
Gene Therapy for Neuronopathic Mucopolysaccharidoses: State of the Art

 ,
, 

Abstract
:1. Introduction
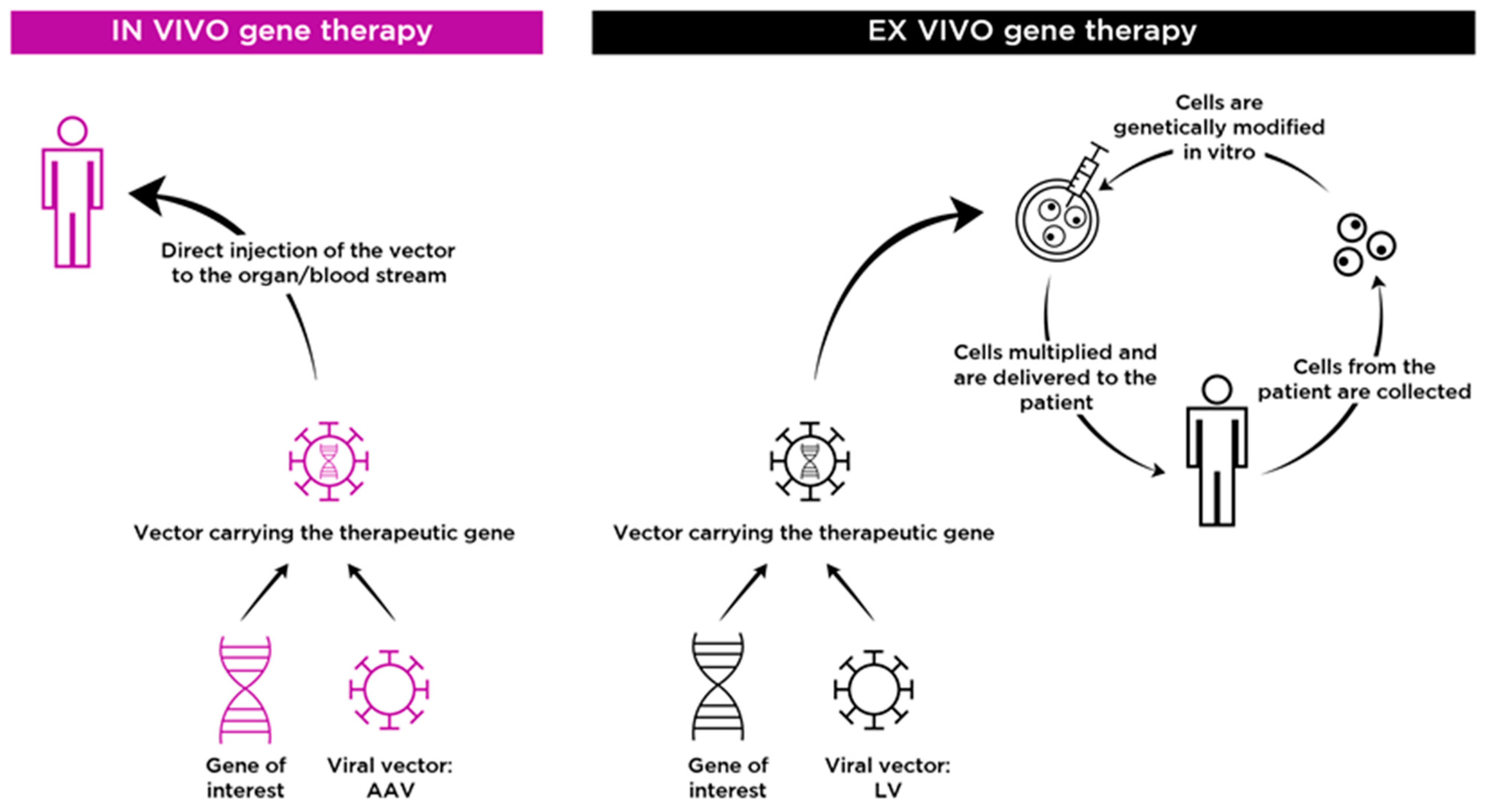
2. In Vivo Gene Therapy for Neuronopathic MPS
2.1. Adeno-Associated Virus (AAV)
2.2. Humoral and Cellular Immunity to AAV Vectors
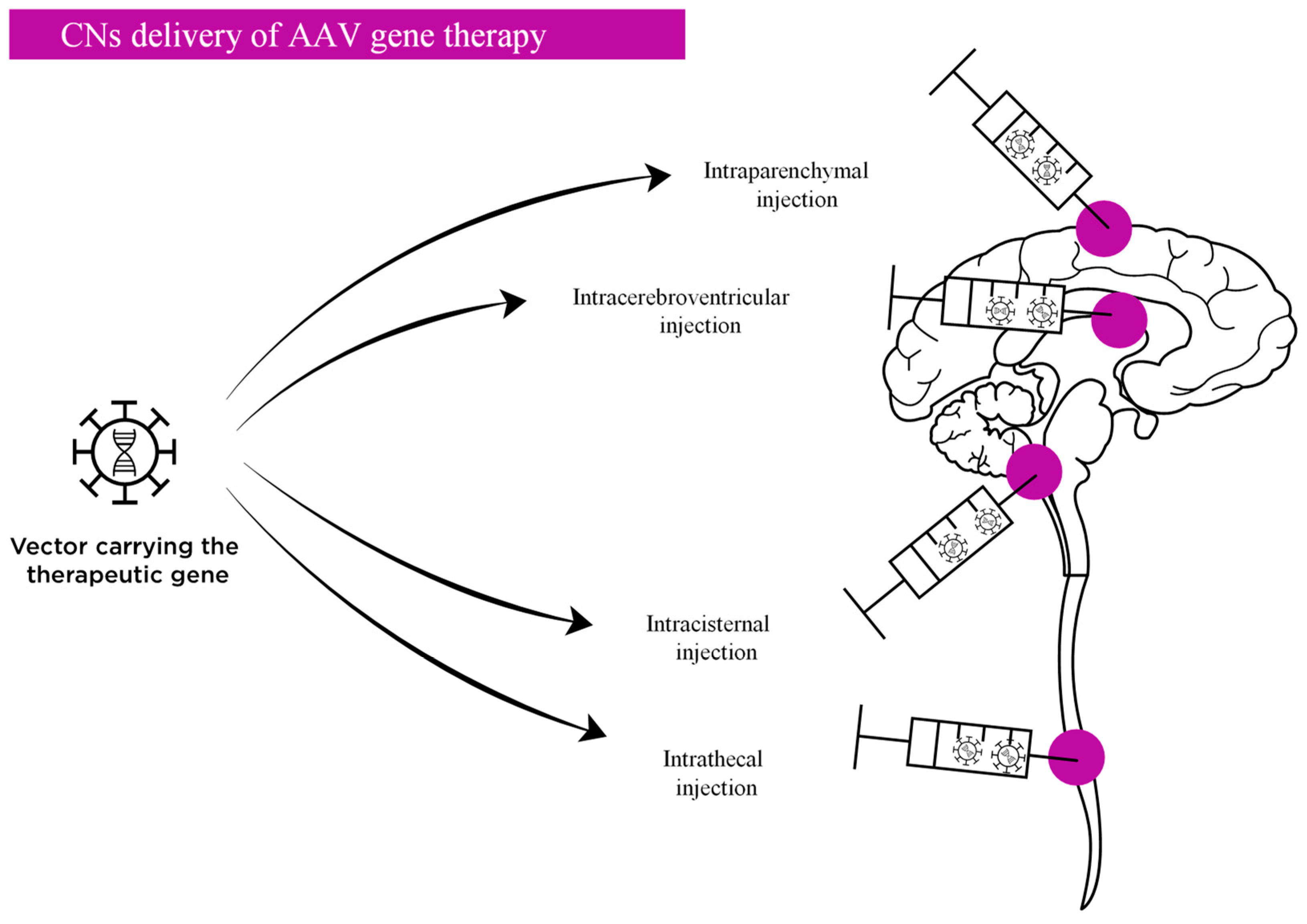
2.3. Routes of Administration AAV Vectors Targeting the CNS
2.3.1. Direct Delivery into the Central Nervous System
2.3.2. Systemic Delivery
2.4. Toxicity of AAV Vectors
2.4.1. Toxicity Related to CNS Delivery
2.4.2. Toxicity Related to Systemic Delivery
2.5. Clinical Trials of AAV Vectors to Treat Neuronopathic MPS
2.5.1. The Following Is a List of Clinical Trials of Gene Therapy Approaches Involving CNS Delivery of AAV Vectors to Treat Neuronopathic MPS
2.5.2. Clinical Trials of Gene Therapy Approaches Involving Systemic Delivery of AAV Vectors to Treat Neuronopathic MPS Are Listed Below
3. Ex Vivo Gene Therapy for Neuronopathic MPS
3.1. Cross Correction Mechanism
3.2. Lentiviral Vectors
3.3. Ex Vivo Gene Therapy for the Treatment of Neuropathic MPS
3.4. Clinical Trials of Ex Vivo Gene Therapy
4. Gene Editing for Neuronopathic MPS
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muenzer, J.; Wraith, J.E.; Clarke, L.A. International Consensus Panel on Management and Treatment of Mucopolysaccharidosis I. Management and treatment guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Almássy, Z.; Beck, M.; Bodamer, O.; Bruce, I.A.; De Meirleir, L.; Guffon, N.; Guillén-Navarro, E.; Hensman, P.; Jones, S.; et al. Hunter Syndrome European Expert Council.Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet. J. Rare. Dis. 2011, 6, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriksz, C.J.; Berger, K.I.; Giugliani, R.; Harmatz, P.; Kampmann, C.; Mackenzie, W.G.; Raiman, J.; Solano Villarreal, M.; Savarirayan, R. International guidelines for the management and treatment of Morquio A syndrome. Am. J. Med. Genet. A 2015, 167, 11–25. [Google Scholar] [CrossRef]
- Giugliani, R.; Harmatz, P.; Wraith, J.E. Management guidelines for mucopolysaccharidosis VI. Pediatrics 2007, 120, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, E.G.; Jones, S.A.; Escolar, M.L. Developmental and behavioral aspects of mucopolysaccharidoses with brain manifestations neurological signs and symptoms. Mol. Genet. Metab. 2017, 122S, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Pellico, A.; Pittalà, A.; Gasperini, S. Neurobehavioral phenotypes of neuronopathic mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44 (Suppl. S2), 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, D.M.; Mitchell, J.J. Pathophysiology, evaluation, and management of sleep disorders in the mucopolysaccharidoses. Mol. Genet. Metab. 2017, 122S, 49–54. [Google Scholar] [CrossRef]
- Zafeiriou, D.I.; Batzios, S.P. Brain and Spinal MR Imaging Findings in Mucopolysaccharidoses: A Review. AJNR Am. J. Neuroradiol. 2013, 34, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Heon-Roberts, R.; Nguyen, A.L.A.; Pshezhetsky, A.V. Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. J. Clin. Med. 2020, 9, 344. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.H.; Sawamoto, K.; Mason, R.W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; Tomatsu, S. Enzyme replacement therapy for mucopolysaccharidoses; past, present, and future. J. Hum Genet. 2019, 64, 1153–1171. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow. Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef] [PubMed]
