
Key Points in Remote-Controlled Drug Delivery: From the Carrier Design to Clinical Trials



Abstract
:1. Introduction
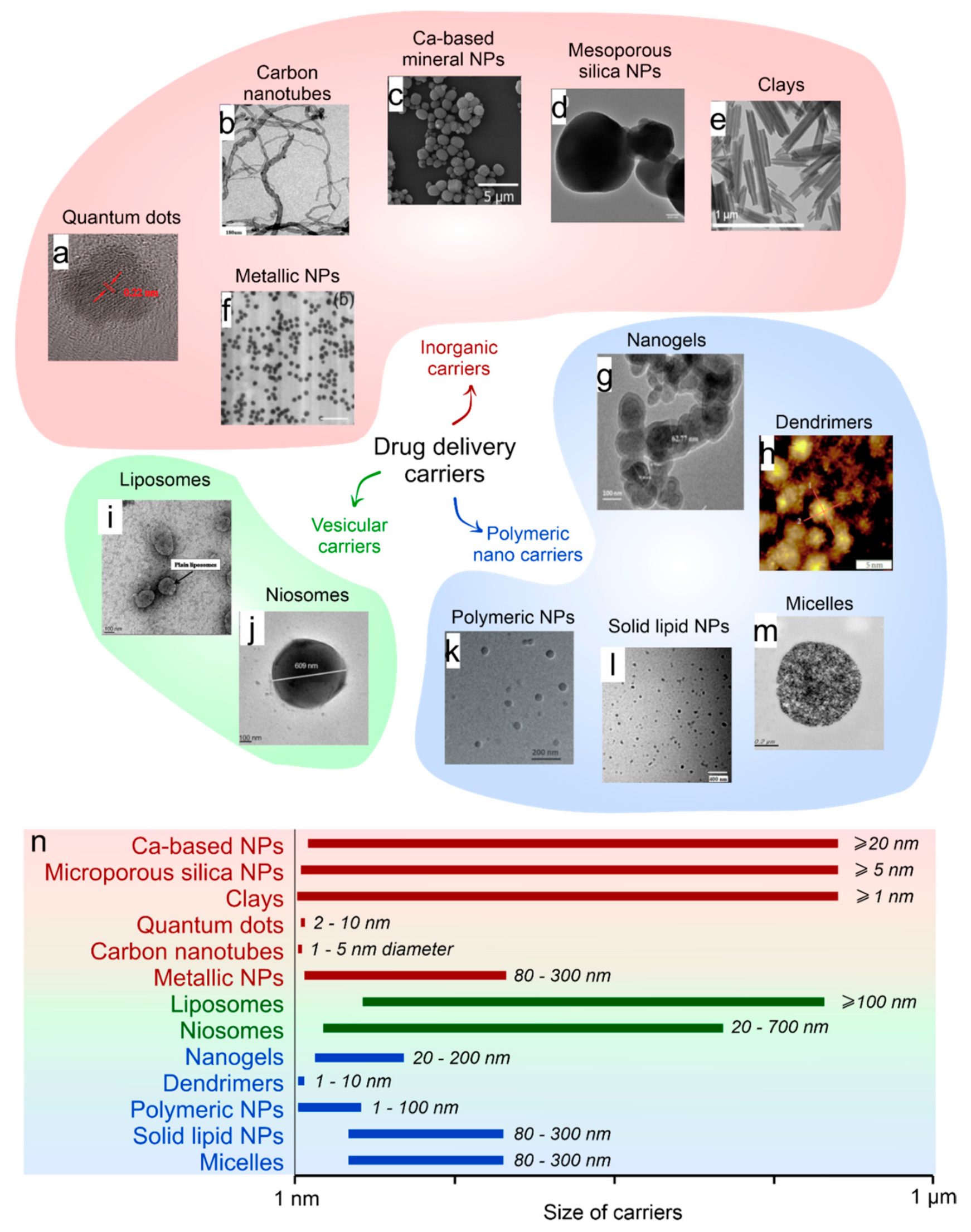
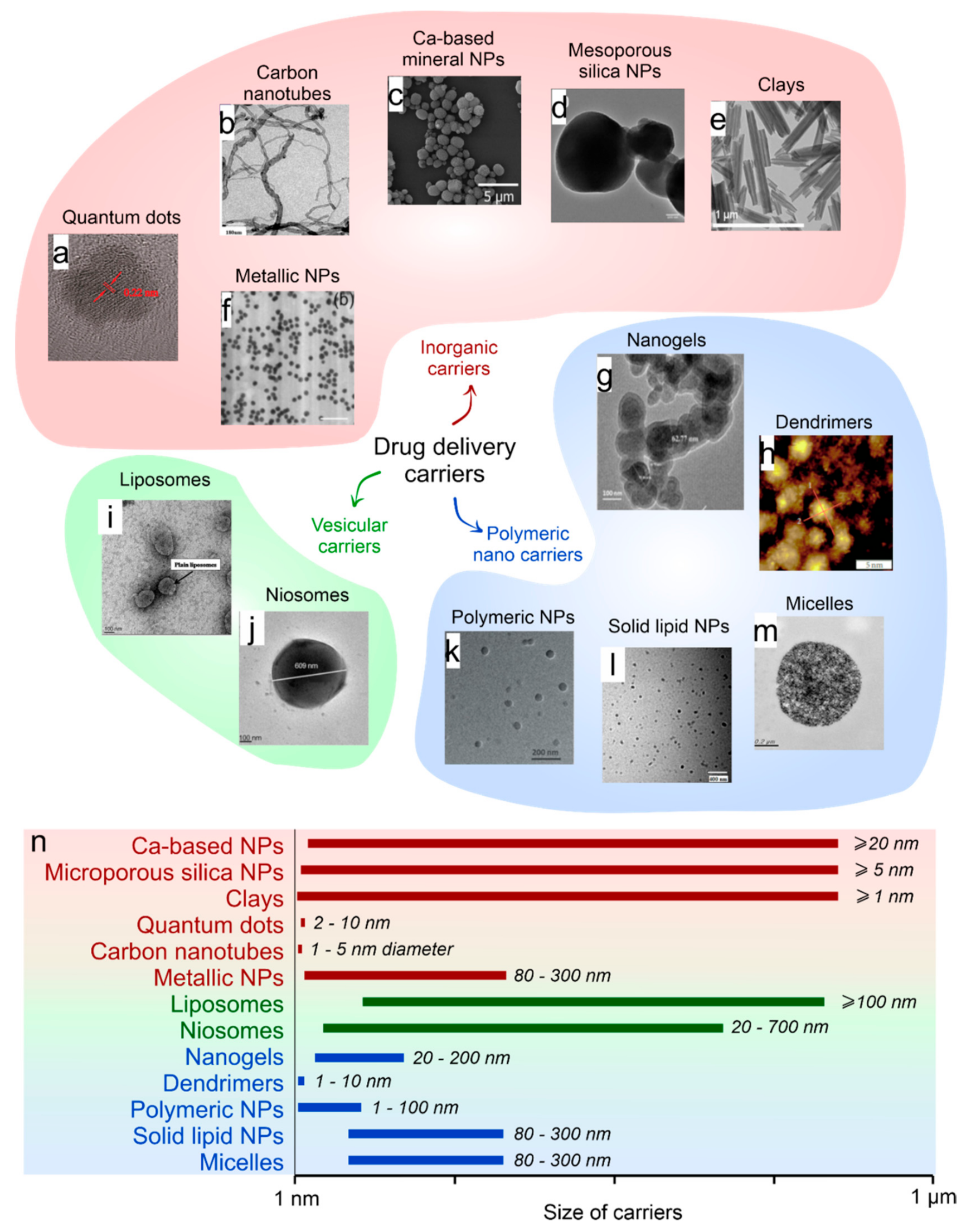
2. Selection of the Drug Carriers Depending on the Type of Encapsulating Substances
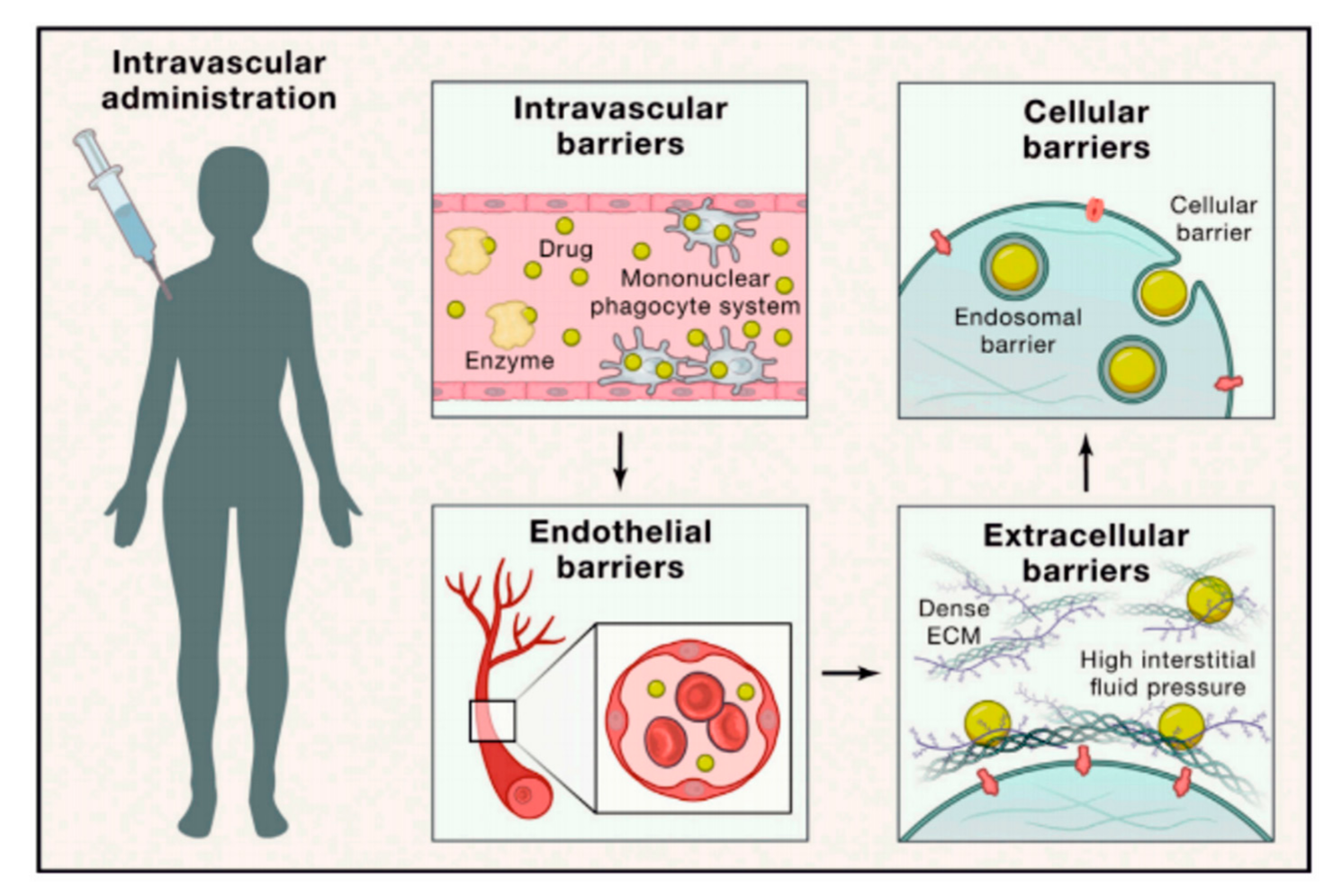
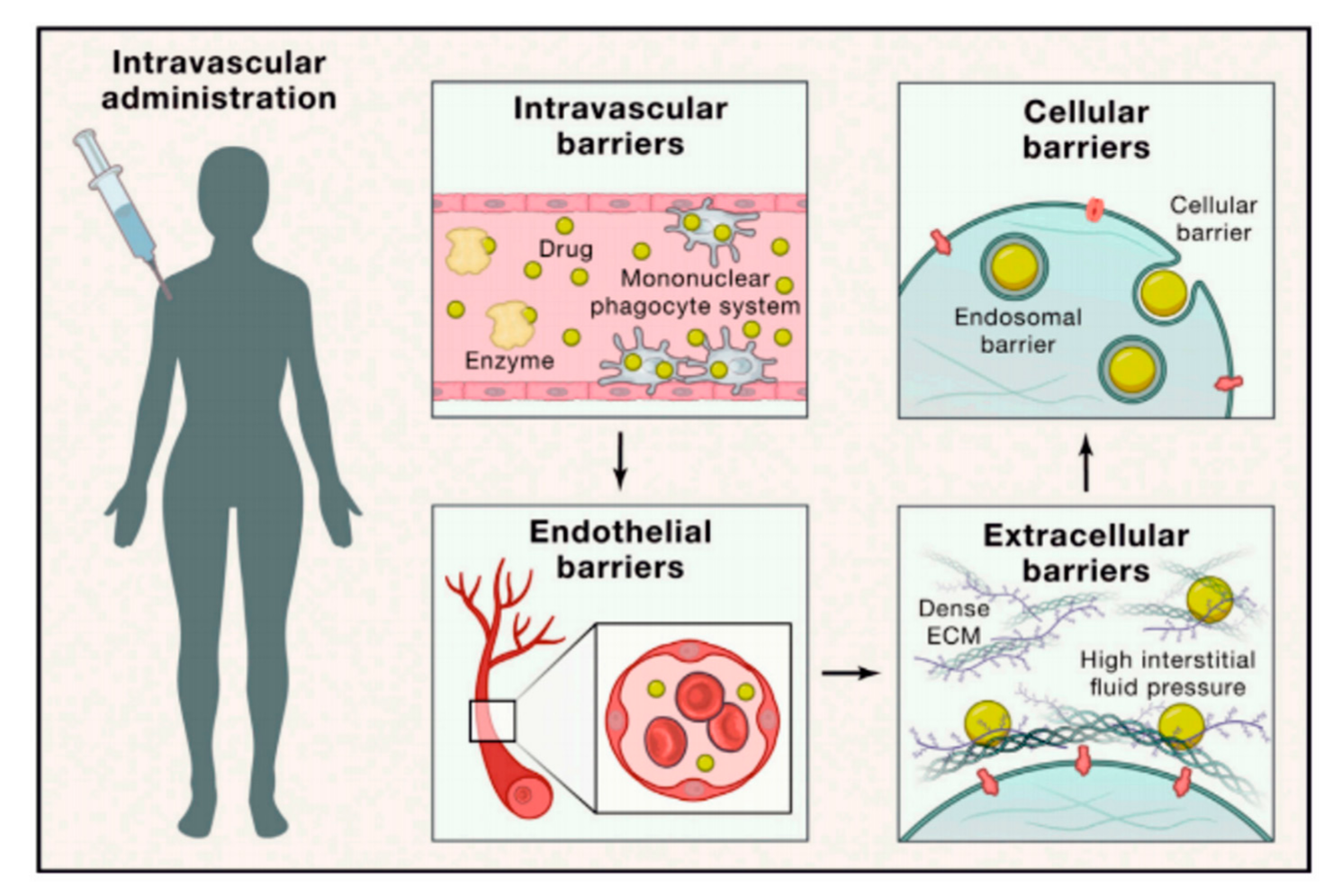
3. Barriers and Limitations Associated with Targeted Drug Delivery
4. Physical Addressing and Release of Encapsulated Drugs by External Stimuli In Vivo. Principles and Safety Considerations
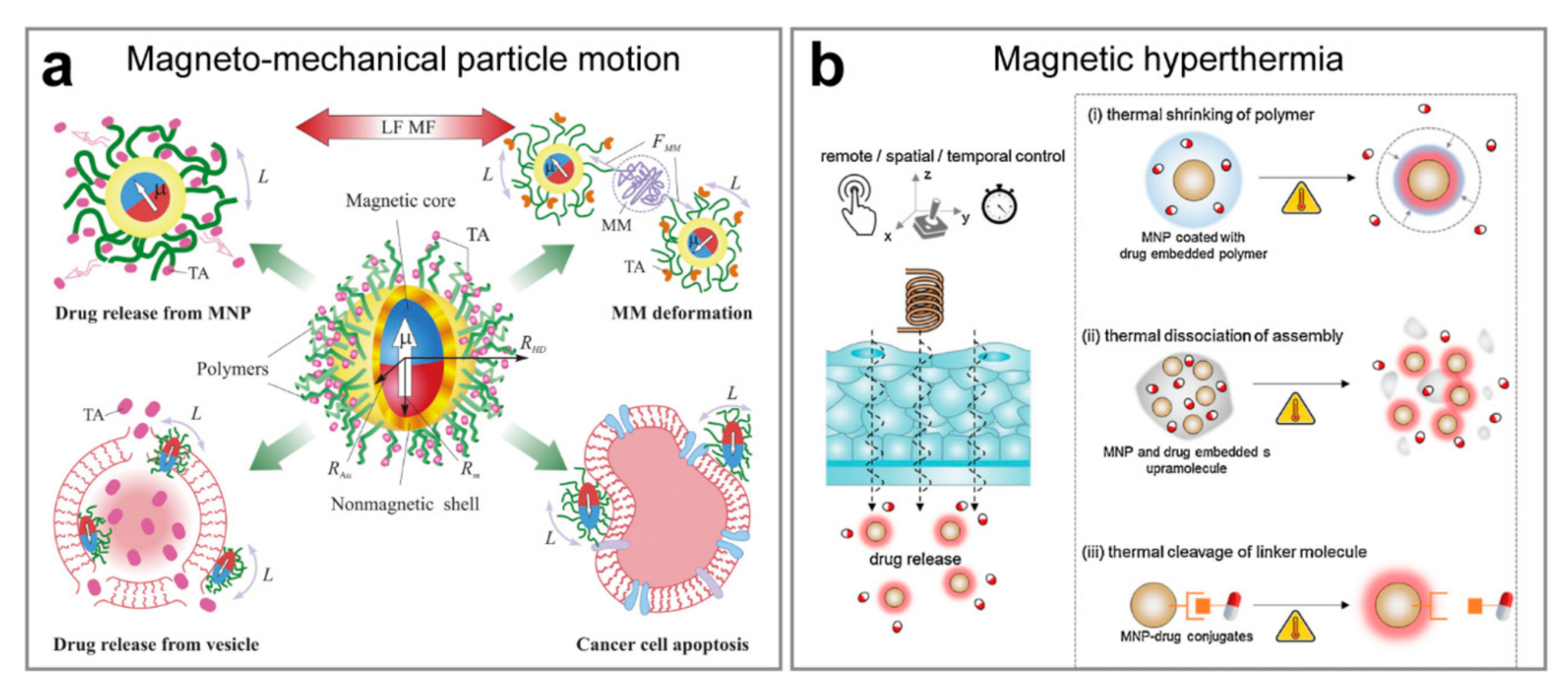
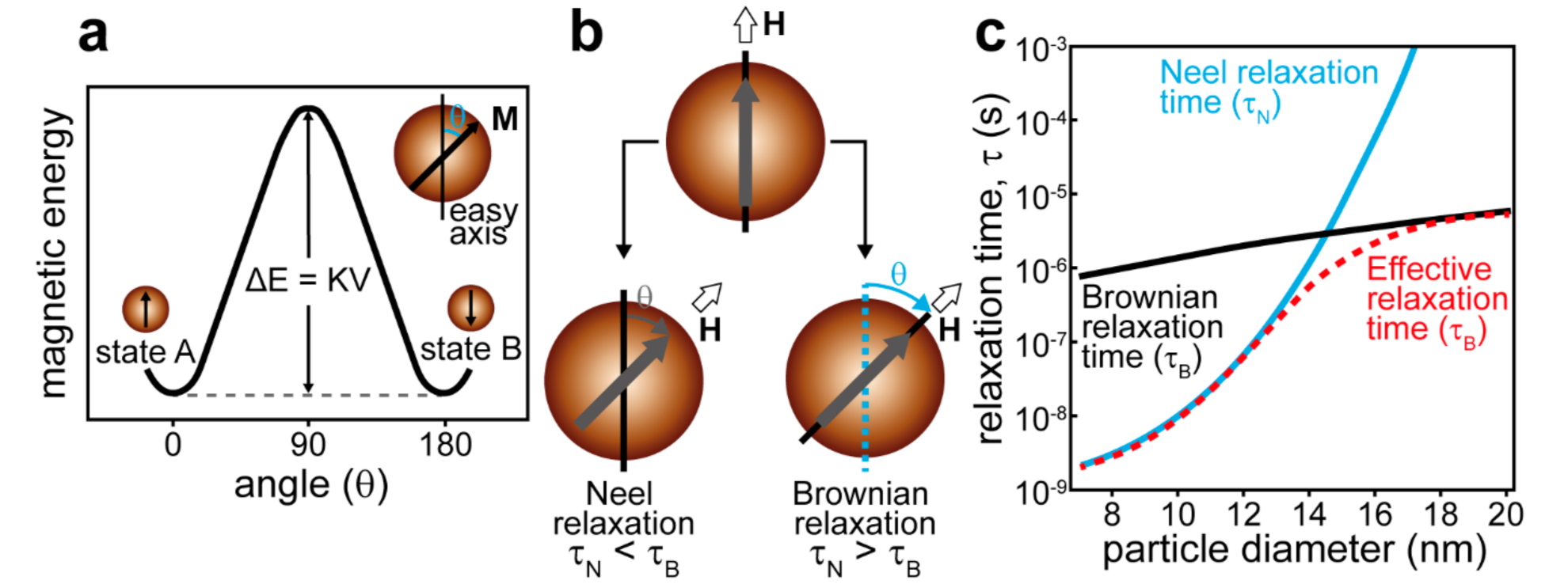
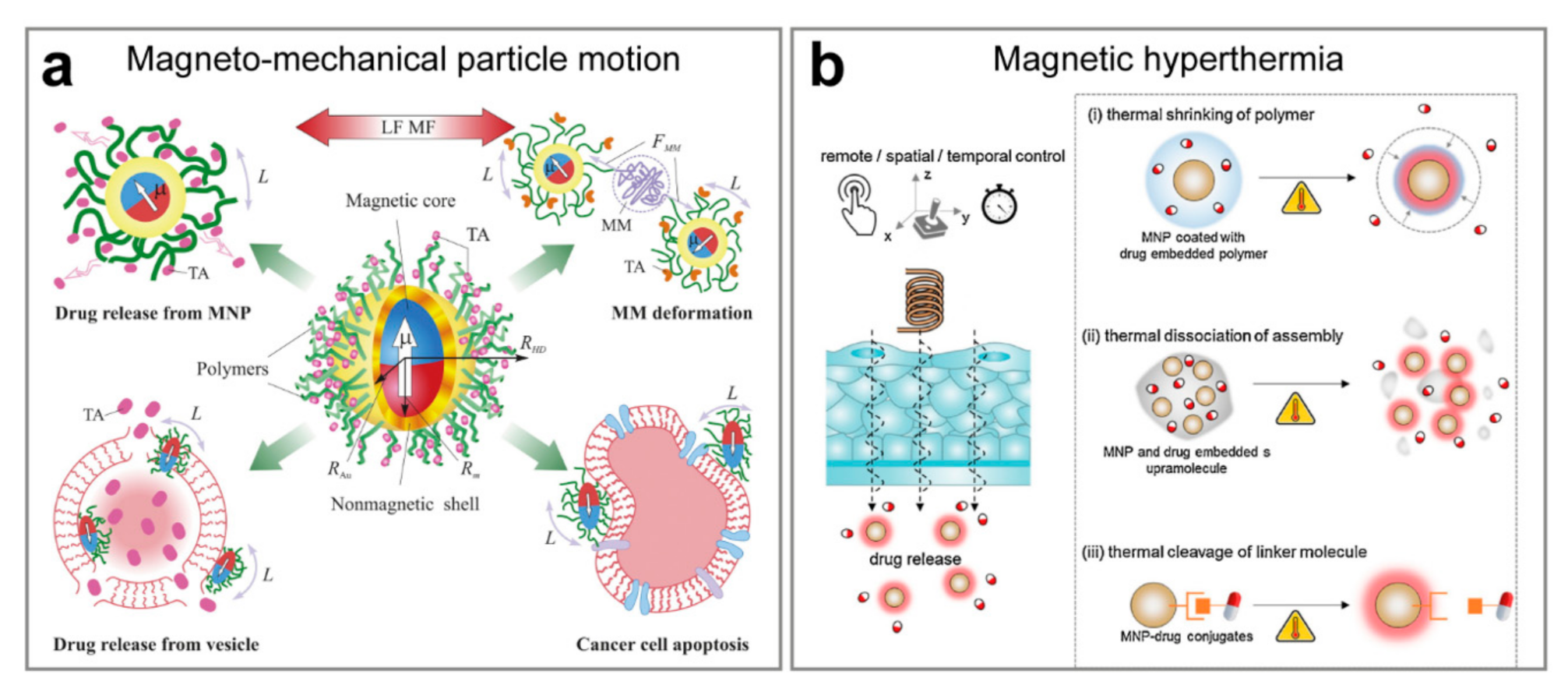
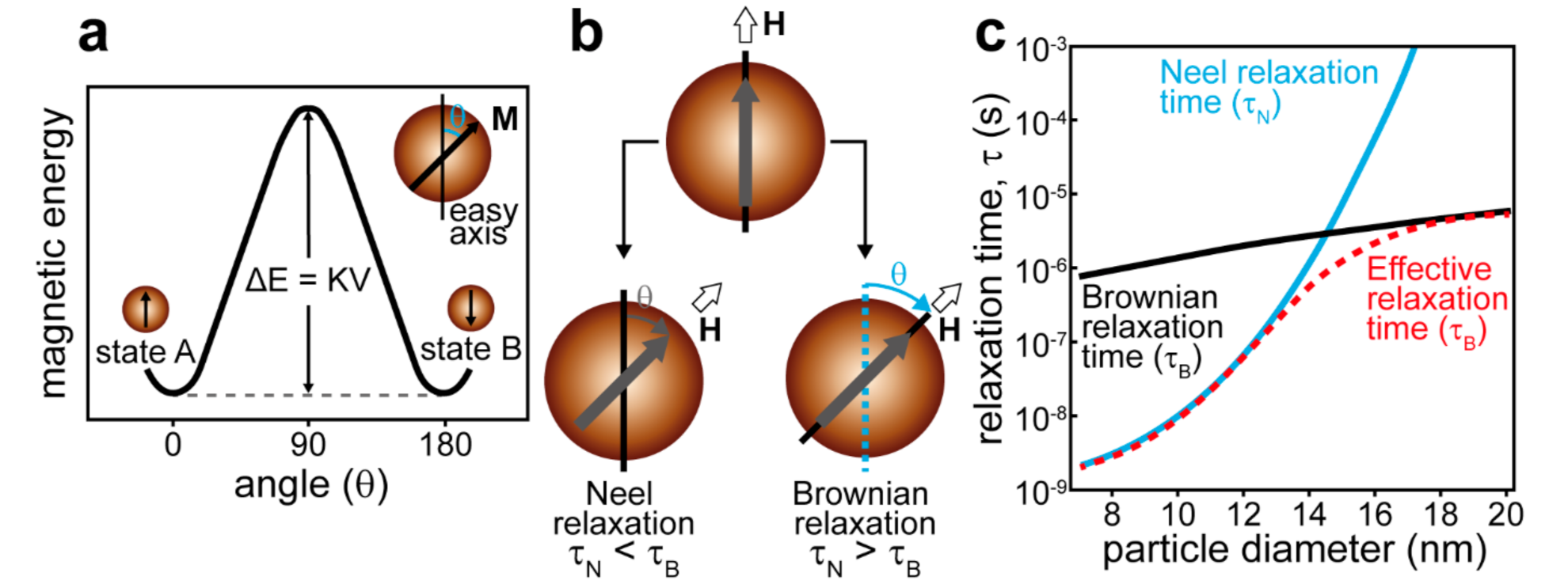
4.1. Remote Navigation and Triggered Release Mediated by the Magnetic Field
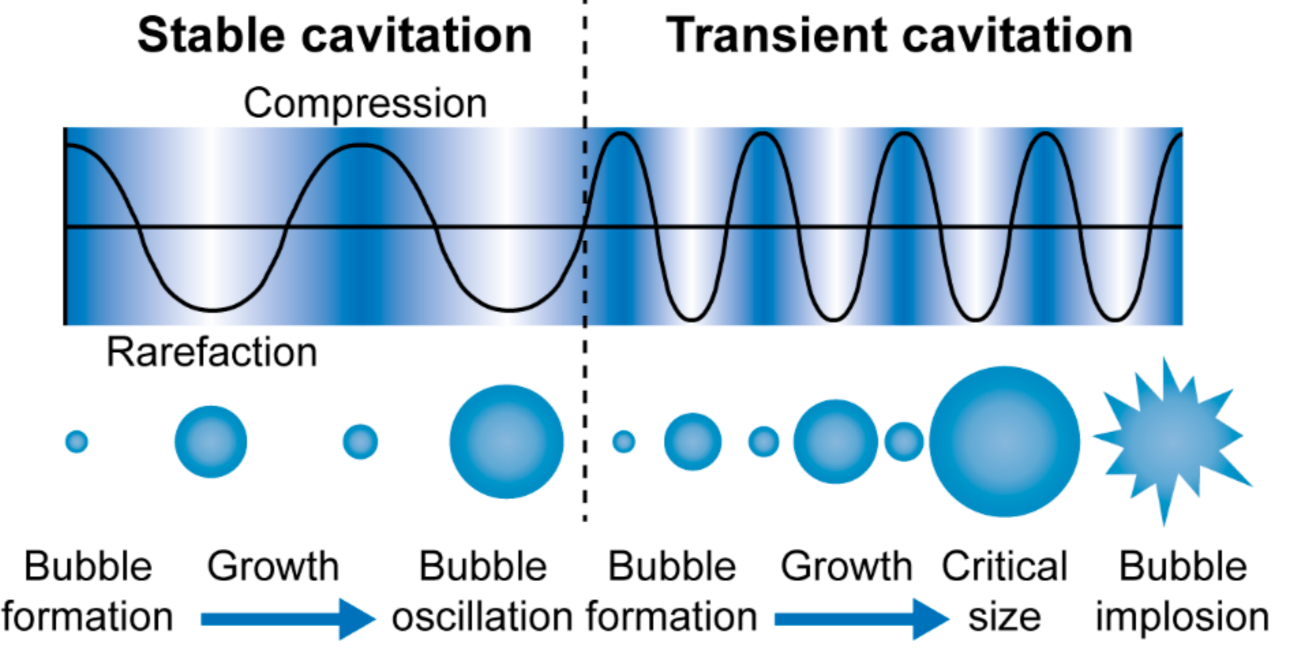
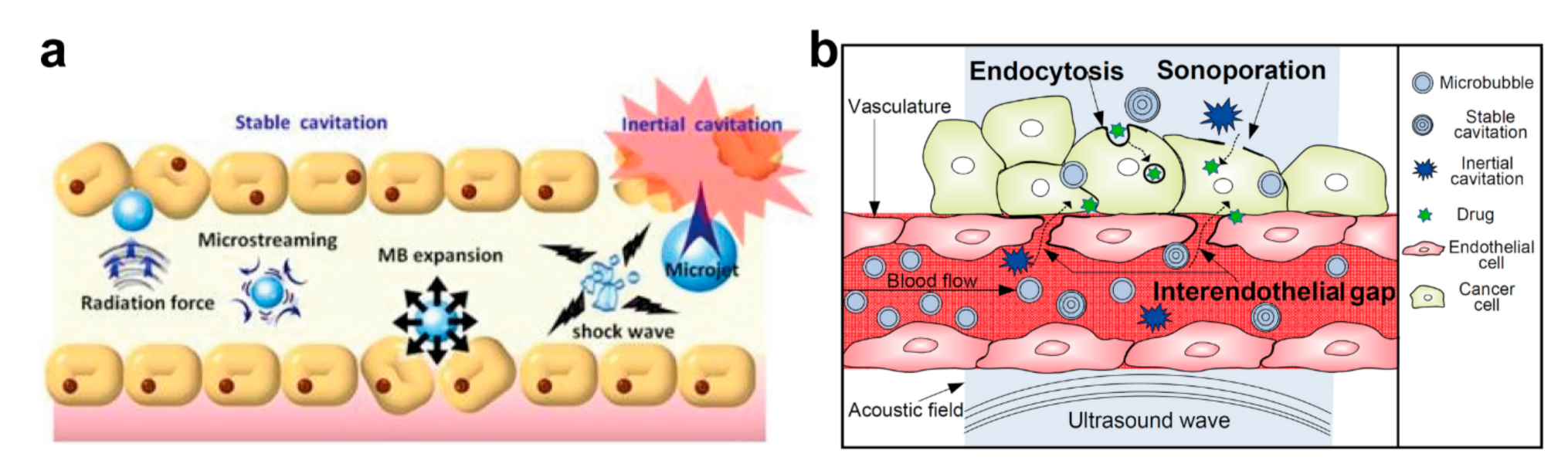
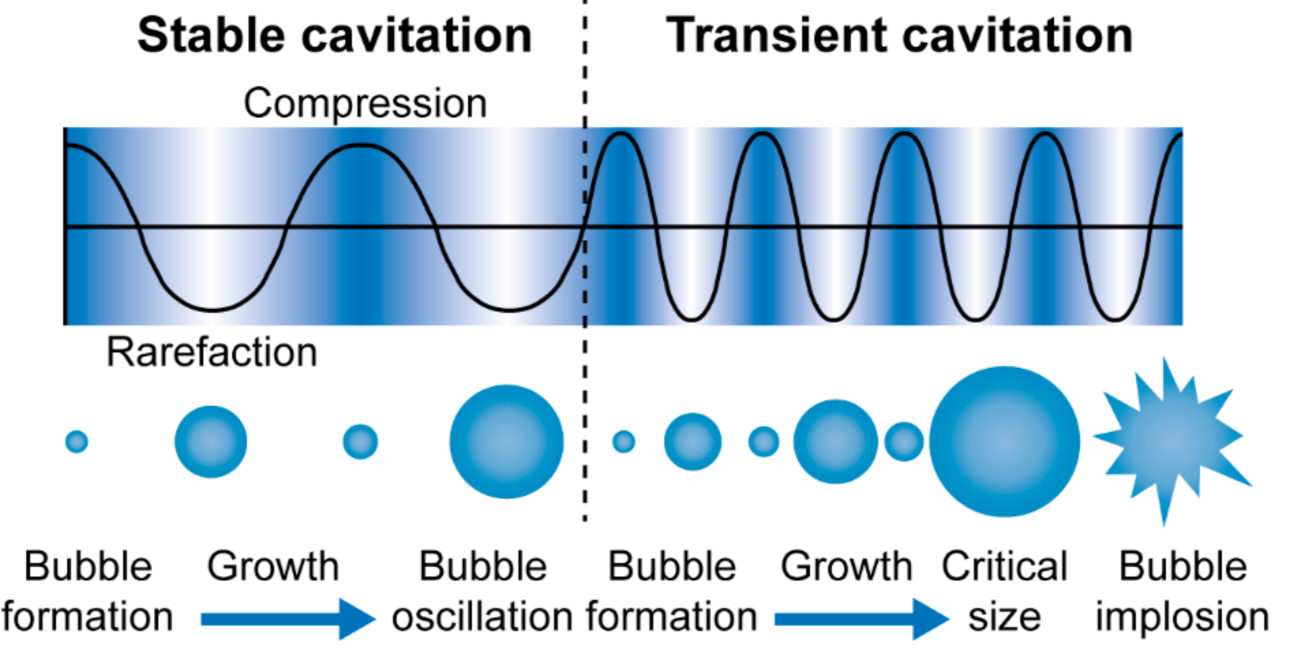
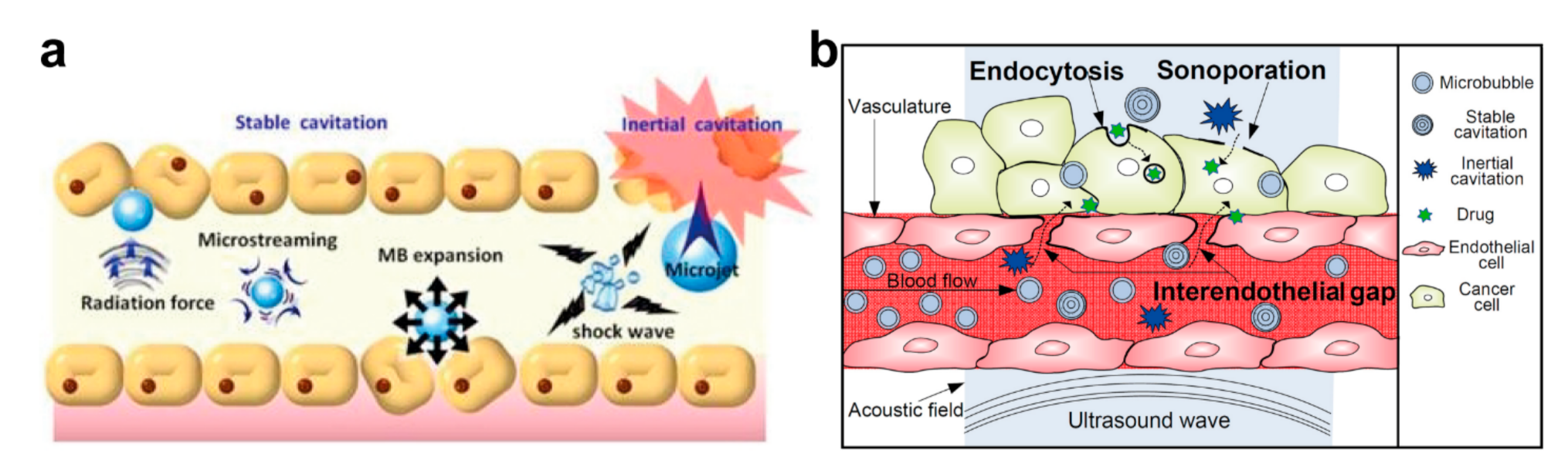
4.2. Enhancement of Site-Specific Drug Delivery with Ultrasound
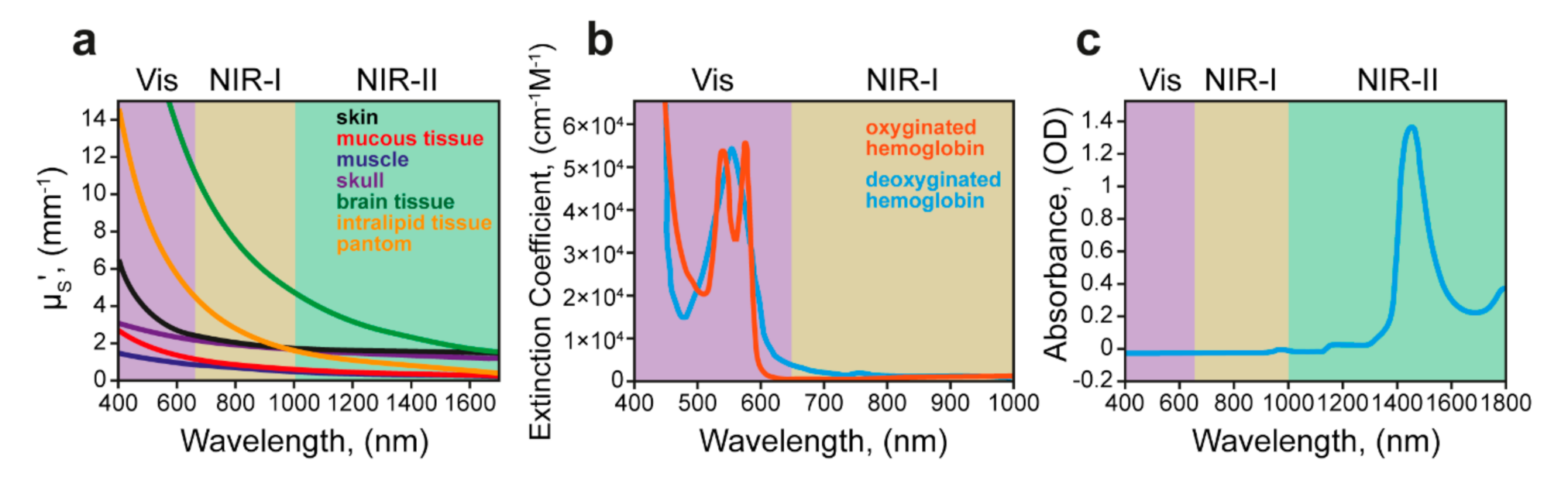
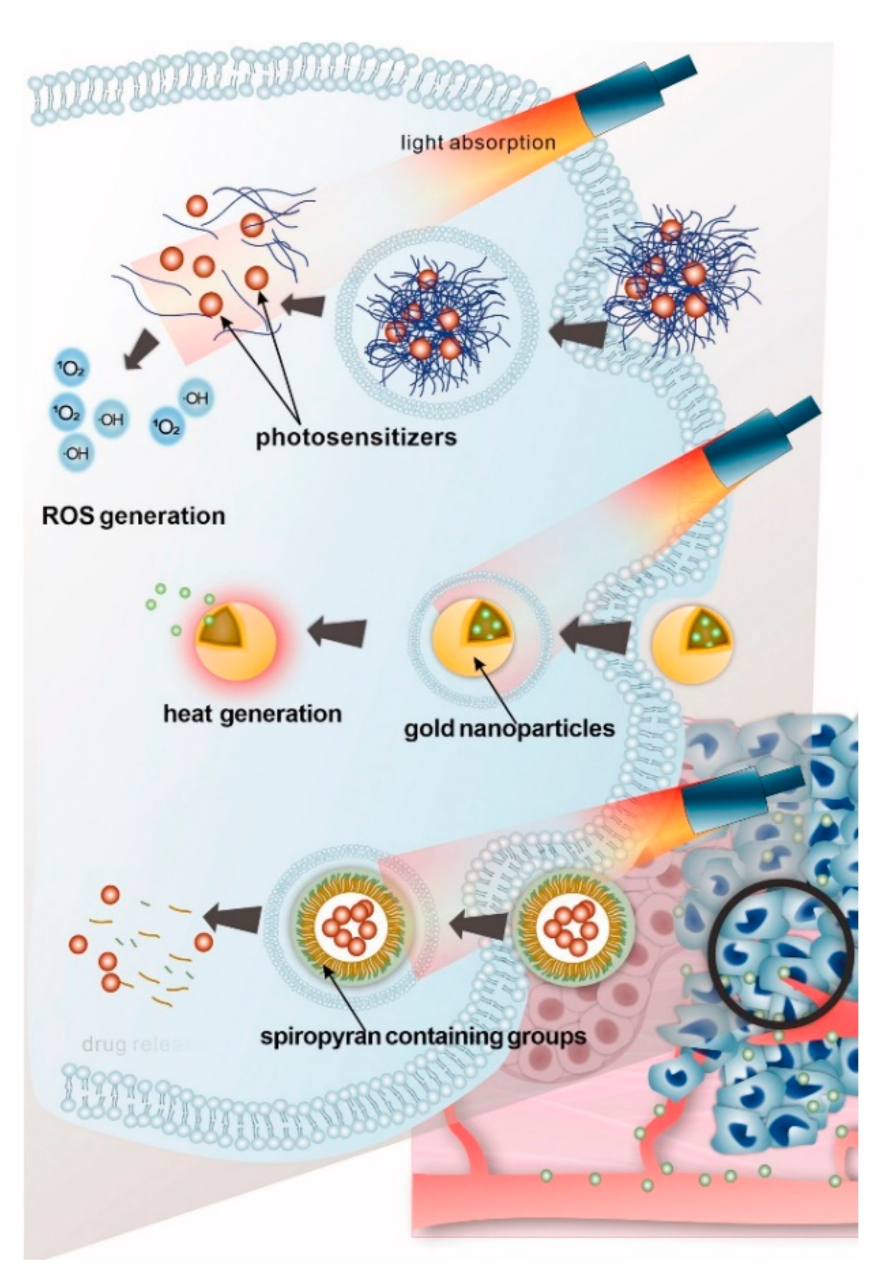
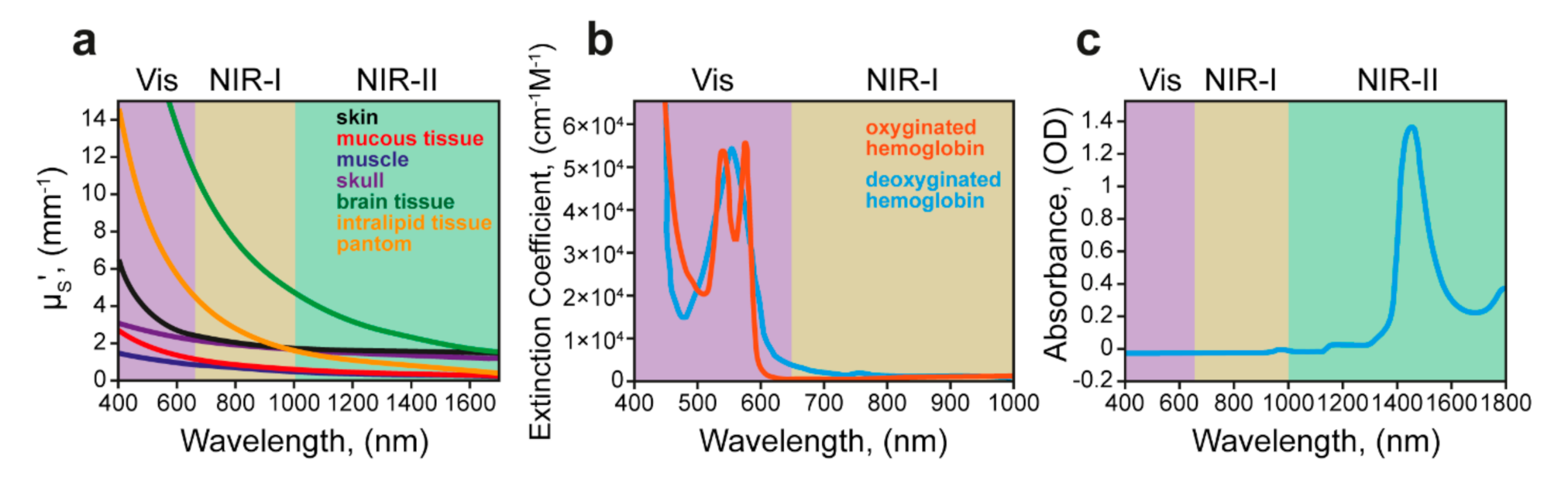
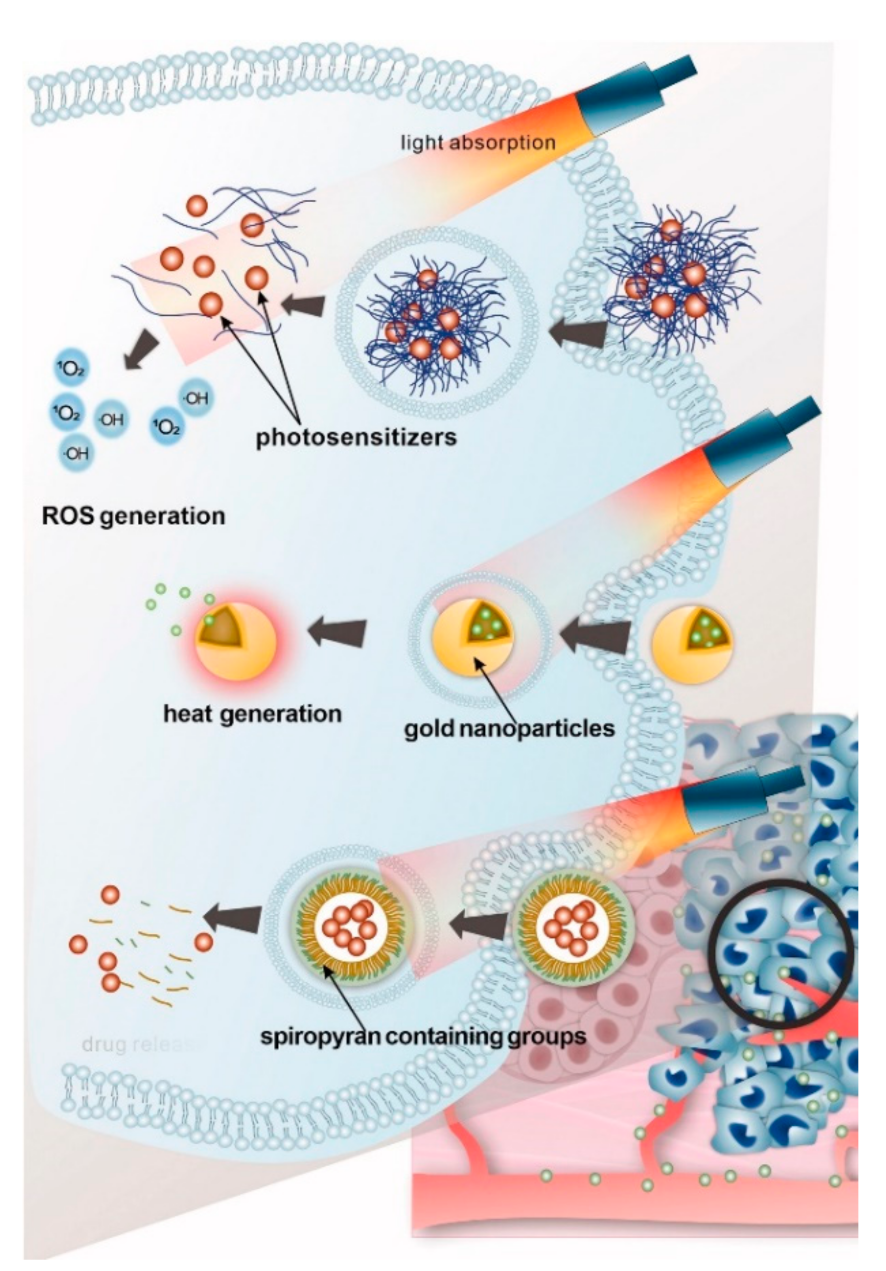
4.3. Light-Responsive Delivery Systems
4.4. Electric Fields in Targeted Drug Delivery
5. Clinical Translation of Drug Delivery Systems: Key Parameters, Challenges, and Successful Examples
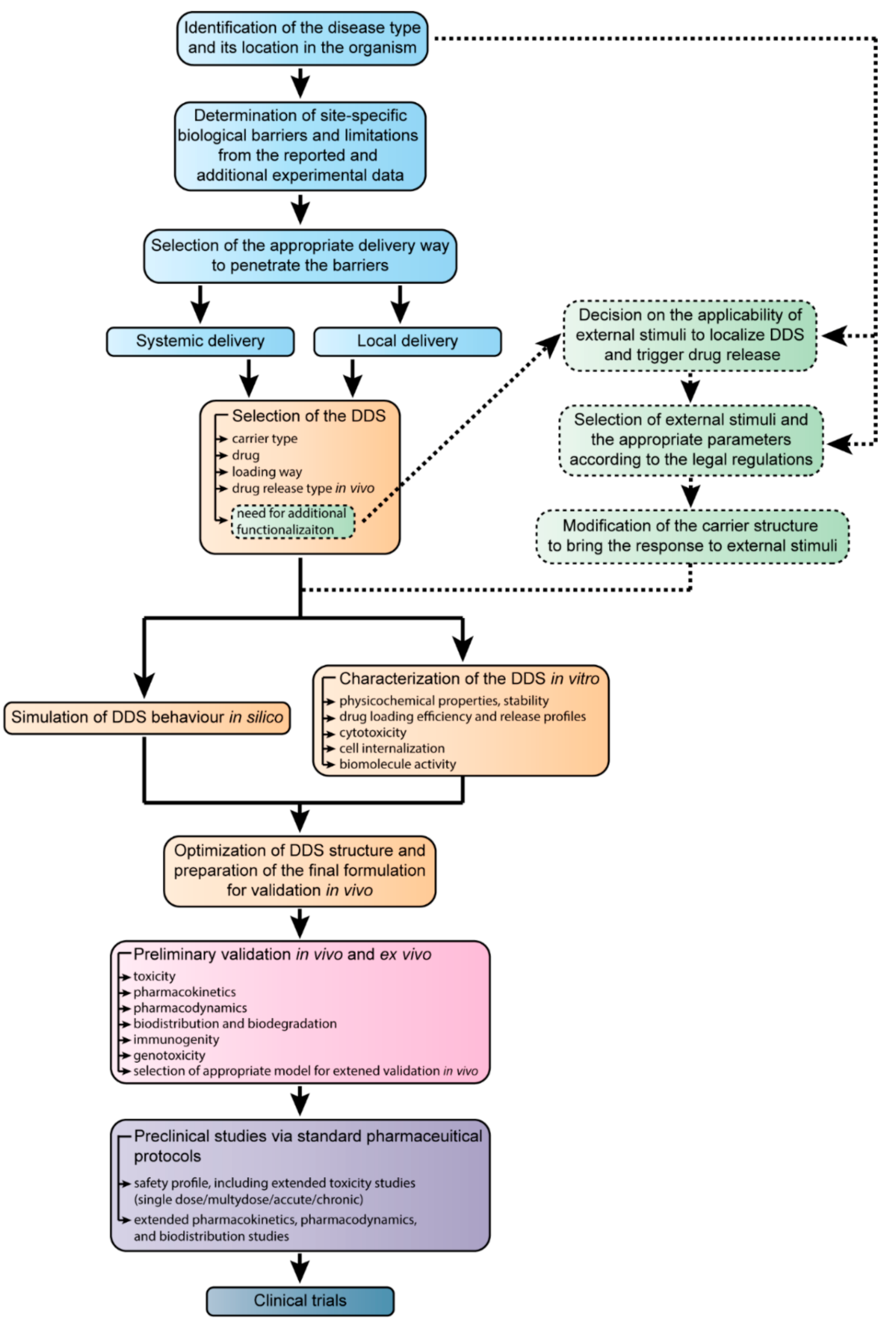
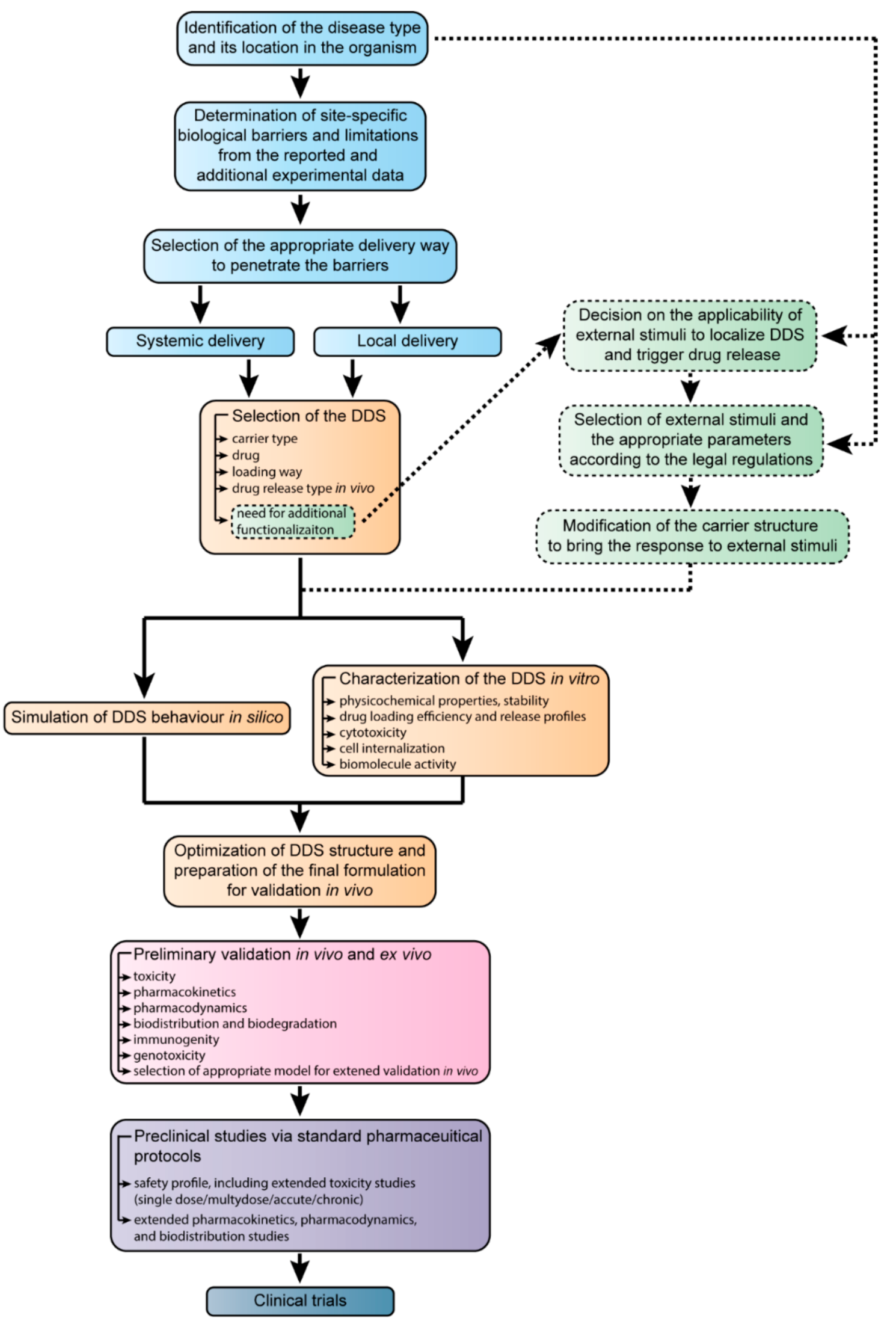
6. The Development of Targeted Drug Delivery Systems from the Design to Clinical Trials
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An Overview of Active and Passive Targeting Strategies to Improve the Nanocarriers Efficiency to Tumour Sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargason, A.M.; Anselmo, A.C.; Mitragotri, S. The Evolution of Commercial Drug Delivery Technologies. Nat. Biomed. Eng. 2021, 1–17. [Google Scholar] [CrossRef]
- Prikhozhdenko, E.S.; Gusliakova, O.I.; Kulikov, O.A.; Mayorova, O.A.; Shushunova, N.A.; Abdurashitov, A.S.; Bratashov, D.N.; Pyataev, N.A.; Tuchin, V.V.; Gorin, D.A.; et al. Target Delivery of Drug Carriers in Mice Kidney Glomeruli via Renal Artery. Balance between Efficiency and Safety. J. Control. Release 2021, 329, 175–190. [Google Scholar] [CrossRef]
- Lammari, N.; Tarhini, M.; Miladi, K.; Louaer, O.; Meniai, A.H.; Sfar, S.; Fessi, H.; Elaissari, A. Encapsulation Methods of Active Molecules for Drug Delivery. In Drug Delivery Devices and Therapeutic Systems; Elsevier: Amsterdam, The Netherlands, 2021; pp. 289–306. [Google Scholar] [CrossRef]
- Kakkar, A.; Traverso, G.; Farokhzad, O.C.; Weissleder, R.; Langer, R. Evolution of Macromolecular Complexity in Drug Delivery Systems. Nat. Rev. Chem. 2017, 1, 0063. [Google Scholar] [CrossRef] [PubMed]
- Mehryab, F.; Rabbani, S.; Shahhosseini, S.; Shekari, F.; Fatahi, Y.; Baharvand, H.; Haeri, A. Exosomes as a Next-Generation Drug Delivery System: An Update on Drug Loading Approaches, Characterization, and Clinical Application Challenges. Acta Biomater. 2020, 113, 42–62. [Google Scholar] [CrossRef]
- Damanik, F.F.R.; Brunelli, M.; Pastorino, L.; Ruggiero, C.; van Blitterswijk, C.; Rotmans, J.; Moroni, L. Sustained Delivery of Growth Factors with High Loading Efficiency in a Layer by Layer Assembly. Biomater. Sci. 2020, 8, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Demina, P.A.; Abalymov, A.A.; Voronin, D.V.; Sadovnikov, A.V.; Lomova, M.V. Highly-Magnetic Mineral Protein–Tannin Vehicles with Anti-Breast Cancer Activity. Mater. Chem. Front. 2021, 5, 2007–2018. [Google Scholar] [CrossRef]
- Svenskaya, Y.I.; Genina, E.A.; Parakhonskiy, B.V.; Lengert, E.V.; Talnikova, E.E.; Terentyuk, G.S.; Utz, S.R.; Gorin, D.A.; Tuchin, V.V.; Sukhorukov, G.B. A Simple Non-Invasive Approach toward Efficient Transdermal Drug Delivery Based on Biodegradable Particulate System. ACS Appl. Mater. Interfaces 2019, 11, 17270–17282. [Google Scholar] [CrossRef]
- Gusliakova, O.; Verkhovskii, R.; Abalymov, A.; Lengert, E.; Kozlova, A.; Atkin, V.; Nechaeva, O.; Morrison, A.; Tuchin, V.; Svenskaya, Y. Transdermal Platform for the Delivery of the Antifungal Drug Naftifine Hydrochloride Based on Porous Vaterite Particles. Mater. Sci. Eng. C 2021, 119, 111428. [Google Scholar] [CrossRef]
- Kozlova, A.A.; German, S.V.; Atkin, V.S.; Zyev, V.V.; Astle, M.A.; Bratashov, D.N.; Svenskaya, Y.I.; Gorin, D.A. Magnetic Composite Submicron Carriers with Structure-Dependent MRI Contrast. Inorganics 2020, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Novoselova, M.V.; Voronin, D.V.; Abakumova, T.O.; Demina, P.A.; Petrov, A.V.; Petrov, V.V.; Zatsepin, T.S.; Sukhorukov, G.B.; Gorin, D.A. Focused Ultrasound-Mediated Fluorescence of Composite Microcapsules Loaded with Magnetite Nanoparticles: In Vitro and in Vivo Study. Colloids Surf. B Biointerfaces 2019, 181, 680–687. [Google Scholar] [CrossRef]
- Inozemtseva, O.A.; Voronin, D.V.; Petrov, A.V.; Petrov, V.V.; Lapin, S.A.; Kozlova, A.A.; Bratashov, D.N.; Zakharevich, A.M.; Gorin, D.A. Disruption of Polymer and Composite Microcapsule Shells under High-Intensity Focused Ultrasound. Colloid J. 2018, 80, 771–782. [Google Scholar] [CrossRef]
- Lomova, M.V.; Sukhorukov, G.B.; Antipina, M.N. Antioxidant Coating of Micronsize Droplets for Prevention of Lipid Peroxidation in Oil-in-Water Emulsion. ACS Appl. Mater. Interfaces 2010, 2, 3669–3676. [Google Scholar] [CrossRef] [PubMed]
- Filipczak, N.; Pan, J.; Yalamarty, S.S.K.; Torchilin, V.P. Recent Advancements in Liposome Technology. Adv. Drug Deliv. Rev. 2020, 156, 4–22. [Google Scholar] [CrossRef]
- Zhao, Z.; Ukidve, A.; Kim, J.; Mitragotri, S. Targeting Strategies for Tissue-Specific Drug Delivery. Cell 2020, 181, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Wolfram, J. Extracellular Vesicles versus Synthetic Nanoparticles for Drug Delivery. Nat. Rev. Mater. 2021, 6, 103–106. [Google Scholar] [CrossRef]
- Zhang, Q.; Liang, J.; Zhao, L.; Wang, Y.; Zheng, Y.; Wu, Y.; Jiang, L. Synthesis of Novel Fluorescent Carbon Quantum Dots From Rosa Roxburghii for Rapid and Highly Selective Detection of O-Nitrophenol and Cellular Imaging. Front. Chem. 2020, 8, 665. [Google Scholar] [CrossRef]
- Shi, C.; Qian, X.; Jing, J.; Che, H. Functionalized CNTs with DOPO and Silicon Containing Agents: Effective Reinforcer for Thermal and Flame Retardant Properties of Polystyrene Nanocomposites. Front. Chem. 2021, 8. [Google Scholar] [CrossRef]
