Interleukin-15 Levels Are Associated with Severity of Pain in Patients with Knee Osteoarthritis. Dis. Markers 2013, 35, 203–206. [Google Scholar] [CrossRef]
- Tao, Y.; Qiu, X.; Xu, C.; Sun, B.; Shi, C. Expression and correlation of matrix metalloproteinase-7 and interleukin-15 in human osteoarthritis. Int. J. Clin. Exp. Pathol. 2015, 8, 9112–9118. [Google Scholar] [PubMed]
- Constantinescu, C.S.; Grygar, C.; Kappos, L.; Leppert, D. Interleukin 15 stimulates production of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by human peripheral blood mononuclear cells. Cytokine 2001, 13, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Warner, S.C.; Nair, A.; Marpadga, R.; Chubinskaya, S.; Doherty, M.; Valdes, A.M.; Scanzello, C.R. IL-15 and IL15RA in Osteoarthritis: Association With Symptoms and Protease Production, but Not Structural Severity. Front. Immunol. 2020, 11, 1385. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Dong, C. IL-17 cytokines in immunity and inflammation. Emerg. Microbes Infect. 2013, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mimpen, J.Y.; Baldwin, M.J.; Cribbs, A.P.; Philpott, M.; Carr, A.J.; Dakin, S.G.; Snelling, S.J.B. Interleukin-17A causes osteoarthritis-like transcriptional changes in human osteoarthritis-derived chondrocytes and synovial fibroblasts in vitro. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wu, X.; Tian, J.; Wang, S. Insight Into Non-Pathogenic Th17 Cells in Autoimmune Diseases. Front. Immunol. 2018, 9, 1112. [Google Scholar] [CrossRef]
- Goldberg, M.; Nadiv, O.; Luknar-Gabor, N.; Agar, G.; Beer, Y.; Katz, Y. Synergism between tumor necrosis factor alpha and interleukin-17 to induce IL-23 p19 expression in fibroblast-like synoviocytes. Mol. Immunol. 2009, 46, 1854–1859. [Google Scholar] [CrossRef]
- Ribot, J.C.; Lopes, N.; Silva-Santos, B. γδ T cells in tissue physiology and surveillance. Nat. Rev. Immunol. 2021, 21, 221–232. [Google Scholar] [CrossRef]
- Ono, T.; Okamoto, K.; Nakashima, T.; Nitta, T.; Hori, S.; Iwakura, Y.; Takayanagi, H. IL-17-producing γδ T cells enhance bone regeneration. Nat. Commun. 2016, 7, 10928. [Google Scholar] [CrossRef]
- Li, Y.; Luo, W.; Zhu, S.; Lei, G. T Cells in Osteoarthritis: Alterations and Beyond. Front. Immunol. 2017, 8, 356. [Google Scholar] [CrossRef] [PubMed]
- Huss, R.S.; Huddleston, J.I.; Goodman, S.B.; Butcher, E.C.; Zabel, B.A. Synovial tissue-infiltrating natural killer cells in osteoarthritis and periprosthetic inflammation. Arthritis Rheum. 2010, 62, 3799–3805. [Google Scholar] [CrossRef] [PubMed]
- Jaime, P.; García-Guerrero, N.; Estella, R.; Pardo, J.; García-Álvarez, F.; Martinez-Lostao, L. CD56+/CD16− Natural Killer cells expressing the inflammatory protease granzyme A are enriched in synovial fluid from patients with osteoarthritis. Osteoarthr. Cartil. 2017, 25, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Gundle, R.; Davies, R.J.O.; Lee, Y.C.G.; McMichael, A.J.; Callan, M.F.C. CD56 bright NK Cells Are Enriched at Inflammatory Sites and Can Engage with Monocytes in a Reciprocal Program of Activation. J. Immunol. 2004, 173, 6418–6426. [Google Scholar] [CrossRef]
- Gu, C.; Wu, L.; Li, X. IL-17 family: Cytokines, receptors and signaling. Cytokine 2013, 64, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Mimpen, J.Y.; Carr, A.J.; Dakin, S.G.; Snelling, S.J. Inhibition of interleukin-17-induced effects in osteoarthritis—An in vitro study. Osteoarthr. Cartil. 2018, 26, S118. [Google Scholar] [CrossRef]
- Na, H.S.; Park, J.-S.; Cho, K.-H.; Kwon, J.Y.; Choi, J.; Jhun, J.; Kim, S.J.; Park, S.-H.; Cho, M.-L. Interleukin-1-Interleukin-17 Signaling Axis Induces Cartilage Destruction and Promotes Experimental Osteoarthritis. Front. Immunol. 2020, 11, 730. [Google Scholar] [CrossRef]