- Concolino, D.; Deodato, F.; Parini, R. Enzyme replacement therapy: Efficacy and limitations. Ital. J. Pediatr. 2018, 44 (Suppl. S2), 120. [Google Scholar] [CrossRef]
- Parini, R.; Deodato, F. Intravenous Enzyme Replacement Therapy in Mucopolysaccharidoses: Clinical Effectiveness and Limitations. Int. J. Mol. Sci. 2020, 21, 2975. [Google Scholar] [CrossRef]
- Sato, Y.; Okuyama, T. Novel Enzyme Replacement Therapies for Neuropathic Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampe, S.C.; Wesley, J.; Lund, T.C.; Orchard, P.J.; Polgreen, L.E.; Eisengart, J.B.; McLoon, L.K.; Cureoglu, S.; Schachern, P.; McIvor, R.S. Mucopolysaccharidosis Type I: Current Treatments, Limitations, and Prospects for Improvement. Biomolecules 2021, 11, 189. [Google Scholar] [CrossRef]
- Shapiro, E.; Guler, O.E.; Rudser, K.; Delaney, K.; Bjoraker, K.; Whitley, C.; Tolar, J.; Orchard, P.; Provenzale, J.; Thomas, K.M. An exploratory study of brain function and structure in Mucopolysaccharidosis Type I: Long term observations following hematopoietic cell transplantation (HCT). Mol. Genet. Metab. 2012, 107, 116–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubaski, F.; Yabe, H.; Suzuki, Y.; Seto, T.; Hamazaki, T.; Mason, R.W.; Xie, L.; Onsten, G.U.; Leistner-Segal, S.; Giugliani, R. Hematopoietic Stem Cell Transplantation for Patients with Mucopolysaccharidosis II. Biol. Blood Marrow. Transplant. 2017, 23, 1795–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, A.L.; de Magalhães, T.S.P.C.; Reis, A.B.R.; de Oliveira, M.L.; Scalco, F.B.; Cavalcanti, N.C.; Silva, D.S.E.; Torres, D.A.; Costa, A.A.P.; Bonfim, C.; et al. Early hematopoietic stem cell transplantation in a patient with severe mucopolysaccharidosis II: A 7 years follow-up. Mol. Genet. Metab. Rep. 2017, 12, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Meadows, A.S.; Ware, T.; Mohney, R.P.; McCarty, D. Near-Complete Correction of Profound Metabolomic Impairments Corresponding to Functional Benefit in MPS IIIB Mice after IV rAAV9-hNAGLU Gene Delivery and clinical settings with the aim of treating CNS disease in MPS. Mol. Ther. 2017, 25, 792–802. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Zaraspe, K.; Murakami, N.; Meadows, A.S.; Pineda, R.J.; McCarty, D.; Muenzer, J. Targeting Root Cause by Systemic scAAV9-hIDS Gene Delivery: Functional Correction and Reversal of Severe MPS II in Mice. Mol. Ther. Methods Clin. Dev. 2018, 10, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Ou, L.; Przybilla, M.J.; Ahlat, O.; Kim, S.; Overn, O.; Jarnes, J.; O’Sullivan, M.G.; Whitley, C.B. A Highly Efficacious PS Gene Editing System Corrects Metabolic and Neurological Complications of Mucopolysaccharidosis Type I. Mol. Ther. 2020, 28, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, M.; Zérah, M.; Gougeon, M.L.; Ausseil, J.; de Bournonville, S.; Husson, B.; Zafeiriou, D.; Parenti, G.; Bourget, P.; Poirier, B.; et al. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: An uncontrolled phase 1/2 clinical trial. Lancet. Neurol. 2017, 16, 681–682. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2017, 8, 87–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef] [Green Version]