- Abalymov, A.; Van Poelvoorde, L.; Atkin, V.; Skirtach, A.G.; Konrad, M.; Parakhonskiy, B. Alkaline Phosphatase Delivery System Based on Calcium Carbonate Carriers for Acceleration of Ossification. ACS Appl. Bio Mater. 2020, 3, 2985–2996. [Google Scholar] [CrossRef]
- Kaczmarek, A.M.; Maegawa, Y.; Abalymov, A.; Skirtach, A.G.; Inagaki, S.; Van Der Voort, P. Lanthanide-Grafted Bipyridine Periodic Mesoporous Organosilicas (BPy-PMOs) for Physiological Range and Wide Temperature Range Luminescence Thermometry. ACS Appl. Mater. Interfaces 2020, 12, 13540–13550. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.Y.; Prajapati, N.; DeCoster, M.A.; Lvov, Y. Tagged Halloysite Nanotubes as a Carrier for Intercellular Delivery in Brain Microvascular Endothelium. Front. Bioeng. Biotechnol. 2020, 8, 451. [Google Scholar] [CrossRef]
- Abalymov, A.A.; Parakhonskiy, B.V.; Skirtach, A.G. Colloids-at-Surfaces: Physicochemical Approaches for Facilitating Cell Adhesion on Hybrid Hydrogels. Colloids Surf. A Physicochem. Eng. Asp. 2020, 603, 125185. [Google Scholar] [CrossRef]
- Vashist, A.; Atluri, V.; Raymond, A.; Kaushik, A.; Parira, T.; Huang, Z.; Durygin, A.; Tomitaka, A.; Nikkhah-Moshaie, R.; Vashist, A.; et al. Development of Multifunctional Biopolymeric Auto-Fluorescent Micro- and Nanogels as a Platform for Biomedical Applications. Front. Bioeng. Biotechnol. 2020, 8, 315. [Google Scholar] [CrossRef]
- Chhibber, S.; Kaur, J.; Kaur, S. Liposome Entrapment of Bacteriophages Improves Wound Healing in a Diabetic Mouse MRSA Infection. Front. Microbiol. 2018, 9, 561. [Google Scholar] [CrossRef]
- Csongradi, C.; du Plessis, J.; Aucamp, M.E.; Gerber, M. Topical Delivery of Roxithromycin Solid-State Forms Entrapped in Vesicles. Eur. J. Pharm. Biopharm. 2017, 114, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Alifu, N.; Wu, Z.; Chen, R.; Wang, X.; Ji, G.; Li, Q.; Qian, J.; Xu, B.; Song, D. Encapsulation-Dependent Enhanced Emission of Near-Infrared Nanoparticles Using in Vivo Three-Photon Fluorescence Imaging. Front. Bioeng. Biotechnol. 2020, 8, 1029. [Google Scholar] [CrossRef] [PubMed]
- Gaur, P.K.; Mishra, S.; Purohit, S. Solid Lipid Nanoparticles of Guggul Lipid as Drug Carrier for Transdermal Drug Delivery. Biomed Res. Int. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-J.; Huang, Y.-C.; Su, C.-M.; Ger, T.-R. Multi-Functional Drug Carrier Micelles with Anti-Inflammatory Drug. Front. Chem. 2019, 7, 93. [Google Scholar] [CrossRef]
- Sur, S.; Rathore, A.; Dave, V.; Reddy, K.R.; Chouhan, R.S.; Sadhu, V. Recent Developments in Functionalized Polymer Nanoparticles for Efficient Drug Delivery System. Nano-Struct. Nano-Objects 2019, 20, 100397. [Google Scholar] [CrossRef]
- Dawidczyk, C.M.; Kim, C.; Park, J.H.; Russell, L.M.; Lee, K.H.; Pomper, M.G.; Searson, P.C. State-of-the-Art in Design Rules for Drug Delivery Platforms: Lessons Learned from FDA-Approved Nanomedicines. J. Control. Release 2014, 187, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Torchilin, V. Tumor Delivery of Macromolecular Drugs Based on the EPR Effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Bharate, G.Y.; Daruwalla, J. Polymeric Drugs for Efficient Tumor-Targeted Drug Delivery Based on EPR-Effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of Exosome-Encapsulated Paclitaxel to Overcome MDR in Cancer Cells. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A Doxorubicin Delivery Platform Using Engineered Natural Membrane Vesicle Exosomes for Targeted Tumor Therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, Y.; Gong, C.; Wang, Z.; Xia, Q.; Gu, F.; Hu, C.; Zhang, L.; Guo, H.; Gao, S. A33 Antibody-Functionalized Exosomes for Targeted Delivery of Doxorubicin against Colorectal Cancer. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1973–1985. [Google Scholar] [CrossRef]
- Wang, Q.-S.; Gao, L.-N.; Zhu, X.-N.; Zhang, Y.; Zhang, C.-N.; Xu, D.; Cui, Y.-L. Co-Delivery of Glycyrrhizin and Doxorubicin by Alginate Nanogel Particles Attenuates the Activation of Macrophage and Enhances the Therapeutic Efficacy for Hepatocellular Carcinoma. Theranostics 2019, 9, 6239–6255. [Google Scholar] [CrossRef]
- Jiang, S.; Hua, L.; Guo, Z.; Sun, L. One-Pot Green Synthesis of Doxorubicin Loaded-Silica Nanoparticles for in vivo Cancer Therapy. Mater. Sci. Eng. C 2018, 90, 257–263. [Google Scholar] [CrossRef]
- Liu, Y.; Ding, X.; Li, J.; Luo, Z.; Hu, Y.; Liu, J.; Dai, L.; Zhou, J.; Hou, C.; Cai, K. Enzyme Responsive Drug Delivery System Based on Mesoporous Silica Nanoparticles for Tumor Therapy in vivo. Nanotechnology 2015, 26, 145102. [Google Scholar] [CrossRef]
- Dong, Z.; Feng, L.; Zhu, W.; Sun, X.; Gao, M.; Zhao, H.; Chao, Y.; Liu, Z. CaCO3 Nanoparticles as an Ultra-Sensitive Tumor-PH-Responsive Nanoplatform Enabling Real-Time Drug Release Monitoring and Cancer Combination Therapy. Biomaterials 2016, 110, 60–70. [Google Scholar] [CrossRef]
- Xu, C.; Yan, Y.; Tan, J.; Yang, D.; Jia, X.; Wang, L.; Xu, Y.; Cao, S.; Sun, S. Biodegradable Nanoparticles of Polyacrylic Acid–Stabilized Amorphous CaCO3 for Tunable PH-Responsive Drug Delivery and Enhanced Tumor Inhibition. Adv. Funct. Mater. 2019, 29, 1808146. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of SiRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes Facilitate Therapeutic Targeting of Oncogenic KRAS in Pancreatic Cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.-Y.; Liu, X.-Y.; Chen, C.-J.; Wang, J.-C.; Feng, Q.; Yu, M.-Z.; Ma, X.-F.; Pei, X.-W.; Niu, Y.-J.; Qiu, C.; et al. Core-Shell Type Lipid/RPAA-Chol Polymer Hybrid Nanoparticles for in Vivo SiRNA Delivery. Biomaterials 2014, 35, 2066–2078. [Google Scholar] [CrossRef]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Qiao, H.; Su, Z.; Chen, M.; Ping, Q.; Sun, M. PEGylated Carboxymethyl Chitosan/Calcium Phosphate Hybrid Anionic Nanoparticles Mediated HTERT SiRNA Delivery for Anticancer Therapy. Biomaterials 2014, 35, 7978–7991. [Google Scholar] [CrossRef]
- Kim, S.M.; Yang, Y.; Oh, S.J.; Hong, Y.; Seo, M.; Jang, M. Cancer-Derived Exosomes as a Delivery Platform of CRISPR/Cas9 Confer Cancer Cell Tropism-Dependent Targeting. J. Control. Release 2017, 266, 8–16. [Google Scholar] [CrossRef]