- Sinkeviciute, D.; Aspberg, A.; He, Y.; Bay-Jensen, A.-C.; Önnerfjord, P. Characterization of the interleukin-17 effect on articular cartilage in a translational model: An explorative study. BMC Rheumatol. 2020, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Huebner, J.L.; Kraus, V.B. Assessment of the utility of biomarkers of osteoarthritis in the guinea pig. Osteoarthr. Cartil. 2006, 14, 923–930. [Google Scholar] [CrossRef]
- Huebner, J.L.; Seifer, D.R.; Kraus, V.B. A longitudinal analysis of serum cytokines in the Hartley guinea pig model of osteoarthritis. Osteoarthr. Cartil. 2007, 15, 354–356. [Google Scholar] [CrossRef][Green Version]
- Chabaud, M.; Lubberts, E.; Joosten, L.; van Den Berg, W.; Miossec, P. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res. 2001, 3, 168–177. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Li, Y.; Qi, L.; Liu, X.; Yuan, C.; Hu, N.; Ma, D.; Li, Z.; Yang, Q.; et al. Increased Frequencies of Th22 Cells as well as Th17 Cells in the Peripheral Blood of Patients with Ankylosing Spondylitis and Rheumatoid Arthritis. PLoS ONE 2012, 7, e31000. [Google Scholar] [CrossRef]
- Zhang, L.; Li, J.; Liu, X.; Ma, D.; Hu, N.; Li, Y.; Li, W.; Hu, Y.; Yu, S.; Qu, X.; et al. Elevated Th22 Cells Correlated with Th17 Cells in Patients with Rheumatoid Arthritis. J. Clin. Immunol. 2011, 31, 606–614. [Google Scholar] [CrossRef]
- Qi, C.; Shan, Y.; Wang, J.; Ding, F.; Zhao, D.; Yang, T.; Jiang, Y. Circulating T helper 9 cells and increased serum interleukin-9 levels in patients with knee osteoarthritis. Clin. Exp. Pharmacol. Physiol. 2016, 43, 528–534. [Google Scholar] [CrossRef]
- Wei, M. Correlation of IL-17 Level in Synovia and Severity of Knee Osteoarthritis. Med. Sci. Monit. 2015, 21, 1732–1736. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Deng, Y.; Tan, Y.; Qin, J.; Chen, L.-B. Bin Association between severity of knee osteoarthritis and serum and synovial fluid interleukin 17 concentrations. J. Int. Med. Res. 2014, 42, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Askari, A.; Naghizadeh, M.M.; Homayounfar, R.; Shahi, A.; Afsarian, M.H.; Paknahad, A.; Kennedy, D.; Ataollahi, M.R. Increased Serum Levels of IL-17A and IL-23 Are Associated with Decreased Vitamin D3 and Increased Pain in Osteoarthritis. PLoS ONE 2016, 11, e0164757. [Google Scholar] [CrossRef]
- Jiang, L.; Zhou, X.; Xiong, Y.; Bao, J.; Xu, K.; Wu, L. Association between interleukin-17A/F single nucleotide polymorphisms and susceptibility to osteoarthritis in a Chinese population. Medicine 2019, 98, e14944. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Xu, J.; Cai, J.; Zheng, S.; Han, W.; Antony, B.; Ding, C. Serum levels of interleukin-17 and adiponectin are associated with infrapatellar fat pad volume and signal intensity alteration in patients with knee osteoarthritis. Arthritis Res. Ther. 2016, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Babaei, M.; Javadian, Y.; Narimani, H.; Ranaei, M.; Heidari, B.; Basereh, H.; Gholinia, H.; Firouzjahi, A. Correlation between systemic markers of inflammation and local synovitis in knee osteoarthritis. Casp. J. Intern. Med. 2019, 10, 383–387. [Google Scholar] [CrossRef]
- Snelling, S.J.B.B.; Bas, S.; Puskas, G.J.; Dakin, S.G.; Suva, D.; Finckh, A.; Gabay, C.; Hoffmeyer, P.; Carr, A.J.; Lübbeke, A. Presence of IL-17 in synovial fluid identifies a potential inflammatory osteoarthritic phenotype. PLoS ONE 2017, 12, e0175109. [Google Scholar] [CrossRef]
- Wang, K.; Xu, J.; Cai, J.; Zheng, S.; Yang, X.; Ding, C. Serum levels of resistin and interleukin-17 are associated with increased cartilage defects and bone marrow lesions in patients with knee osteoarthritis. Mod. Rheumatol. 2017, 27, 339–344. [Google Scholar] [CrossRef]