- Hoy, S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs 2019, 79, 1255–1262. [Google Scholar] [CrossRef]
- Morgan, R.A.; Gray, D.; Lomova, A.; Kohn, D.B. Hematopoietic Stem Cell Gene Therapy: Progress and Lessons Learned. Cell Stem Cell 2017, 21, 574–590. [Google Scholar] [CrossRef] [Green Version]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Meier, A.F.; Fraefel, C.; Seyffert, M. The Interplay between Adeno-Associated Virus and Its Helper Viruses. Viruses 2020, 12, 662. [Google Scholar] [CrossRef]
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–756. [Google Scholar] [CrossRef]
- Scott, L.J. Alipogene tiparvovec: A review of its use in adults with familial lipoprotein lipase deficiency. Drugs 2015, 75, 175–182. [Google Scholar] [CrossRef]
- Ronzitti, G.; Gross, D.A.; Mingozzi, F. Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front. Immunol. 2020, 11, 670. [Google Scholar] [CrossRef]
- Asokan, A.; Schaffer, D.V.; Samulski, J.R. The AAV Vector Toolkit: Poised at the Clinical Crossroads. Mol. Ther. 2012, 20, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Hocquemiller, M.; Giersch, L.; Audrain, M.; Parker, S.; Cartier, N. AAV- based gene therapy for CNS diseases. Hum. Gene. Ther. 2016, 27, 478–496. [Google Scholar] [CrossRef] [Green Version]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors:overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Costa Verdera, H.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Mingozzi, F.; Maus, M.V.; Hui, D.J.; Sabatino, D.E.; Murphy, S.L.; Rasko, J.E.J.; Ragni, M.R.; Manno, C.S.; Sommer, J.; Jiang, J.; et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007, 13, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Kruzik, A.; Fetahagic, D.; Hartlieb, B.; Dorn, S.; Koppensteiner, H.; Horling, F.M.; Scheiflinger, F.; Reipert, B.M.; de la Rosa, M.; et al. Prevalence of Anti-Adeno-Associated Virus Immune Responses in International Cohorts of Healthy Donors. Mol. Ther. Methods Clin. Dev. 2019, 14, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Meadows, A.S.; Pineda, R.J.; Kunkler, K.L.; Truxal, K.V.; McBride, K.L.; Flanigan, K.M.; McCarty, D.M. Differential Prevalence of Antibodies against Adeno-Associated Virus in Healthy Children and Patients with Mucopolysaccharidosis III: Perspective for AAV-Mediated Gene Therapy. Hum. Gene. Ther. Clin. Dev. 2017, 28, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Monahan, P.; Négrier, C.; Tarantino, M.; Valentino, L.A.; Mingozzi, F. Emerging Immunogenicity and Genotoxicity Considerations of Adeno-Associated Virus Vector Gene Therapy for Hemophilia. J. Clin. Med. 2021, 10, 2471. [Google Scholar] [CrossRef]
- Masat, E.; Pavani, G.; Mingozzi, F. Humoral immunity to AAV vectors in gene therapy: Challenges and potential solutions. Discov. Med. 2013, 15, 379–389. [Google Scholar]
- Corti, M.; Elder, M.E.; Falk, D.J.; Lawson, L.; Smith, B.K.; Nayak, S.; Conlon, T.J.; Clément, N.; Erger, K.; Lavassani, E.; et al. B-cell depletion is protective against anti-AAV capsid immune response: A human subject case study. Mol. Ther. Methods Clin. Dev. 2014, 1, 14033. [Google Scholar] [CrossRef] [PubMed]