- Nelson, C.E.; Kim, A.J.; Adolph, E.J.; Gupta, M.K.; Yu, F.; Hocking, K.M.; Davidson, J.M.; Guelcher, S.A.; Duvall, C.L. Tunable Delivery of SiRNA from a Biodegradable Scaffold to Promote Angiogenesis In Vivo. Adv. Mater. 2014, 26, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, B.; Kang, L.; Chen, L.; Sun, P.; Jin, M.; Wang, Q.; Bae, Y.H.; Huang, W.; Gao, Z. Systemic SiRNA Delivery with a Dual PH-Responsive and Tumor-Targeted Nanovector for Inhibiting Tumor Growth and Spontaneous Metastasis in Orthotopic Murine Model of Breast Carcinoma. Theranostics 2017, 7, 357–376. [Google Scholar] [CrossRef] [Green Version]
- Yoon, H.Y.; Kim, H.R.; Saravanakumar, G.; Heo, R.; Chae, S.Y.; Um, W.; Kim, K.; Kwon, I.C.; Lee, J.Y.; Lee, D.S.; et al. Bioreducible Hyaluronic Acid Conjugates as SiRNA Carrier for Tumor Targeting. J. Control. Release 2013, 172, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, H.; Wang, K.; Guo, Q.; Li, C.; Jiang, H.; Hu, Y.; Oupicky, D.; Sun, M. Bioreducible Cross-Linked Hyaluronic Acid/Calcium Phosphate Hybrid Nanoparticles for Specific Delivery of SiRNA in Melanoma Tumor Therapy. ACS Appl. Mater. Interfaces 2017, 9, 14576–14589. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as Drug Delivery Vehicles for Parkinson’s Disease Therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Yuan, D.; Zhao, Y.; Banks, W.A.; Bullock, K.M.; Haney, M.; Batrakova, E.; Kabanov, A.V. Macrophage Exosomes as Natural Nanocarriers for Protein Delivery to Inflamed Brain. Biomaterials 2017, 142, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sajeesh, S.; Vauthier, C.; Gueutin, C.; Ponchel, G.; Sharma, C.P. Thiol Functionalized Polymethacrylic Acid-Based Hydrogel Microparticles for Oral Insulin Delivery. Acta Biomater. 2010, 6, 3072–3080. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, V.; Shi, Z.; Huang, S.; Du, Y.; Kopp, M.; Frede, A.; Knuschke, T.; Buer, J.; Yang, D.; Wu, J.; et al. Delivery of the TLR Ligand Poly(I:C) to Liver Cells in Vitro and in Vivo by Calcium Phosphate Nanoparticles Leads to a Pronounced Immunostimulation. Acta Biomater. 2017, 64, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.; Yin, N.; Zhang, Y.; Gou, J.; Yin, T.; He, H.; Ding, H.; Zhang, Y.; Tang, X. Sialic Acid-Modified Dexamethasone Lipid Calcium Phosphate Gel Core Nanoparticles for Target Treatment of Kidney Injury. Biomater. Sci. 2020, 8, 3871–3884. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.X.; Chung, E.P.; Bowser, R.; Sirianni, R.W. Lipid and Polymer Blended Polyester Nanoparticles Loaded with Adapalene for Activation of Retinoid Signaling in the CNS Following Intravenous Administration. J. Drug Deliv. Sci. Technol. 2019, 52, 927–933. [Google Scholar] [CrossRef]
- Su, C.-W.; Chiang, C.-S.; Li, W.-M.; Hu, S.-H.; Chen, S.-Y. Multifunctional Nanocarriers for Simultaneous Encapsulation of Hydrophobic and Hydrophilic Drugs in Cancer Treatment. Nanomedicine 2014, 9, 1499–1515. [Google Scholar] [CrossRef]
- Xu, J.; Zhao, Q.; Jin, Y.; Qiu, L. High Loading of Hydrophilic/Hydrophobic Doxorubicin into Polyphosphazene Polymersome for Breast Cancer Therapy. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 349–358. [Google Scholar] [CrossRef]
- Kita, K.; Dittrich, C. Drug Delivery Vehicles with Improved Encapsulation Efficiency: Taking Advantage of Specific Drug–Carrier Interactions. Expert Opin. Drug Deliv. 2011, 8, 329–342. [Google Scholar] [CrossRef]
- Som, A.; Raliya, R.; Paranandi, K.; High, R.A.; Reed, N.; Beeman, S.C.; Brandenburg, M.; Sudlow, G.; Prior, J.L.; Akers, W.; et al. Calcium Carbonate Nanoparticles Stimulate Tumor Metabolic Reprogramming and Modulate Tumor Metastasis. Nanomedicine 2019, 14, 169–182. [Google Scholar] [CrossRef]
- Khalifehzadeh, R.; Arami, H. Biodegradable Calcium Phosphate Nanoparticles for Cancer Therapy. Adv. Colloid Interface Sci. 2020, 279, 102157. [Google Scholar] [CrossRef]
- Wang, N.; Cheng, X.; Li, N.; Wang, H.; Chen, H. Nanocarriers and Their Loading Strategies. Adv. Healthc. Mater. 2019, 8, 1801002. [Google Scholar] [CrossRef]
- Rudge, S.; Peterson, C.; Vessely, C.; Koda, J.; Stevens, S.; Catterall, L. Adsorption and Desorption of Chemotherapeutic Drugs from a Magnetically Targeted Carrier (MTC). J. Control. Release 2001, 74, 335–340. [Google Scholar] [CrossRef]
- Radtchenko, I.L.; Sukhorukov, G.B.; Leporatti, S.; Khomutov, G.B.; Donath, E.; Möhwald, H. Assembly of Alternated Multivalent Ion/Polyelectrolyte Layers on Colloidal Particles. Stability of the Multilayers and Encapsulation of Macromolecules into Polyelectrolyte Capsules. J. Colloid Interface Sci. 2000, 230, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhang, S.; Tong, W.; Gao, C.; Shen, J. Polyelectrolyte Microcapsules Templated on Poly(Styrene Sulfonate)-Doped CaCO3 Particles for Loading and Sustained Release of Daunorubicin and Doxorubicin. Eur. Polym. J. 2006, 42, 3341–3351. [Google Scholar] [CrossRef]
- Vergaro, V.; Papadia, P.; Petrini, P.; Fanizzi, F.P.; De Pascali, S.A.; Baldassarre, F.; Pastorino, L.; Ciccarella, G. Nanostructured Polysaccharidic Microcapsules for Intracellular Release of Cisplatin. Int. J. Biol. Macromol. 2017, 99, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Donath, E.; Möhwald, H. Permeability of Ibuprofen in Various Polyelectrolyte Multilayers. Macromol. Mater. Eng. 2001, 286, 591. [Google Scholar] [CrossRef]
- Prabu, C.; Latha, S.; Selvamani, P.; Ahrentorp, F.; Johansson, C.; Takeda, R.; Takemura, Y.; Ota, S. Layer-by-Layer Assembled Magnetic Prednisolone Microcapsules (MPC) for Controlled and Targeted Drug Release at Rheumatoid Arthritic Joints. J. Magn. Magn. Mater. 2017, 427, 258–267. [Google Scholar] [CrossRef]
- Ivanov, A.N.; Chibrikova, Y.A.; Saveleva, M.S.; Rogozhina, A.S.; Norkin, I.A. Biocompatibility of Polycaprolactone Scaffold Providing Targeting Delivery of Alkaline Phosphatase. Cell Tissue Biol. 2021, 15, 301–309. [Google Scholar] [CrossRef]
- Huang, L.; Zhou, J.; Chen, Y.; Li, W.; Han, X.; Wang, L. Engineering Microcapsules for Simultaneous Delivery of Combinational Therapeutics. Adv. Mater. Technol. 2020, 5, 2000623. [Google Scholar] [CrossRef]