- Lee, Y.H.; Song, G.G. Association between IL-17 gene polymorphisms and circulating IL-17 levels in osteoarthritis: A meta-analysis. Z. Rheumatol. 2020, 79, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-Y.; Liu, Y.-Z.; Zhou, X.-D.; Huang, Y.; Xu, N.-W. Role of IL-17 gene polymorphisms in osteoarthritis: A meta-analysis based on observational studies. World J. Clin. Cases 2020, 8, 2280–2293. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, D.; Long, L.; Deng, X.; Tao, R.; Huang, G. Correlation between plasma, synovial fluid and articular cartilage Interleukin-18 with radiographic severity in 33 patients with osteoarthritis of the knee. Clin. Exp. Med. 2014, 14, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Hiraoka, K.; Hoshino, T.; Okamoto, M.; Iwanaga, T.; Zenmyo, M.; Shoda, T.; Aizawa, H.; Nagata, K. High levels of serum IL-18 promote cartilage loss through suppression of aggrecan synthesis. Bone 2008, 42, 1102–1110. [Google Scholar] [CrossRef]
- Kaplanski, G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol. Rev. 2018, 281, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Panina, S.B.; Krolevets, I.V.; Milyutina, N.P.; Sagakyants, A.B.; Kornienko, I.V.; Ananyan, A.A.; Zabrodin, M.A.; Plotnikov, A.A.; Vnukov, V.V. Circulating levels of proinflammatory mediators as potential biomarkers of post-traumatic knee osteoarthritis development. J. Orthop. Traumatol. 2017, 18, 349–357. [Google Scholar] [CrossRef]
- Fu, Z.; Liu, P.; Yang, D.; Wang, F.; Yuan, L.; Lin, Z.; Jiang, J. Interleukin-18-induced inflammatory responses in synoviocytes and chondrocytes from osteoarthritic patients. Int. J. Mol. Med. 2012, 30, 805–810. [Google Scholar] [CrossRef]
- Dai, S.-M. Implication of interleukin 18 in production of matrix metalloproteinases in articular chondrocytes in arthritis: Direct effect on chondrocytes may not be pivotal. Ann. Rheum. Dis. 2005, 64, 735–742. [Google Scholar] [CrossRef]
- Olee, T.; Hashimoto, S.; Quach, J.; Lotz, M. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J. Immunol. 1999, 162, 1096–1100. [Google Scholar]
- Futani, H.; Okayama, A.; Matsui, K.; Kashiwamura, S.; Sasaki, T.; Hada, T.; Nakanishi, K.; Tateishi, H.; Maruo, S.; Okamura, H. Relation Between Interleukin-18 and PGE2 in Synovial Fluid of Osteoarthritis: A Potential Therapeutic Target of Cartilage Degradation. J. Immunother. 2002, 25, S61–S64. [Google Scholar] [CrossRef]
- Lee, J.-K.; Kim, S.-H.; Lewis, E.C.; Azam, T.; Reznikov, L.L.; Dinarello, C.A. Differences in signaling pathways by IL-1 and IL-18. Proc. Natl. Acad. Sci. USA 2004, 101, 8815–8820. [Google Scholar] [CrossRef]
- Miyaura, C.; Inada, M.; Suzawa, T.; Sugimoto, Y.; Ushikubi, F.; Ichikawa, A.; Narumiya, S.; Suda, T. Impaired bone resorption to prostaglandin E2 in prostaglandin E receptor EP4-knockout mice. J. Biol. Chem. 2000, 275, 19819–19823. [Google Scholar] [CrossRef] [PubMed]
- Scharstuhl, A.; Glansbeek, H.L.; van Beuningen, H.M.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Inhibition of Endogenous TGF-β During Experimental Osteoarthritis Prevents Osteophyte Formation and Impairs Cartilage Repair. J. Immunol. 2002, 169, 507–514. [Google Scholar] [CrossRef]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef]
- Xing, R.; Yang, L.; Jin, Y.; Sun, L.; Li, C.; Li, Z.; Zhao, J.; Liu, X. Interleukin-21 Induces Proliferation and Proinflammatory Cytokine Profile of Fibroblast-like Synoviocytes of Patients with Rheumatoid Arthritis. Scand. J. Immunol. 2016, 83, 64–71. [Google Scholar] [CrossRef]
- Li, J.; Shen, W.; Kong, K.; Liu, Z. Interleukin-21 Induces T-cell Activation and Proinflammatory Cytokine Secretion in Rheumatoid Arthritis. Scand. J. Immunol. 2006, 64, 515–522. [Google Scholar] [CrossRef]
- Shan, Y.; Qi, C.; Liu, Y.; Gao, H.; Zhao, D.; Jiang, Y. Increased frequency of peripheral blood follicular helper T cells and elevated serum IL-21 levels in patients with knee osteoarthritis. Mol. Med. Rep. 2017, 15, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Dudakov, J.A.; Hanash, A.M.; van den Brink, M.R.M. Interleukin-22: Immunobiology and Pathology. Annu. Rev. Immunol. 2015, 33, 747–785. [Google Scholar] [CrossRef]