- Nidetz, N.F.; McGee, M.; Tse, L.V.; Li, C.; Cong, L.; Li, Y.; Huang, W. Adeno-associated viral vector-mediated immune responses: Understanding barriers to gene delivery. Pharmacol. Ther. 2020, 207, 107453. [Google Scholar] [CrossRef] [PubMed]
- Leborgne, C.; Barbon, E.; Alexander, J.M.; Hanby, H.; Delignat, S.; Cohen, D.M.; Collaud, F.; Muraleetharan, S.; Lupo, D.; Silverberg, J.; et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 2020, 26, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Piguet, F.; Alves, S.; Cartier, N. Clinical Gene Therapy for Neurodegenerative Diseases: Past, Present, and Future. Hum. Gene. Ther. 2017, 28, 988–1003. [Google Scholar] [CrossRef] [PubMed]
- Pérez, B.A.; Shutterly, A.; Chan, Y.K.; Byrne, B.J.; Corti, M. Management of Neuroinflammatory Responses to AAV-Mediated Gene Therapies for Neurodegenerative Diseases. Brain Sci. 2020, 10, 119. [Google Scholar] [CrossRef] [Green Version]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef] [Green Version]
- Desmaris, N.; Verot, L.; Puech, J.P.; Caillaud, C.; Vanier, M.T.; Heard, J.M. Prevention of neuropathology in the mouse model of Hurler syndrome. Ann. Neurol. 2004, 56, 68–76. [Google Scholar] [CrossRef]
- Cressant, A.; Desmaris, N.; Verot, L.; Brejot, T.; Froissart, R.; Vanier, M.T.; Maire, I.; Heard, J.M. Improved behavior and neuropathology in the mouse model of Sanfilippo type IIIB disease after adeno-associated virus-mediated gene transfer in the striatum. J. Neurosci. 2004, 24, 10229–10239. [Google Scholar] [CrossRef] [Green Version]
- Winner, L.K.; Beard, H.; Hassiotis, S.; Lau, A.A.; Luck, A.J.; Hopwood, J.J.; Hemsley, K.M. A preclinical study evaluating AAVrh10-based gene therapy for Sanfilippo syndrome. Hum. Gene. Ther. 2016, 27, 363–375. [Google Scholar] [CrossRef]
- Ellinwood, N.M.; Ausseil, J.; Desmaris, N.; Bigou, S.; Song, L.; Jens, J.K.; Snella, E.M.; Mohammed, E.E.A.; Thomson, C.B.; Raoul, S.; et al. Safe, Efficient, and Reproducible Gene Therapy of the Brain in the Dog Models of Sanfilippo and Hurler Syndromes. Mol. Ther. 2011, 19, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Chen, Y.H.; He, X.; Martins, I.; Heth, J.A.; Chiorini, J.A.; Davidson, B.E. Adeno-associated virus type 5 reduces learning deficits and restores glutamate receptor subunit levels in MPS VII mice CNS. Mol. Ther. 2007, 15, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Fraldi, A.; Serafini, M.; Sorrentino, N.C.; Gentner, B.; Aiuti, A.; Bernardo, M.E. Gene therapy for mucopolysaccharidoses: In vivo and ex vivo approaches. Ital. J. Pediatr. 2018, 44 (Suppl. S2), 130. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.A.; Lenander, A.W.; Nan, Z.; Belur, L.R.; Whitley, C.B.; Gupta, P.; Low, W.C.; McIvor, R.S. Direct gene transfer to the CNS prevents emergence of neurologic disease in a murine model of mucopolysaccharidosis type I. Neurobiol. Dis. 2011, 43, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraldi, A.; Hemsley, K.; Crawley, A.; Lombardi, A.; Lau, A.; Sutherland, L.; Auricchio, A.; Ballabio, A.; Hopwood, J.L. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum. Mol. Genet. 2007, 16, 2693–2702. [Google Scholar] [CrossRef] [Green Version]