- Kumar, C.S.S.R.; Mohammad, F. Magnetic Nanomaterials for Hyperthermia-Based Therapy and Controlled Drug Delivery. Adv. Drug Deliv. Rev. 2011, 63, 789–808. [Google Scholar] [CrossRef] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Langermans, J.A.M.; Hazenbos, W.L.W.; van Furth, R. Antimicrobial Functions of Mononuclear Phagocytes. J. Immunol. Methods 1994, 174, 185–194. [Google Scholar] [CrossRef]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Li, Y. Physicochemical Characteristics of Nanoparticles Affect Circulation, Biodistribution, Cellular Internalization, and Trafficking. Small 2013, 9, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Arvizo, R.R.; Miranda, O.R.; Moyano, D.F.; Walden, C.A.; Giri, K.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Reid, J.M.; Mukherjee, P. Modulating Pharmacokinetics, Tumor Uptake and Biodistribution by Engineered Nanoparticles. PLoS ONE 2011, 6, e24374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundqvist, M.; Stigler, J.; Elia, G.; Lynch, I.; Cedervall, T.; Dawson, K.A. Nanoparticle Size and Surface Properties Determine the Protein Corona with Possible Implications for Biological Impacts. Proc. Natl. Acad. Sci. USA 2008, 105, 14265–14270. [Google Scholar] [CrossRef] [Green Version]
- Tenzer, S.; Docter, D.; Rosfa, S.; Wlodarski, A.; Kuharev, J.; Rekik, A.; Knauer, S.K.; Bantz, C.; Nawroth, T.; Bier, C.; et al. Nanoparticle Size Is a Critical Physicochemical Determinant of the Human Blood Plasma Corona: A Comprehensive Quantitative Proteomic Analysis. ACS Nano 2011, 5, 7155–7167. [Google Scholar] [CrossRef] [PubMed]
- Decuzzi, P.; Lee, S.; Bhushan, B.; Ferrari, M. A Theoretical Model for the Margination of Particles within Blood Vessels. Ann. Biomed. Eng. 2005, 33, 179–190. [Google Scholar] [CrossRef]
- Decuzzi, P.; Pasqualini, R.; Arap, W.; Ferrari, M. Intravascular Delivery of Particulate Systems: Does Geometry Really Matter? Pharm. Res. 2009, 26, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Narayanaswamy, R.; Attia, S.A.; Torchilin, V.P. Parameters and Strategies to Overcome Barriers to Systemic Delivery; Springer: Cham, Switzerland, 2020; pp. 447–475. [Google Scholar] [CrossRef]
- Cuggino, J.C.; Blanco, E.R.O.; Gugliotta, L.M.; Alvarez Igarzabal, C.I.; Calderón, M. Crossing Biological Barriers with Nanogels to Improve Drug Delivery Performance. J. Control. Release 2019, 307, 221–246. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. An Overview of Clinical and Commercial Impact of Drug Delivery Systems. J. Control. Release 2014, 190, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of Nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal Escape Pathways for Delivery of Biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Khan, J.M.; Haque, S. Strategies in the Design of Endosomolytic Agents for Facilitating Endosomal Escape in Nanoparticles. Biochimie 2019, 160, 61–75. [Google Scholar] [CrossRef]
- Bus, T.; Traeger, A.; Schubert, U.S. The Great Escape: How Cationic Polyplexes Overcome the Endosomal Barrier. J. Mater. Chem. B 2018, 6, 6904–6918. [Google Scholar] [CrossRef]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.-Y.; Pellois, J.-P. Improving the Endosomal Escape of Cell-Penetrating Peptides and Their Cargos: Strategies and Challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef] [PubMed]
- Selbo, P.K.; Weyergang, A.; Høgset, A.; Norum, O.-J.; Berstad, M.B.; Vikdal, M.; Berg, K. Photochemical Internalization Provides Time- and Space-Controlled Endolysosomal Escape of Therapeutic Molecules. J. Control. Release 2010, 148, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Peeler, D.J.; Sellers, D.L.; Pun, S.H. PH-Sensitive Polymers as Dynamic Mediators of Barriers to Nucleic Acid Delivery. Bioconjug. Chem. 2019, 30, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Linko, V. Challenges and Perspectives of DNA Nanostructures in Biomedicine. Angew. Chemie Int. Ed. 2020, 59, 15818–15833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wu, F.; Ji, Y.; Yin, L. Recent Advances in Anti-Cancer Protein/Peptide Delivery. Bioconjug. Chem. 2019, 30, 305–324. [Google Scholar] [CrossRef]
- Torchilin, V.P.; Lukyanov, A.N. Peptide and Protein Drug Delivery to and into Tumors: Challenges and Solutions. Drug Discov. Today 2003, 8, 259–266. [Google Scholar] [CrossRef]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the Challenges in Administering Biopharmaceuticals: Formulation and Delivery Strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef] [Green Version]
- Barua, S.; Mitragotri, S. Challenges Associated with Penetration of Nanoparticles across Cell and Tissue Barriers: A Review of Current Status and Future Prospects. Nano Today 2014, 9, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Zborowski, M.; Chalmers, J.J.; Lowrie, W.G. Magnetic Cell Manipulation and Sorting; Springer: Cham, Switzerland, 2017; pp. 15–55. [Google Scholar] [CrossRef]
- Schenck, J.F. Physical Interactions of Static Magnetic Fields with Living Tissues. Prog. Biophys. Mol. Biol. 2005, 87, 185–204. [Google Scholar] [CrossRef]
- Voronin, D.V.; Sindeeva, O.A.; Kurochkin, M.A.; Mayorova, O.; Fedosov, I.V.; Semyachkina-Glushkovskaya, O.; Gorin, D.A.; Tuchin, V.V.; Sukhorukov, G.B. In Vitro and in Vivo Visualization and Trapping of Fluorescent Magnetic Microcapsules in a Bloodstream. ACS Appl. Mater. Interfaces 2017, 9, 6885–6893. [Google Scholar] [CrossRef] [PubMed]
- International Commission on Non-Ionizing Radiation Protection. Guidelines on limits of exposure to static magnetic fields. Health Phys. 2009, 96, 504–514. [Google Scholar] [CrossRef]
- Holden, A.V. The Sensitivity of the Heart to Static Magnetic Fields. Prog. Biophys. Mol. Biol. 2005, 87, 289–320. [Google Scholar] [CrossRef]
- World Health Organization. Environmental Health Criteria 232: Static Fields; World Health Organization: Geneva, Switzerland, 2006. [Google Scholar]
- Atef, M.M.; Abd Ei-Baset, M.S.; Ell-Kareem, A.; Aida, S.; Fadel, M.A. Effects of a Static Magnetic Field on Haemoglobin Structure and Function. Int. J. Biol. Macromol. 1995, 17, 105–111. [Google Scholar] [CrossRef]
- Mayda, S.; Kandemir, Z.; Bulut, N.; Maekawa, S. Magnetic Mechanism for the Biological Functioning of Hemoglobin. Sci. Rep. 2020, 10, 8569. [Google Scholar] [CrossRef]
- Zaremba, L.; Phillips, R. CE—MRI 3: FDA Guidelines for Magnetic Resonance Equipment Safety; Food and Drug Administration: Rockville, MD, USA, 2002. [Google Scholar]
- International Non-Ionizing Radiation Committee of the International Radiation Protection Association (IRPA/INIRC). Protection of the Patient Undergoing a Magnetic Resonance Examination. Health Phys. 1991, 61, 923–928. [Google Scholar]