- Deligne, C.; Casulli, S.; Pigenet, A.; Bougault, C.; Campillo-Gimenez, L.; Nourissat, G.; Berenbaum, F.; Elbim, C.; Houard, X. Differential expression of interleukin-17 and interleukin-22 in inflamed and non-inflamed synovium from osteoarthritis patients. Osteoarthr. Cartil. 2015, 23, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Yi, Y.; Wei, J.; Jin, Q.; Li, J.; Sacitharan, P.K. Targeting IL-22 and IL-22R protects against experimental osteoarthritis. Cell. Mol. Immunol. 2021, 18, 1329–1331. [Google Scholar] [CrossRef] [PubMed]
- Carrión, M.; Juarranz, Y.; Martínez, C.; González-Álvaro, I.; Pablos, J.L.; Gutiérrez-Cañas, I.; Gomariz, R.P. IL-22/IL-22R1 axis and S100A8/A9 alarmins in human osteoarthritic and rheumatoid arthritis synovial fibroblasts. Rheumatology 2013, 52, 2177–2186. [Google Scholar] [CrossRef]
- Brown, M.A.; Hural, J. Functions of IL-4 and control of its expression. Crit. Rev. Immunol. 2017, 37, 181–212. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Shukla, M.; Yakubenko, V.P.; Mulya, A.; Kundu, S.; Cathcart, M.K. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic. Biol. Med. 2013, 54, 1–16. [Google Scholar] [CrossRef]
- Forster, T.; Chapman, K.; Loughlin, J. Common variants within the interleukin 4 receptor α gene (IL4R) are associated with susceptibility to osteoarthritis. Hum. Genet. 2004, 114, 391–395. [Google Scholar] [CrossRef]
- Schlaak, J.F.; Pfers, I.; Meyer Zum Büschenfelde, K.H.; Märker-Hermann, E. Different cytokine profiles in the synovial fluid of patients with osteoarthritis, rheumatoid arthritis and seronegative spondylarthropathies. Clin. Exp. Rheumatol. 1996, 14, 155–162. [Google Scholar]
- Wagner, S.; Fritz, P.; Einsele, H.; Sell, S.; Saal, J.G. Evaluation of synovial cytokine patterns in rheumatoid arthritis and osteoarthritis by quantitative reverse transcription polymerase chain reaction. Rheumatol. Int. 1997, 16, 191–196. [Google Scholar] [CrossRef]
- Ishii, H.; Tanaka, H.; Katoh, K.; Nakamura, H.; Nagashima, M.; Yoshino, S. Characterization of infiltrating T cells and Th1/Th2-type cytokines in the synovium of patients with osteoarthritis. Osteoarthr. Cartil. 2002, 10, 277–281. [Google Scholar] [CrossRef]
- Yeh, L.A.; Augustine, A.J.; Lee, P.; Riviere, L.R.; Sheldon, A. Interleukin-4, an inhibitor of cartilage breakdown in bovine articular cartilage explants. J. Rheumatol. 1995, 22, 1740–1746. [Google Scholar]
- Doi, H.; Nishida, K.; Yorimitsu, M.; Komiyama, T.; Kadota, Y.; Tetsunaga, T.; Yoshida, A.; Kubota, S.; Takigawa, M.; Ozaki, T. Interleukin-4 downregulates the cyclic tensile stress-induced matrix metalloproteinases-13 and cathepsin B expression by rat normal chondrocytes. Acta Med. Okayama 2008, 62, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Van Meegeren, M.E.R.; Roosendaal, G.; Jansen, N.W.D.; Wenting, M.J.G.; Van Wesel, A.C.W.; Van Roon, J.A.G.; Lafeber, F.P.J.G. IL-4 alone and in combination with IL-10 protects against blood-induced cartilage damage. Osteoarthr. Cartil. 2012, 20, 764–772. [Google Scholar] [CrossRef]
- Schuerwegh, A.J.; Dombrecht, E.J.; Stevens, W.J.; Van Offel, J.F.; Bridts, C.H.; De Clerck, L.S. Influence of pro-inflammatory (IL-1α, IL-6, TNF-α, IFN-γ) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthr. Cartil. 2003, 11, 681–687. [Google Scholar] [CrossRef]
- Schulze-Tanzil, G.; Zreiqat, H.; Sabat, R.; Kohl, B.; Halder, A.; Muller, R.; John, T. Interleukin-10 and Articular Cartilage: Experimental Therapeutical Approaches in Cartilage Disorders. Curr. Gene Ther. 2009, 9, 306–315. [Google Scholar] [CrossRef]
- Mostafa, E.; Chollet-martin, S.; Oudghiri, M.; Laquay, N.; Jacob, M.; Michel, J.; Feldman, L.J. Effects of interleukin-10 on monocyte / endothelial cell adhesion and MMP-9/TIMP-1 secretion. Cardiovasc. Res. 2001, 49, 882–890. [Google Scholar] [CrossRef]
- Wang, Y.S.; Wang, Y.H.; Zhao, G.Q.; Li, Y.B. Osteogenic potential of human calcitonin gene-related peptide alpha gene-modified bone marrow mesenchymal stem cells. Chin. Med. J. 2011, 124, 3976–3981. [Google Scholar] [CrossRef]