- Haurigot, V.; Marcó, S.; Ribera, A.; Garcia, M.; Ruzo, A.; Villacampa, P.; Ayuso, E.; Añor, S.; Andaluz, A.; Pineda, M.; et al. Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J. Clin. Investig. 2013, 123, 3254–3271. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; DiRosario, J.; Kang, L.; Muenzer, J.; McCarty, D.M. Restoration of central nervous system alpha-N-acetylglucosaminidase activity and therapeutic benefits in mucopolysaccharidosis IIIB mice by a single intracisternal recombinant adeno-associated viral type 2 vector delivery. J. Gene. Med. 2010, 12, 624–633. [Google Scholar] [CrossRef]
- Chuster, D.J.; Dykstra, J.A.; Riedl, M.S.; Kitto, K.F.; Belur, L.R.; McIvor, R.S.; Elde, R.P.; Fairbanks, C.A.; Vulchanova, L. Biodistribution of adeno-associated virus serotype 9 (AAV9) vector after intrathecal and intravenous delivery in mouse. Front. Neuroanat. 2014, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Hinderer, C.; Bell, P.; Vite, C.H.; Louboutin, J.P.; Grant, R.; Bote, E.; Yu, H.; Pukenas, B.; Hurst, R.; Wilson, J.M. Widespread gene transfer in the central nervous system of cynomolgus macaques following delivery of AAV9 into the cisterna magna. Mol. Ther. Methods Clin. Dev. 2014, 1, 14051. [Google Scholar] [CrossRef]
- Mijanović, O.; Branković, A.; Borovjagin, A.; Butnaru, D.V.; Bezrukov, E.A.; Sukhanov, R.B.; Shpichka, A.; Timashev, P.; Ulasov, I. Battling Neurodegenerative Diseases with Adeno-Associated Virus-Based Approaches. Viruses 2020, 12, 460. [Google Scholar] [CrossRef] [Green Version]
- Belur, L.R.; Podetz-Pedersen, K.M.; Tran, T.A.; Mesick, J.A.; Singh, N.M.; Riedl, M.; Vulchanova, L.; Kozarsky, K.F.; McIvor, R.S. Intravenous delivery for treatment of mucopolysaccharidosis type I: A comparison of AAV serotypes 9 and rh10. Mol. Genet. Metab. Rep. 2020, 24, 100604. [Google Scholar] [CrossRef] [PubMed]
- Duncan, F.J.; Naughton, B.J.; Zaraspe, K.; Murrey, D.A.; Meadows, A.S.; Klark, K.R.; Newsom, D.E.; White, P.; Fu, H.; McCarty, D.M. Broad functional correction of molecular impairments by systemic delivery of scAAVrh74-hSGSH gene delivery in MPS IIIA mice. Mol. Ther. 2015, 23, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Gurda, B.L.; De Guilhem De Lataillade, A.; Bell, P.; Zhu, Y.; Yu, H.; Bagel, J.; Vite, C.H.; Sikora, T.; Hinderer, C.; Calcedo, R.; et al. Evaluation of AAV-mediated Gene Therapy for Central Nervous System Disease in Canine Mucopolysaccharidosis VII. Mol. Ther. 2016, 24, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wan, J.; Wu, Y.; Tian, Y.; Yao, Y.; Yao, S.; Ji, X.; Wang, S.; Su, X.; Xu, H.; et al. Challenges in adeno-associated virus-based treatment of central nervous system diseases through systemic injection. Life Sci. 2021, 270, 119142. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, D.A.; Shutova, M.V.; Johnston, N.R.; Smith, O.P.; Fedorin, V.V.; Kukushkin, Y.S.; van der Loo, J.C.M.; Johnstone, E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug. Discov. 2021, 20, 173–174. [Google Scholar] [CrossRef]
- Hordeaux, J.; Buza, E.L.; Dyer, C.; Goode, T.; Mitchell, T.W.; Richman, L.; Denton, N.; Hinderer, C.; Katz, N.; Schmid, R.; et al. Adeno-Associated Virus-Induced Dorsal Root Ganglion Pathology. Hum. Gene Ther. 2020, 31, 808–818. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.M.; Flotte, T.R. Moving forward after Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum. Gene Ther. 2020, 31, 695–696. [Google Scholar] [CrossRef]