- International Commission on Non-Ionizing Radiation Protection. Amendment to the ICNIRP “Statement on medical magnetic resonance (MR) procedures: Protection of patients”. Health Phys. 2009, 97, 259–261. [Google Scholar] [CrossRef]
- Medicines and Healthcare Products Regulatory Agency. Magnetic Resonance Imaging Equipment in Clinical Use: Safety Guidelines. Relevant Safety Information for Users of Magnetic Resonance Imaging (MRI) Equipment in Clinical Use; Medicines and Healthcare Products Regulatory Agency: London, UK, 2021. [Google Scholar]
- Golovin, Y.I.; Klyachko, N.L.; Zhigachev, A.O.; Gribanovskii, S.L.; Efremova, M.V.; Majouga, A.G.; Kabanov, A.V. Selective Deformation of Single Macromolecules and Biomolecular Structures as a Method for Remote Control of Their Properties and Functions for Next-Generation Medicine. Russ. Metall. 2019, 2019, 374–384. [Google Scholar] [CrossRef]
- Noh, S.; Moon, S.H.; Shin, T.-H.; Lim, Y.; Cheon, J. Recent Advances of Magneto-Thermal Capabilities of Nanoparticles: From Design Principles to Biomedical Applications. Nano Today 2017, 13, 61–76. [Google Scholar] [CrossRef]
- Golovin, Y.I.; Gribanovsky, S.L.; Golovin, D.Y.; Klyachko, N.L.; Majouga, A.G.; Master, A.M.; Sokolsky, M.; Kabanov, A.V. Towards Nanomedicines of the Future: Remote Magneto-Mechanical Actuation of Nanomedicines by Alternating Magnetic Fields. J. Control. Release 2015, 219, 43–60. [Google Scholar] [CrossRef] [Green Version]
- Hergt, R.; Dutz, S.; Müller, R.; Zeisberger, M. Magnetic Particle Hyperthermia: Nanoparticle Magnetism and Materials Development for Cancer Therapy. J. Phys. Condens. Matter 2006, 18, S2919–S2934. [Google Scholar] [CrossRef]
- Deatsch, A.E.; Evans, B.A. Heating Efficiency in Magnetic Nanoparticle Hyperthermia. J. Magn. Magn. Mater. 2014, 354, 163–172. [Google Scholar] [CrossRef]
- International Commission on Non-Ionizing Radiation Protection. Guidelines for Limiting Exposure to Electromagnetic Fields (100 KHz to 300 GHz). Health Phys. 2020, 118, 483–524. [Google Scholar] [CrossRef]
- International Commission on Non-Ionizing Radiation Protection. Guidelines for Limiting Exposure to Time Varying Electric, Magnetic, and Electromagnetic Fields (up to 300 GHz). Health Phys. 1998, 74, 494–522. [Google Scholar]
- Golovin, Y.I.; Klyachko, N.L.; Majouga, A.G.; Sokolsky, M.; Kabanov, A.V. Theranostic Multimodal Potential of Magnetic Nanoparticles Actuated by Non-Heating Low Frequency Magnetic Field in the New-Generation Nanomedicine. J. Nanopart. Res. 2017, 19, 63. [Google Scholar] [CrossRef]
- Dutz, S.; Hergt, R. Magnetic Nanoparticle Heating and Heat Transfer on a Microscale: Basic Principles, Realities and Physical Limitations of Hyperthermia for Tumour Therapy. Int. J. Hyperth. 2013, 29, 790–800. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Guo, Y.; Jiao, W.; Gao, X.; Lee, W.S.V.; Wang, Y.; Deng, X.; He, Y.; Jiao, J.; et al. Electromagnetic Field-Programmed Magnetic Vortex Nanodelivery System for Efficacious Cancer Therapy. Adv. Sci. 2021, 2100950. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.-T.; Hsu, R.-S.; Fang, J.-H.; Hu, P.-F.; Chiang, C.-S.; Hu, S.-H. Marginative Delivery-Mediated Extracellular Leakiness and T Cell Infiltration in Lung Metastasis by a Biomimetic Nanoraspberry. Nano Lett. 2021, 21, 1375–1383. [Google Scholar] [CrossRef]
- Liu, S.; Shi, D.; Chen, L.; Yan, Y.; Wang, X.; Song, Y.; Pu, S.; Liang, Y.; Zhao, Y.; Zhang, Y.; et al. Paclitaxel-Loaded Magnetic Nanocrystals for Tumor Neovascular-Targeted Theranostics: An Amplifying Synergistic Therapy Combining Magnetic Hyperthermia with Chemotherapy. Nanoscale 2021, 13, 3613–3626. [Google Scholar] [CrossRef] [PubMed]
- Leighton, T.G. What Is Ultrasound? Prog. Biophys. Mol. Biol. 2007, 93, 3–83. [Google Scholar] [CrossRef]
- International Commission on Non-Ionizing Radiation Protection. Statement on Diagnostic Devices Using Non-Ionizing Radiation. Health Phys. 2017, 112, 305–321. [Google Scholar] [CrossRef]
- Duan, L.; Yang, L.; Jin, J.; Yang, F.; Liu, D.; Hu, K.; Wang, Q.; Yue, Y.; Gu, N. Micro/Nano-Bubble-Assisted Ultrasound to Enhance the EPR Effect and Potential Theranostic Applications. Theranostics 2020, 10, 462–483. [Google Scholar] [CrossRef]
- Vyas, N.; Manmi, K.; Wang, Q.; Jadhav, A.J.; Barigou, M.; Sammons, R.L.; Kuehne, S.A.; Walmsley, A.D. Which Parameters Affect Biofilm Removal with Acoustic Cavitation? A Review. Ultrasound Med. Biol. 2019, 45, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Pirsaheb, M.; Moradi, N. Sonochemical Degradation of Pesticides in Aqueous Solution: Investigation on the Influence of Operating Parameters and Degradation Pathway—A Systematic Review. RSC Adv. 2020, 10, 7396–7423. [Google Scholar] [CrossRef]
- Yusof, N.S.M.; Babgi, B.; Alghamdi, Y.; Aksu, M.; Madhavan, J.; Ashokkumar, M. Physical and Chemical Effects of Acoustic Cavitation in Selected Ultrasonic Cleaning Applications. Ultrason. Sonochem. 2016, 29, 568–576. [Google Scholar] [CrossRef] [PubMed]
- van Wijngaarden, L. Mechanics of Collapsing Cavitation Bubbles. Ultrason. Sonochem. 2016, 29, 524–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roovers, S.; Segers, T.; Lajoinie, G.; Deprez, J.; Versluis, M.; De Smedt, S.C.; Lentacker, I. The Role of Ultrasound-Driven Microbubble Dynamics in Drug Delivery: From Microbubble Fundamentals to Clinical Translation. Langmuir 2019, 35, 10173–10191. [Google Scholar] [CrossRef]
- Lentacker, I.; De Cock, I.; Deckers, R.; De Smedt, S.C.; Moonen, C.T.W. Understanding Ultrasound Induced Sonoporation: Definitions and Underlying Mechanisms. Adv. Drug Deliv. Rev. 2014, 72, 49–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, P.; Han, T.; Yu, A.C.H.; Xu, L. Mechanistic Understanding the Bioeffects of Ultrasound-Driven Microbubbles to Enhance Macromolecule Delivery. J. Control. Release 2018, 272, 169–181. [Google Scholar] [CrossRef]
- Ferrara, K.; Pollard, R.; Borden, M. Ultrasound Microbubble Contrast Agents: Fundamentals and Application to Gene and Drug Delivery. Annu. Rev. Biomed. Eng. 2007, 9, 415–447. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-L.; Fan, C.-H.; Ting, C.-Y.; Yeh, C.-K. Combining Microbubbles and Ultrasound for Drug Delivery to Brain Tumors: Current Progress and Overview. Theranostics 2014, 4, 432–444. [Google Scholar] [CrossRef]
- World Health Organization. Ultrasound; World Health Organization: Geneva, Switzerland, 1982. [Google Scholar]