- Behrendt, P.; Feldheim, M.; Preusse-Prange, A.; Weitkamp, J.T.; Haake, M.; Eglin, D.; Rolauffs, B.; Fay, J.; Seekamp, A.; Grodzinsky, A.J.; et al. Chondrogenic potential of IL-10 in mechanically injured cartilage and cellularized collagen ACI grafts. Osteoarthr. Cartil. 2018, 26, 264–275. [Google Scholar] [CrossRef]
- Barker, T.; Rogers, V.E.; Henriksen, V.T.; Trawick, R.H.; Momberger, N.G.; Lynn Rasmussen, G. Circulating IL-10 is compromised in patients predisposed to developing and in patients with severe knee osteoarthritis. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Helmark, I.C.; Mikkelsen, U.R.; Børglum, J.; Rothe, A.; Petersen, M.C.; Andersen, O.; Langberg, H.; Kjaer, M. Exercise increases interleukin-10 levels both intraarticularly and peri-synovially in patients with knee osteoarthritis: A randomized controlled trial. Arthritis Res. Ther. 2010, 12, R126. [Google Scholar] [CrossRef]
- Fernandes, T.L.; Gomoll, A.H.; Lattermann, C.; Hernandez, A.J.; Bueno, D.F.; Amano, M.T. Macrophage: A Potential Target on Cartilage Regeneration. Front. Immunol. 2020, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Rong, Y.; Luo, C.; Cui, W. Bone marrow mesenchymal stem cell-derived exosomes prevent osteoarthritis by regulating synovial macrophage polarization. Aging 2020, 12, 25138–25152. [Google Scholar] [CrossRef]
- Watkins, L.R.; Chavez, R.A.; Landry, R.; Fry, M.; Green-Fulgham, S.M.; Coulson, J.D.; Collins, S.D.; Glover, D.K.; Rieger, J.; Forsayeth, J.R. Targeted interleukin-10 plasmid DNA therapy in the treatment of osteoarthritis: Toxicology and pain efficacy assessments. Brain. Behav. Immun. 2020, 90, 155–166. [Google Scholar] [CrossRef]
- Borish, L.C.; Steinke, J.W. 2. Cytokines and chemokines. J. Allergy Clin. Immunol. 2003, 111, S460–S475. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta- Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interf. Cytokine Res. 2009, 29, 313–325. [Google Scholar] [CrossRef]
- Monibi, F.; Roller, B.L.; Stoker, A.; Garner, B.; Bal, S.; Cook, J.L. Identification of Synovial Fluid Biomarkers for Knee Osteoarthritis and Correlation with Radiographic Assessment. J. Knee Surg. 2016, 29, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.E.; Paterson, E.; Freidin, A.; Kenny, M.; Judge, A.; Saklatvala, J.; Williams, A.; Vincent, T.L. Acute Molecular Changes in Synovial Fluid Following Human Knee Injury: Association With Early Clinical Outcomes. Arthritis Rheumatol. 2016, 68, 2129–2140. [Google Scholar] [CrossRef]
- Miller, R.E.; Tran, P.B.; Das, R.; Ghoreishi-Haack, N.; Ren, D.; Miller, R.J.; Malfait, A.M. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc. Natl. Acad. Sci. USA 2012, 109, 20602–20607. [Google Scholar] [CrossRef]
- Miller, R.E.; Belmadani, A.; Ishihara, S.; Tran, P.B.; Ren, D.; Miller, R.J.; Malfait, A.M. Damage-associated molecular patterns generated in osteoarthritis directly excite murine nociceptive neurons through toll-like receptor 4. Arthritis Rheumatol. 2015, 67, 2933–2943. [Google Scholar] [CrossRef] [PubMed]
- Hulin-Curtis, S.L.; Bidwell, J.L.; Perry, M.J. Association between CCL2 haplotypes and knee osteoarthritis. Int. J. Immunogenet. 2013, 40, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Borzí, R.M.; Mazzetti, I.; Cattini, L.; Uguccioni, M.; Baggiolini, M.; Facchini, A. Human chondrocytes express functional chemokine receptors and release matrix-degrading enzymes in response to C-X-C and C-C chemokines. Arthritis Rheum. 2000, 43, 1734–1741. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Yang, Z.B.; Zhang, Z.J.; Zhang, Z.Q.; Kang, Y.; Huang, G.X.; Wang, S.W.; Huang, H.; Liao, W.M. CCL3 serves as a potential plasma biomarker in knee degeneration (osteoarthritis). Osteoarthr. Cartil. 2015, 23, 1405–1411. [Google Scholar] [CrossRef]