- Chand, D.; Mohr, F.; McMillan, H.; Tukov, F.F.; Montgomery, K.; Sun, R.; Tauscher-Wisniewski, S.; Kaufmann, P.; Kullak-Ublick, G. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J. Hepatol. 2021, 74, 560–566. [Google Scholar] [CrossRef]
- Giugliani, R. RGX-121 Gene Therapy for the Treatment of Severe Mucopolysaccha-ridosis Type II: Interim Analysis of the First in Human Study. In Proceedings of the American Society of Gene and Cell Therapy’s 24th Annual Meeting, Virtual. 11–14 May 2021. [Google Scholar]
- Harmatz, P. RGX-121 Gene Therapy for the Treatment of Severe Mucopolysaccharidosis Type II: Interim Analysis of the First. In Proceedings of the Human Study 16th International Symposium on MPS and Related Diseases, Virtual. 23–25 July 2021. [Google Scholar]
- Laufer, R. Clinical trial MPSIIIA: Intraparenchymal administration. In Proceedings of the 16th International Symposium on MPS and Related Diseases, Virtual. 23–25 July 2021. [Google Scholar]
- Pineda, M. Safety and tolerability after intracerebroventricular (ICV) administration of adeno-associated viral vector serotype 9 containing the human sulfamidase gene: Ongoing Phase I/II Clinical Trial in Sanfilippo Syndrome Type, A. In Proceedings of the 16th International Symposium on MPS and Related Diseases, Virtual. 23–25 July 2021. [Google Scholar]
- Flanigan, K. Updated Results of Transpher A, a Multicenter, Single-Dose, Phase 1/2 Clinical Trial of ABO-102 Gene Therapy for Sanfilippo Syndrome Type A (Mucopolysaccharidosis IIIA). In Proceedings of the American Society of Gene and Cell Therapy’s 24th Annual Meeting, Virtual. 16–19 May 2021. [Google Scholar]
- Ruiz, J. Developing Gene Therapies for Sanfilippo Syndrome: Studies for MPSIIIA and IIIB. In Proceedings of the 16th International Symposium on MPS and Related Diseases, Virtual. 23–25 July 2021. [Google Scholar]
- de Castro, M.J. Updated Results of Transpher B, a Multicenter, Single-Dose, Phase 1/2 Clinical Trial of ABO-101 Gene Therapy for Sanfilippo Syndrome Type B (Mucopolysaccharidosis IIIB). In Proceedings of the American Society of Gene and Cell Therapy’s 24th Annual Meeting, Virtual. 16–19 May 2021. [Google Scholar]
- Fratantoni, J.C.; Hall, C.W.; Neufeld, E.F. Hurler and Hunter syndromes: Mutual correction of the defect in cultured fibroblasts. Science 1968, 162, 570–572. [Google Scholar] [CrossRef]
- Hasilik, A.; Klein, U.; Waheed, A.; Strecker, G.; Von Figura, K. Phosphorylated oligosaccharides in lysosomal enzymes: Identification of alpha-N-acetylglucosamine(1)phospho(6)mannose diester groups. Proc. Natl. Acad. Sci. USA 1980, 77, 7074–7078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orchard, P.J.; Blazar, B.R.; Wagner, J.; Charnas, L.; Krivit, W.; Tolar, J. Hematopoietic cell therapy for metabolic disease. J. Pediatr. 2007, 151, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A. Hematopoietic Stem Cell Gene Therapy for Storage Disease: Current and New Indications. Mol. Ther. 2017, 25, 1155–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Rozengurt, N.; Ryazantsev, S.; Kohn, D.B.; Satake, N.; Neufeld, E.F. Treatment of the mouse model of Mucopolysaccharidosis I with retrovirally transduced bone marrow. Mol. Genet. Metab. 2003, 79, 233–244. [Google Scholar] [CrossRef]