- Nelson, T.R.; Fowlkes, J.B.; Abramowicz, J.S.; Church, C.C. Ultrasound Biosafety Considerations for the Practicing Sonographer and Sonologist. J. Ultrasound Med. 2009, 28, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Center for Devices and Radiological Health. Marketing Clearance of Diagnostic Ultrasound Systems and Transducers Guidance for Industry and Food and Drug Administration Staff; Center for Devices and Radiological Health: Rockville, MD, USA, 2019. [Google Scholar]
- Miller, D.L.; Smith, N.B.; Bailey, M.R.; Czarnota, G.J.; Hynynen, K.; Makin, I.R.S. Overview of Therapeutic Ultrasound Applications and Safety Considerations. J. Ultrasound Med. 2012, 31, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Government of Canada. Radiation Emitting Devices Regulations, CRC, c 1370; Government of Canada: Ottawa, ON, Canada, 2014. [Google Scholar]
- Li, D.; Lin, L.; Fan, Y.; Liu, L.; Shen, M.; Wu, R.; Du, L.; Shi, X. Ultrasound-Enhanced Fluorescence Imaging and Chemotherapy of Multidrug-Resistant Tumors Using Multifunctional Dendrimer/Carbon Dot Nanohybrids. Bioact. Mater. 2021, 6, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wan, Q.; Zou, C.; Chen, M.; Wan, G.; Liu, X.; Chen, H. Stepwise Drug Release from a Nanoplatform under MR-Assisted Focused Ultrasound Stimulation. Chem. Eng. J. 2021, 417, 128004. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, T.; Sun, S.; Ren, W.; Wu, A.; Xu, H. Ultrasound-Mediated Cavitation Enhances EGFR-Targeting PLGA-PEG Nano-Micelle Delivery for Triple-Negative Breast Cancer Treatment. Cancers 2021, 13, 3383. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.; Antaris, A.L.; Dai, H. Near-Infrared Fluorophores for Biomedical Imaging. Nat. Biomed. Eng. 2017, 1, 0010. [Google Scholar] [CrossRef]
- Tong, R.; Kohane, D.S. New Strategies in Cancer Nanomedicine. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 41–57. [Google Scholar] [CrossRef]
- Son, J.; Yi, G.; Yoo, J.; Park, C.; Koo, H.; Choi, H.S. Light-Responsive Nanomedicine for Biophotonic Imaging and Targeted Therapy. Adv. Drug Deliv. Rev. 2019, 138, 133–147. [Google Scholar] [CrossRef]
- Cai, Y.; Wei, Z.; Song, C.; Tang, C.; Han, W.; Dong, X. Optical Nano-Agents in the Second near-Infrared Window for Biomedical Applications. Chem. Soc. Rev. 2019, 48, 22–37. [Google Scholar] [CrossRef]
- Miao, Q.; Pu, K. Organic Semiconducting Agents for Deep-Tissue Molecular Imaging: Second Near-Infrared Fluorescence, Self-Luminescence, and Photoacoustics. Adv. Mater. 2018, 30, 1801778. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Song, J.; Qu, J.; Cheng, Z. Crucial Breakthrough of Second Near-Infrared Biological Window Fluorophores: Design and Synthesis toward Multimodal Imaging and Theranostics. Chem. Soc. Rev. 2018, 47, 4258–4278. [Google Scholar] [CrossRef]
- Hong, G.; Diao, S.; Antaris, A.L.; Dai, H. Carbon Nanomaterials for Biological Imaging and Nanomedicinal Therapy. Chem. Rev. 2015, 115, 10816–10906. [Google Scholar] [CrossRef]
- Gai, S.; Yang, G.; Yang, P.; He, F.; Lin, J.; Jin, D.; Xing, B. Recent Advances in Functional Nanomaterials for Light–Triggered Cancer Therapy. Nano Today 2018, 19, 146–187. [Google Scholar] [CrossRef]
- Li, X.; Lovell, J.F.; Yoon, J.; Chen, X. Clinical Development and Potential of Photothermal and Photodynamic Therapies for Cancer. Nat. Rev. Clin. Oncol. 2020, 17, 657–674. [Google Scholar] [CrossRef]
- Liang, J.; Yang, B.; Zhou, X.; Han, Q.; Zou, J.; Cheng, L. Stimuli-Responsive Drug Delivery Systems for Head and Neck Cancer Therapy. Drug Deliv. 2021, 28, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Khlebtsov, B.N.; Khanadeev, V.A.; Burov, A.M.; Le Ru, E.C.; Khlebtsov, N.G. Reexamination of Surface-Enhanced Raman Scattering from Gold Nanorods as a Function of Aspect Ratio and Shape. J. Phys. Chem. C 2020, 124, 10647–10658. [Google Scholar] [CrossRef]
- Liu, Y.; Bhattarai, P.; Dai, Z.; Chen, X. Photothermal Therapy and Photoacoustic Imaging via Nanotheranostics in Fighting Cancer. Chem. Soc. Rev. 2019, 48, 2053–2108. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhang, E.; Su, Y.; Cheng, T.; Shi, C. A Review of NIR Dyes in Cancer Targeting and Imaging. Biomaterials 2011, 32, 7127–7138. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Wang, D.; Liang, W.; Liu, L.; Zhang, Y.; Chen, X.; Sang, D.K.; Xing, C.; Li, Z.; Dong, B.; et al. Novel Concept of the Smart NIR-Light–Controlled Drug Release of Black Phosphorus Nanostructure for Cancer Therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 501–506. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Yu, W.; Chen, X.; Gao, H. Self-Propelled Micro/Nanomotors for Tumor Targeting Delivery and Therapy. Adv. Healthc. Mater. 2021, 10, 2001212. [Google Scholar] [CrossRef]
- International Commission on Non-Ionizing Radiation Protection. Guidelines on Limits of Exposure to Laser Radiation of Wavelengths between 180 Nm and 1000 Μm. Health Phys. 2013, 105, 271–295. [Google Scholar] [CrossRef] [Green Version]
- Intended Human Exposure to Non-Ionizing Radiation for Cosmetic Purposes. Health Phys. 2020, 118, 562–579. [CrossRef] [Green Version]
- Niu, S.; Zhang, X.; Williams, G.R.; Wu, J.; Gao, F.; Fu, Z.; Chen, X.; Lu, S.; Zhu, L.-M. Hollow Mesoporous Silica Nanoparticles Gated by Chitosan-Copper Sulfide Composites as Theranostic Agents for the Treatment of Breast Cancer. Acta Biomater. 2021, 126, 408–420. [Google Scholar] [CrossRef]
- Amatya, R.; Hwang, S.; Park, T.; Min, K.A.; Shin, M.C. In Vitro and In Vivo Evaluation of PEGylated Starch-Coated Iron Oxide Nanoparticles for Enhanced Photothermal Cancer Therapy. Pharmaceutics 2021, 13, 871. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhang, J.; Wu, Y.-P.; Yang, X.; Kuang, X.-P.; Li, W.-X.; Li, Y.-F.; He, R.-R.; Liu, M. Multifunctional HNT@Fe3O4@PPy@DOX Nanoplatform for Effective Chemo-Photothermal Combination Therapy of Breast Cancer with MR Imaging. ACS Biomater. Sci. Eng. 2020, 6, 3361–3374. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, D.A.; Fernandes, D.D.; Malik, A.; Gomes, G.-N.W.; Appak-Baskoy, S.; Berndl, E.; Gradinaru, C.C.; Kolios, M.C. Multifunctional Nanoparticles as Theranostic Agents for Therapy and Imaging of Breast Cancer. J. Photochem. Photobiol. B Biol. 2021, 218, 112110. [Google Scholar] [CrossRef]
- Novickij, V.; Malyško, V.; Želvys, A.; Balevičiūtė, A.; Zinkevičienė, A.; Novickij, J.; Girkontaitė, I. Electrochemotherapy Using Doxorubicin and Nanosecond Electric Field Pulses: A Pilot in vivo Study. Molecules 2020, 25, 4601. [Google Scholar] [CrossRef]