- Takebe, K.; Rai, M.F.; Schmidt, E.J.; Sandell, L.J. The chemokine receptor CCR5 plays a role in post-traumatic cartilage loss in mice, but does not affect synovium and bone. Osteoarthr. Cartil. 2015, 23, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.J.; Xing, X.; Franz, C.; Davies, S.; Chang, L.W.; Patra, D. Exuberant expression of chemokine genes by adult human articular chondrocytes in response to IL-1β. Osteoarthr. Cartil. 2008, 16, 1560–1571. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef]
- Merz, D.; Liu, R.; Johnson, K.; Terkeltaub, R. IL-8/CXCL8 and Growth-Related Oncogene α/CXCL1 Induce Chondrocyte Hypertrophic Differentiation. J. Immunol. 2003, 171, 4406–4415. [Google Scholar] [CrossRef] [PubMed]
- Galliera, E.; Locati, M.; Mantovani, A.; Corsi, M.M. Chemokines and bone remodeling. Int. J. Immunopathol. Pharmacol. 2008, 21, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Zhang, Y.; Li, Q. Therapeutic mechanisms of ibuprofen, prednisone and betamethasone in osteoarthritis. Mol. Med. Rep. 2017, 15, 981–987. [Google Scholar] [CrossRef]
- Rasheed, Z.; Akhtar, N.; Haqqi, T.M. Advanced glycation end products induce the expression of interleukin-6 and interleukin-8 by receptor for advanced glycation end product-mediated activation of mitogen-activated protein kinases and nuclear factor-κB in human osteoarthritis chondrocytes. Rheumatology 2011, 50, 838–851. [Google Scholar] [CrossRef]
- Takahashi, A.; de Andrés, M.C.; Hashimoto, K.; Itoi, E.; Oreffo, R.O.C. Epigenetic regulation of interleukin-8, an inflammatory chemokine, in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1946–1954. [Google Scholar] [CrossRef]
- Frommer, K.W.; Hasseli, R.; Schäffler, A.; Lange, U.; Rehart, S.; Steinmeyer, J.; Rickert, M.; Sarter, K.; Zaiss, M.M.; Culmsee, C.; et al. Free Fatty Acids in Bone Pathophysiology of Rheumatic Diseases. Front. Immunol. 2019, 10, 2757. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, S.G.; Zhang, F.J.; Luo, W.; Xue, J.X.; Lei, G.H. Effects of osteopontin on the expression of IL-6 and IL-8 inflammatory factors in human knee osteoarthritis chondrocytes. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3580–3586. [Google Scholar]
- Sakao, K.; Takahashi, K.A.; Arai, Y.; Saito, M.; Honjo, K.; Hiraoka, N.; Asada, H.; Shin-Ya, M.; Imanishi, J.; Mazda, O.; et al. Osteoblasts derived from osteophytes produce interleukin-6, interleukin-8, and matrix metalloproteinase-13 in osteoarthritis. J. Bone Miner. Metab. 2009, 27, 412–423. [Google Scholar] [CrossRef]
- Remick, D.G.; DeForge, L.E.; Sullivan, J.F.; Showell, H.J. Profile of cytokines in synovial fluid specimens from patients with arthritis. Immunol. Invest. 1992, 21, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.A.; Choi, H.M.; Lee, S.H.; Yang, H.I.; Yoo, M.C.; Hong, S.J.; Kim, K.S. Synergy between adiponectin and interleukin-1β on the expression of interleukin-6, interleukin-8, and cyclooxygenase-2 in fibroblast-like synoviocytes. Exp. Mol. Med. 2012, 44, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Furuzawa-Carballeda, J.; Alcocer-Varela, J. Interleukin-8, interleukin- 10, intercellular adhesion molecule- 1 and vascular cell adhesion molecule-1 expression levels are higher in synovial tissue from patients with rheumatoid arthritis than in osteoarthritis. Scand. J. Immunol. 1999, 50, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Bertazzolo, N.; Punzi, L.; Stefani, M.P.; Cesaro, G.; Pianon, M.; Finco, B.; Todesco, S. Interrelationships between interleukin (IL)-1, IL-6 and IL-8 in synovial fluid of various arthropathies. Agents Actions 1994, 41, 90–92. [Google Scholar] [CrossRef]
- Valcamonica, E.; Chighizola, C.B.; Comi, D.; De Lucia, O.; Pisoni, L.; Murgo, A.; Salvi, V.; Sozzani, S.; Meroni, P.L. Levels of chemerin and interleukin 8 in the synovial fluid of patients with inflammatory arthritides and osteoarthritis. Clin. Exp. Rheumatol. 2014, 32, 243–250. [Google Scholar]
- Kaneko, S.; Satoh, T.; Chiba, J.; Ju, C.; Inoue, K.; Kagawa, J. Interleukin-6 and interleukin-8 levels in serum and synovial fluid of patients with osteoarthritis. Cytokines Cell. Mol. Ther. 2000, 6, 71–79. [Google Scholar] [CrossRef]