- Matzner, U.; Schestag, F.; Hartmann, D.; Lullmann-Rauch, R.; D’Hooge, R.; De Deyn, P.P.; Gieselmann, V. Bone marrow stem cell gene therapy of arylsulfatase A-deficient mice, using an arylsulfatase A mutant that is hypersecreted from retrovirally transduced donor-type cells. Hum. Gene Ther. 2001, 12, 1021–1033. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Shimada, Y.; Akiyama, K.; Higuchi, T.; Fukuda, T.; Kobayashi, H.; Eto, Y.; Ida, H.; Ohashi, T. Hematopoietic stem cell gene therapy corrects neuropathic phenotype in murine model of mucopolysaccharidosis type II. Hum. Gene Ther. 2015, 26, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Langford-Smith, A.; Wilkinson, F.L.; Langford-Smith, K.J.; Holley, R.J.; Sergijenko, A.; Howe, S.J.; Bennett, W.R.; Jones, S.A.; Wraith, J.; Merry, C.L.; et al. Hematopoie-tic stem cell and gene therapy corrects primary neuropathology and behavior in mucopolysaccharidosis IIIA mice. Mol. Ther. 2012, 20, 1610–1621. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Ates, I.; Rathbone, T.; Stuart, C.; Bridges, P.H.; Cottle, R.N. Delivery Approaches for Therapeutic Genome Editing and Challenges. Genes 2020, 23, 1113. [Google Scholar] [CrossRef]
- Sarkis, C.; Philippe, S.; Mallet, J.; Serguera, C. Non-Integrating Lentiviral Vectors. Curr. Gene Ther. 2008, 8, 430–437. [Google Scholar] [CrossRef]
- Luis, A. The Old and the New: Prospects for Non-Integrating Lentiviral Vector Technology. Viruses 2020, 12, 1103. [Google Scholar] [CrossRef]
- Visigalli, I.; Delai, S.; Politi, L.S.; Di Domenico, C.; Cerri, F.; Mrak, E.; D’Isa, R.; Ungaro, D.; Stok, M.; Sanvito, F.; et al. Gene therapy augments the efficacy of hemato-poietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood 2010, 116, 5130–5139. [Google Scholar] [CrossRef] [PubMed]
- Sergijenko, A.; Langford-Smith, A.; Liao, A.Y.; Pickford, C.E.; McDermott, J.; Nowinski, G.; Langford-Smith, K.J.; Merry, C.L.; Jones, S.A.; Wraith, J.E.; et al. Myeloid/Microglial driven au-tologous hematopoietic stem cell gene therapy corrects a neuronopathic lysosomal disease. Mol. Ther. 2013, 21, 1938–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capotondo, A.; Milazzo, R.; Politi, L.S.; Quattrini, A.; Palini, A.; Plati, T.; Merella, S.; Nonis, A.; di Serio, C.; Montini, E.; et al. Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc. Natl. Acad. Sci. USA 2010, 109, 15018–15023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, F.L.; Sergijenko, A.; Langford-Smith, K.J.; Malinowska, M.; Wynn, R.F.; Bigger, B.W. Busulfan conditioning enhances engraftment of hematopoietic donor-derived cells in the brain compared with irradiation. Mol. Ther. 2013, 21, 868–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleitz, H.F.; Liao, A.Y.; Cook, J.R.; Rowlston, S.F.; Forte, G.M.; D’Souza, Z.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 2018, 10, e8730. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Bak, R.O.; Gomez-Ospina, N.; Porteus, M.H. Gene Editing on Center Stage. Trends. Genet. 2018, 34, 600–611. [Google Scholar] [CrossRef]
- Ou, L.; DeKelver, R.C.; Rohde, M.; Tom, S.; Radeke, R.; St Martin, S.J.; Santiago, Y.; Sproul, S.; Przybilla, M.J.; Koniar, B.L.; et al. ZFN-Mediated In vivo Genome Editing Corrects Murine Hurler Syndrome. Mol. Ther. 2019, 27, 178–187. [Google Scholar] [CrossRef]