- Jaroszeski, M.J.; Gilbert, R.A.; Heller, R. In Vivo Antitumor Effects of Electrochemotherapy in a Hepatoma Model. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1997, 1334, 15–18. [Google Scholar] [CrossRef]
- Ho, C.H.; Triolo, R.J.; Elias, A.L.; Kilgore, K.L.; DiMarco, A.F.; Bogie, K.; Vette, A.H.; Audu, M.L.; Kobetic, R.; Chang, S.R.; et al. Functional Electrical Stimulation and Spinal Cord Injury. Phys. Med. Rehabil. Clin. N. Am. 2014, 25, 631–654. [Google Scholar] [CrossRef] [Green Version]
- Nuccitelli, R. A Role for Endogenous Electric Fields in Wound Healing. Curr. Top. Dev. Biol. 2003, 58, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, I.A.; Zaytsev, K.I.; Cherkasova, O.P.; Tuchind, V.V.; Yurchenko, S.O.; Yakovlev, E.V.; Troshina, A.V.; Korsakova, S.A.; Aliev, I.N.; Andronik, M. Colloidal Suspensions in External Rotating Electric Field: Experimental Studies and Prospective Applications in Physics, Material Science, and Biomedicine. In Saratov Fall Meeting 2017: Optical Technologies in Biophysics and Medicine XIX; Tuchin, V.V., Postnov, D.E., Genina, E.A., Derbov, V.L., Eds.; SPIE: Bellingham, WA, USA, 2018; p. 124. [Google Scholar] [CrossRef]
- International Commission on Non-Ionizing Radiation Protection. Guidelines for limiting exposure to time-varying electric and magnetic fields (1 Hz TO 100 KHz). Health Phys. 2010, 99, 818–836. [Google Scholar] [CrossRef]
- Adair, R.K. Biological Effects on the Cellular Level of Electric Field Pulses. Health Phys. 1991, 61, 395–399. [Google Scholar] [CrossRef]
- Leroux, J.-C. Editorial: Drug Delivery: Too Much Complexity, Not Enough Reproducibility? Angew. Chemie Int. Ed. 2017, 56, 15170–15171. [Google Scholar] [CrossRef] [Green Version]
- Cui, P.; Wang, S. Application of Microfluidic Chip Technology in Pharmaceutical Analysis: A Review. J. Pharm. Anal. 2019, 9, 238–247. [Google Scholar] [CrossRef]
- Shepherd, S.J.; Issadore, D.; Mitchell, M.J. Microfluidic Formulation of Nanoparticles for Biomedical Applications. Biomaterials 2021, 274, 120826. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. A Review of Clinical Translation of Inorganic Nanoparticles. AAPS J. 2015, 17, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the Clinic. Bioeng. Transl. Med. 2016, 1, 10–29. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the Clinic: An Update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Liu, L.; Morin, E.E.; Liu, M.; Schwendeman, A. Survey of Clinical Translation of Cancer Nanomedicines—Lessons Learned from Successes and Failures. Acc. Chem. Res. 2019, 52, 2445–2461. [Google Scholar] [CrossRef]
- Hassan, S.; Prakash, G.; Bal Ozturk, A.; Saghazadeh, S.; Farhan Sohail, M.; Seo, J.; Remzi Dokmeci, M.; Zhang, Y.S.; Khademhosseini, A. Evolution and Clinical Translation of Drug Delivery Nanomaterials. Nano Today 2017, 15, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and Challenges towards Targeted Delivery of Cancer Therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [Green Version]
- Pillai, O.; Dhanikula, A.B.; Panchagnula, R. Drug Delivery: An Odyssey of 100 Years. Curr. Opin. Chem. Biol. 2001, 5, 439–446. [Google Scholar] [CrossRef]
- Park, K. Nanotechnology: What It Can Do for Drug Delivery. J. Control. Release 2007, 120, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, A.; Elshaer, A.; Sareh, P.; Elsayed, M.; Hassanin, H. Additive Manufacturing Technologies for Drug Delivery Applications. Int. J. Pharm. 2020, 580, 119245. [Google Scholar] [CrossRef]
- Lecocq, Q.; De Vlaeminck, Y.; Hanssens, H.; D’Huyvetter, M.; Raes, G.; Goyvaerts, C.; Keyaerts, M.; Devoogdt, N.; Breckpot, K. Theranostics in Immuno-Oncology Using Nanobody Derivatives. Theranostics 2019, 9, 7772–7791. [Google Scholar] [CrossRef] [PubMed]
- Langbein, T.; Weber, W.A.; Eiber, M. Future of Theranostics: An Outlook on Precision Oncology in Nuclear Medicine. J. Nucl. Med. 2019, 60, 13S–19S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyner, M.J.; Paneth, N. Promises, Promises, and Precision Medicine. J. Clin. Investig. 2019, 129, 946–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Yang, H.; Zhou, Y.-F.; Hu, B. Dual and Multi-Targeted Nanoparticles for Site-Specific Brain Drug Delivery. J. Control. Release 2020, 317, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Villalba, N.; Baby, S.; Cha, B.J.; Yuan, S.Y. Site-Specific Opening of the Blood-Brain Barrier by Extracellular Histones. J. Neuroinflamm. 2020, 17, 281. [Google Scholar] [CrossRef]
- Raut, S.; Mooberry, L.; Sabnis, N.; Garud, A.; Dossou, A.S.; Lacko, A. Reconstituted HDL: Drug Delivery Platform for Overcoming Biological Barriers to Cancer Therapy. Front. Pharmacol. 2018, 9, 1154. [Google Scholar] [CrossRef]
- Filippi, M.; Rocca, M.A.; Ciccarelli, O.; De Stefano, N.; Evangelou, N.; Kappos, L.; Rovira, A.; Sastre-Garriga, J.; Tintorè, M.; Frederiksen, J.L.; et al. MRI Criteria for the Diagnosis of Multiple Sclerosis: MAGNIMS Consensus Guidelines. Lancet Neurol. 2016, 15, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Gurrera, R.J.; Caroff, S.N.; Cohen, A.; Carroll, B.T.; DeRoos, F.; Francis, A.; Frucht, S.; Gupta, S.; Levenson, J.L.; Mahmood, A.; et al. An International Consensus Study of Neuroleptic Malignant Syndrome Diagnostic Criteria Using the Delphi Method. J. Clin. Psychiatry 2011, 72, 1222–1228. [Google Scholar] [CrossRef]
- Dougan, M.; Dougan, S.K. Targeting Immunotherapy to the Tumor Microenvironment. J. Cell. Biochem. 2017, 118, 3049–3054. [Google Scholar] [CrossRef]
- Deckers, R.; Rome, C.; Moonen, C.T.W. The Role of Ultrasound and Magnetic Resonance in Local Drug Delivery. J. Magn. Reson. Imaging 2008, 27, 400–409. [Google Scholar] [CrossRef]
- Liang, J.; Peng, X.; Zhou, X.; Zou, J.; Cheng, L. Emerging Applications of Drug Delivery Systems in Oral Infectious Diseases Prevention and Treatment. Molecules 2020, 25, 516. [Google Scholar] [CrossRef] [Green Version]
- Abdou, P.; Wang, Z.; Chen, Q.; Chan, A.; Zhou, D.R.; Gunadhi, V.; Gu, Z. Advances in Engineering Local Drug Delivery Systems for Cancer Immunotherapy. WIREs Nanomed. Nanobiotechnol. 2020, 12, e1632. [Google Scholar] [CrossRef]
- Azzi, J.; Jraij, A.; Auezova, L.; Fourmentin, S.; Greige-Gerges, H. Novel Findings for Quercetin Encapsulation and Preservation with Cyclodextrins, Liposomes, and Drug-in-Cyclodextrin-in-Liposomes. Food Hydrocoll. 2018, 81, 328–340. [Google Scholar] [CrossRef]