- Koh, S.M.M.; Chan, C.K.K.; Teo, S.H.H.; Singh, S.; Merican, A.; Ng, W.M.M.; Abbas, A.; Kamarul, T. Elevated plasma and synovial fluid interleukin-8 and interleukin-18 may be associated with the pathogenesis of knee osteoarthritis. Knee 2020, 27, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.I.; Conzemius, M.G.; Evans, R.B.; Kiefer, K. Correlation between synovial fluid cytokine concentrations and limb function in normal dogs and in dogs with lameness from spontaneous osteoarthritis. Vet. Surg. 2019, 48, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Kleine, S.A.; Gogal, R.M.; George, C.; Thaliath, M.; Budsberg, S.C. Elevated Synovial Fluid Concentration of Monocyte Chemoattractant Protein-1 and Interleukin-8 in Dogs with Osteoarthritis of the Stifle. Vet. Comp. Orthop. Traumatol. 2020, 33, 147–150. [Google Scholar] [CrossRef]
- García-Manrique, M.; Calvet, J.; Orellana, C.; Berenguer-Llergo, A.; Garcia-Cirera, S.; Llop, M.; Albiñana-Giménez, N.; Galisteo-Lencastre, C.; Gratacós, J. Synovial fluid but not plasma interleukin-8 is associated with clinical severity and inflammatory markers in knee osteoarthritis women with joint effusion. Sci. Rep. 2021, 11, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.; Xu, J.; Wang, K.; Zheng, S.; Wu, J.; Bian, F.; Chang, B.; Zhang, Y.; Meng, T.; Zhu, Z.; et al. Associations between serum IL-8 and knee symptoms, joint structures, and cartilage or bone biomarkers in patients with knee osteoarthritis. Clin. Rheumatol. 2019, 38, 3609–3617. [Google Scholar] [CrossRef]
- Cecil, D.L.; Rose, D.M.; Terkeltaub, R.; Liu-Bryan, R. Role of interleukin-8 in PiT-1 expression and CXCR1-mediated inorganic phosphate uptake in chondrocytes. Arthritis Rheum. 2005, 52, 144–154. [Google Scholar] [CrossRef]
- Marquez-Curtis, L.A.; Janowska-Wieczorek, A. Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 axis. Biomed Res. Int. 2013, 2013, 1–15. [Google Scholar] [CrossRef]
- Shen, W.; Chen, J.; Zhu, T.; Chen, L.; Zhang, W.; Fang, Z.; Heng, B.C.; Yin, Z.; Chen, X.; Ji, J.; et al. Intra-Articular Injection of Human Meniscus Stem/Progenitor Cells Promotes Meniscus Regeneration and Ameliorates Osteoarthritis Through Stromal Cell-Derived Factor-1/CXCR4-Mediated Homing. Stem Cells Transl. Med. 2014, 3, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Tian, J.; Pan, H.; Gao, J.; Yao, M. Association of CCL13 levels in serum and synovial fluid with the radiographic severity of knee osteoarthritis. J. Investig. Med. 2015, 63, 545–547. [Google Scholar] [CrossRef]
- Wei, F.; Moore, D.C.; Wei, L.; Li, Y.; Zhang, G.; Wei, X.; Lee, J.K.; Chen, Q. Correction: Attenuation of osteoarthritis via blockade of the SDF-1/CXCR4 signaling pathway. Arthritis Res. Ther. 2013, 15, 410. [Google Scholar] [CrossRef]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Primorac, D.; Molnar, V.; Matišić, V.; Hudetz, D.; Jeleč, Ž.; Rod, E.; Čukelj, F.; Vidović, D.; Vrdoljak, T.; Dobričić, B.; et al. Comprehensive Review of Knee Osteoarthritis Pharmacological Treatment and the Latest Professional Societies’ Guidelines. Pharmaceuticals 2021, 14, 205. [Google Scholar] [CrossRef]
- Messina, O.D.; Vidal Wilman, M.; Vidal Neira, L.F. Nutrition, osteoarthritis and cartilage metabolism. Aging Clin. Exp. Res. 2019, 31, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Malorgio, A.; Malorgio, M.; Benedetti, M.; Casarosa, S.; Cannataro, R. High intensity resistance training as intervention method to knee osteoarthritis. Sport. Med. Health Sci. 2021, 3, 46–48. [Google Scholar] [CrossRef]
- Hill, C.L.; March, L.M.; Aitken, D.; Lester, S.E.; Battersby, R.; Hynes, K.; Fedorova, T.; Proudman, S.M.; James, M.; Cleland, L.G.; et al. Fish oil in knee osteoarthritis: A randomised clinical trial of low dose versus high dose. Ann. Rheum. Dis. 2016, 75, 23–29. [Google Scholar] [CrossRef]