- Laoharawee, K.; DeKelver, R.C.; Podetz-Pedersen, K.M.; Rohde, M.; Sproul, S.; Nguyen, H.O.; Nguyen, T.; St Martin, S.J.; Ou, L.; Tom, S.; et al. Dose-Dependent Prevention of Metabolic and Neurologic Disease in Murine MPS II by ZFN-Mediated In vivo Genome Editing. Mol. Ther. 2018, 26, 1127–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poletto, E.; Baldo, B.; Gomez-Ospina, N. Genome Editing for Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 500. [Google Scholar] [CrossRef] [Green Version]
- First in vivo human genome editing trial. Nat. Biotechnol. 2018, 36, 5. [CrossRef] [PubMed]
- Harmatz, P.; Lau, H.E.; Heldermon, C.; Leslie, N.; Wong Po Foo, C.; Vaidya, S.A.; Whitley, C.B. EMPOWERS: A phase 1/2 clinical trial of SB-318 ZFN-mediated in vivo human genome editing for treatment of MPS I (Hurler syndrome). Mol. Genet. Metab. 2019, 126, S68. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Disease | Study Phase | Experimental Product | Route | Sponsor |
|---|---|---|---|---|
| in vivo gene therapy | ||||
| MPS I | I/II | AAV9 vector containing the human α-L-iduronidase gene | Intracisternal | Regenxbio |
| MPS II | I/II | AAV9 vector containing the human iduronate-2-sulfatase gene | Intracisternal | Regenxbio |
| MPS IIIA | II/III | AAV rh10 containing the human N-sulfoglucosamine sulfohydrolase gene | Intraparenchymal | Lysogene |
| MPS IIIA | I/II | AAV9 vector containing the human iduronate-2-sulfatase gene | Intracisternal | Esteve |
| MPS IIIA | I/II | AAV9 containing N-sulfoglucosamine sulfohydrolase expression cassette | Intravenous | Abeona |
| MPS IIIB | I/II | AAV9 containing the human α-N acetylglucosaminidase gene | Intravenous | Abeona |
| ex vivo gene therapy | ||||
| MPS I | I/II | Autologous CD34+ cells transduced with a lentiviral vector containing the human α-L-iduronidase gene | Intravenous | Fondazione Telethon |
| MPS II | I/II | Autologous CD34+ cells transduced with a lentiviral vector containing the human iduronate sufatase gene | Intravenous | Avrobio |
| MPS IIIA | I/II | Autologous CD34+ cells transduced with a lentiviral vector containing the human N-sulfoglucosamine sulfohydrolase gene | Intravenous | Orchard |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Castro, M.J.; del Toro, M.; Giugliani, R.; Couce, M.L. Gene Therapy for Neuronopathic Mucopolysaccharidoses: State of the Art. Int. J. Mol. Sci. 2021, 22, 9200. https://doi.org/10.3390/ijms22179200
de Castro MJ, del Toro M, Giugliani R, Couce ML. Gene Therapy for Neuronopathic Mucopolysaccharidoses: State of the Art. International Journal of Molecular Sciences. 2021; 22(17):9200. https://doi.org/10.3390/ijms22179200
Chicago/Turabian Stylede Castro, María José, Mireia del Toro, Roberto Giugliani, and María Luz Couce. 2021. "Gene Therapy for Neuronopathic Mucopolysaccharidoses: State of the Art" International Journal of Molecular Sciences 22, no. 17: 9200. https://doi.org/10.3390/ijms22179200
APA Stylede Castro, M. J., del Toro, M., Giugliani, R., & Couce, M. L. (2021). Gene Therapy for Neuronopathic Mucopolysaccharidoses: State of the Art. International Journal of Molecular Sciences, 22(17), 9200. https://doi.org/10.3390/ijms22179200