- Schell, J.; Scofield, R.; Barrett, J.; Kurien, B.; Betts, N.; Lyons, T.; Zhao, Y.; Basu, A. Strawberries Improve Pain and Inflammation in Obese Adults with Radiographic Evidence of Knee Osteoarthritis. Nutrients 2017, 9, 949. [Google Scholar] [CrossRef]
- Cohen, S.B.; Proudman, S.; Kivitz, A.J.; Burch, F.X.; Donohue, J.P.; Burstein, D.; Sun, Y.N.; Banfield, C.; Vincent, M.S.; Ni, L.; et al. A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res. Ther. 2011, 13, R125. [Google Scholar] [CrossRef] [PubMed]
- Verbruggen, G.; Wittoek, R.; Vander Cruyssen, B.; Elewaut, D. Tumour necrosis factor blockade for the treatment of erosive osteoarthritis of the interphalangeal finger joints: A double blind, randomised trial on structure modification. Ann. Rheum. Dis. 2012, 71, 891–898. [Google Scholar] [CrossRef]
- Cosenza, S.; Ruiz, M.; Toupet, K.; Jorgensen, C.; Noël, D. Mesenchymal stem cells derived exosomes and microparticles protect cartilage and bone from degradation in osteoarthritis. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Petryk, N.; Shevchenko, O. Mesenchymal stem cells anti-inflammatory activity in rats: Proinflammatory cytokines. J. Inflamm. Res. 2020, 13, 293–301. [Google Scholar] [CrossRef]
- Kyurkchiev, D. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J. Stem Cells 2014, 6, 552. [Google Scholar] [CrossRef]
- Hudetz, D.; Borić, I.; Rod, E.; Jeleč, Ž.; Radić, A.; Vrdoljak, T.; Skelin, A.; Lauc, G.; Trbojević-Akmačić, I.; Plečko, M.; et al. The effect of intra-articular injection of autologous microfragmented fat tissue on proteoglycan synthesis in patients with knee osteoarthritis. Genes 2017, 8, 270. [Google Scholar] [CrossRef]
- Hudetz, D.; Borić, I.; Rod, E.; Jeleč, Ž.; Kunovac, B.; Polašek, O.; Vrdoljak, T.; Plečko, M.; Skelin, A.; Polančec, D.; et al. Early results of intra-articular micro-fragmented lipoaspirate treatment in patients with late stages knee osteoarthritis: A prospective study. Croat. Med. J. 2019, 60, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Polancec, D.; Zenic, L.; Hudetz, D.; Boric, I.; Jelec, Z.; Rod, E.; Primorac, D. Immunophenotyping of a Stromal Vascular Fraction from Microfragmented Lipoaspirate Used in Osteoarthritis Cartilage Treatment and Its Lipoaspirate Counterpart. Genes 2019, 10, 474. [Google Scholar] [CrossRef] [PubMed]
- Borić, I.; Hudetz, D.; Rod, E.; Jeleč, Ž.; Vrdoljak, T.; Skelin, A.; Polašek, O.; Plečko, M.; Trbojević-Akmačić, I.; Lauc, G.; et al. A 24-month follow-up study of the effect of intra-articular injection of autologous microfragmented fat tissue on proteoglycan synthesis in patients with knee osteoarthritis. Genes 2019, 10, 1051. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molnar, V.; Matišić, V.; Kodvanj, I.; Bjelica, R.; Jeleč, Ž.; Hudetz, D.; Rod, E.; Čukelj, F.; Vrdoljak, T.; Vidović, D.; et al. Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9208. https://doi.org/10.3390/ijms22179208
Molnar V, Matišić V, Kodvanj I, Bjelica R, Jeleč Ž, Hudetz D, Rod E, Čukelj F, Vrdoljak T, Vidović D, et al. Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. International Journal of Molecular Sciences. 2021; 22(17):9208. https://doi.org/10.3390/ijms22179208
Chicago/Turabian StyleMolnar, Vilim, Vid Matišić, Ivan Kodvanj, Roko Bjelica, Željko Jeleč, Damir Hudetz, Eduard Rod, Fabijan Čukelj, Trpimir Vrdoljak, Dinko Vidović, and et al. 2021. "Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis" International Journal of Molecular Sciences 22, no. 17: 9208. https://doi.org/10.3390/ijms22179208
APA StyleMolnar, V., Matišić, V., Kodvanj, I., Bjelica, R., Jeleč, Ž., Hudetz, D., Rod, E., Čukelj, F., Vrdoljak, T., Vidović, D., Starešinić, M., Sabalić, S., Dobričić, B., Petrović, T., Antičević, D., Borić, I., Košir, R., Zmrzljak, U. P., & Primorac, D. (2021). Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. International Journal of Molecular Sciences, 22(17), 9208. https://doi.org/10.3390/ijms22179208