- Wang, C.; Chen, S.; Wang, Y.; Liu, X.; Hu, F.; Sun, J.; Yuan, H. Lipase-Triggered Water-Responsive “Pandora’s Box” for Cancer Therapy: Toward Induced Neighboring Effect and Enhanced Drug Penetration. Adv. Mater. 2018, 30, 1706407. [Google Scholar] [CrossRef]
- Sousa, F.; Cruz, A.; Pinto, I.M.; Sarmento, B. Nanoparticles Provide Long-Term Stability of Bevacizumab Preserving Its Antiangiogenic Activity. Acta Biomater. 2018, 78, 285–295. [Google Scholar] [CrossRef]
- Kudryavtseva, V.; Boi, S.; Read, J.; Gould, D.; Szewczyk, P.K.; Stachewicz, U.; Kiryukhin, M.V.; Pastorino, L.; Sukhorukov, G.B. Micro-Sized “Pelmeni”—A Universal Microencapsulation Approach Overview. Mater. Des. 2021, 202, 109527. [Google Scholar] [CrossRef]
- Linnik, D.S.; Tarakanchikova, Y.V.; Zyuzin, M.V.; Lepik, K.V.; Aerts, J.L.; Sukhorukov, G.; Timin, A.S. Layer-by-Layer Technique as a Versatile Tool for Gene Delivery Applications. Expert Opin. Drug Deliv. 2021, 1–19. [Google Scholar] [CrossRef]
- Song, F.; Gao, H.; Li, D.; Petrov, A.V.; Petrov, V.V.; Wen, D.; Sukhorukov, G.B. Low Intensity Focused Ultrasound Responsive Microcapsules for Non-Ablative Ultrafast Intracellular Release of Small Molecules. J. Mater. Chem. B 2021, 9, 2384–2393. [Google Scholar] [CrossRef]
- Mayorova, O.A.; Sindeeva, O.A.; Lomova, M.V.; Gusliakov, O.I.; Tarakanchikova, Y.V.; Tyutyaev, E.V.; Pinyaev, S.I.; Kulikov, O.A.; German, S.V.; Pyataev, N.A.; et al. Endovascular Addressing Improves the Effectiveness of Magnetic Targeting of Drug Carrier. Comparison with the Conventional Administration Method. Nanomed. Nanotechnol. Biol. Med. 2020, 28, 102184. [Google Scholar] [CrossRef]
- Maleki, R.; Afrouzi, H.H.; Hosseini, M.; Toghraie, D.; Rostami, S. Molecular Dynamics Simulation of Doxorubicin Loading with N-Isopropyl Acrylamide Carbon Nanotube in a Drug Delivery System. Comput. Methods Programs Biomed. 2020, 184, 105303. [Google Scholar] [CrossRef]
- Mollazadeh, S.; Sahebkar, A.; Shahlaei, M.; Moradi, S. Nano Drug Delivery Systems: Molecular Dynamic Simulation. J. Mol. Liq. 2021, 332, 115823. [Google Scholar] [CrossRef]
- Hashida, M. Role of Pharmacokinetic Consideration for the Development of Drug Delivery Systems: A Historical Overview. Adv. Drug Deliv. Rev. 2020, 157, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.M.; Collier, A.D.; Meshalkina, D.A.; Kysil, E.V.; Khatsko, S.L.; Kolesnikova, T.; Morzherin, Y.Y.; Warnick, J.E.; Kalueff, A.V.; Echevarria, D.J. Zebrafish Models in Neuropsychopharmacology and CNS Drug Discovery. Br. J. Pharmacol. 2017, 174, 1925–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löscher, W. Animal Models of Seizures and Epilepsy: Past, Present, and Future Role for the Discovery of Antiseizure Drugs. Neurochem. Res. 2017, 42, 1873–1888. [Google Scholar] [CrossRef]
- Edington, C.D.; Chen, W.L.K.; Geishecker, E.; Kassis, T.; Soenksen, L.R.; Bhushan, B.M.; Freake, D.; Kirschner, J.; Maass, C.; Tsamandouras, N.; et al. Interconnected Microphysiological Systems for Quantitative Biology and Pharmacology Studies. Sci. Rep. 2018, 8, 4530. [Google Scholar] [CrossRef] [Green Version]
- Pashayan, N.; Morris, S.; Gilbert, F.J.; Pharoah, P.D.P. Cost-Effectiveness and Benefit-to-Harm Ratio of Risk-Stratified Screening for Breast Cancer. JAMA Oncol. 2018, 4, 1504. [Google Scholar] [CrossRef] [Green Version]
- Darwich, A.S.; Polasek, T.M.; Aronson, J.K.; Ogungbenro, K.; Wright, D.F.B.; Achour, B.; Reny, J.-L.; Daali, Y.; Eiermann, B.; Cook, J.; et al. Model-Informed Precision Dosing: Background, Requirements, Validation, Implementation, and Forward Trajectory of Individualizing Drug Therapy. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef]
- Singh, S.K.; Singh, S.; Lillard, J.W., Jr.; Singh, R. Drug Delivery Approaches for Breast Cancer. Int. J. Nanomed. 2017, 12, 6205–6218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, S.; DeGiovanni, P.; Piel, B.; Rai, P. Cancer Nanomedicine: A Review of Recent Success in Drug Delivery. Clin. Transl. Med. 2017, 6, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Faheem, A.M.; Abdelkader, D.H. Novel Drug Delivery Systems. In Engineering Drug Delivery Systems; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–16. [Google Scholar] [CrossRef]
- Zavvar, T.; Babaei, M.; Abnous, K.; Taghdisi, S.M.; Nekooei, S.; Ramezani, M.; Alibolandi, M. Synthesis of Multimodal Polymersomes for Targeted Drug Delivery and MR/Fluorescence Imaging in Metastatic Breast Cancer Model. Int. J. Pharm. 2020, 578, 119091. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Drug | Drug | Core/Shell | Targeting/Release | In Vivo Evaluation | Ref |
|---|---|---|---|---|---|
| Anticancer | Paclitaxel, Doxorubicin (DOX) | -/Exosomes released by macrophages | Exosome fusion with target cells occurs more efficiently under acidic conditions, implying that exosomes may be taken up preferentially by tumors/- | Carriers with loaded drugs demonstrated superior inhibition of pulmonary metastases growth in the Lewis lung cancer (LLC) mouse model. Three mechanisms have a significant impact on carriers with loaded drug anticancer activity, i.e., (1) preferential accumulation in cancer cells, (2) efficient delivery of incorporated cargo into target cancer cells, and (3) bypassing Pgp-mediated drug efflux in resistant cancer cells. | [34] |
| DOX | -/Exosomes released by immature dendritic cells (imDCs) | By exosomal membrane protein/- | DOX delivered by the carrier slows tumor growth four-fold without overt toxicity. | [35] | |
| DOX | -/A33 antibody-functionalized exosomes released by LIM1215 cells | By exosomal membrane protein/- | The mean tumor treated by exosomes with A33 antibodies was 3.04- and 2.90-fold lower than in the control DOX and exosomes without antibodies groups on day 16. | [36] | |
| Glycyrrhizin and DOX | -/Alginate nanogel particles | Specific binding of glycyrrhizin and glycyrrhetinic acid GL with cellular membranes of hepatocytes (liver targeting)/- | After 14 days of the experiment, the particles with glycyrrhizin and DOX could inhibit the growth activity of tumor cells and promote apoptotic of tumor cells to enhance antitumor effects. | [37] | |
| DOX loaded to Tween 80 micelles | Tween 80 micelles/Silica nanoparticles | -/- | On the 18th day, the tumor size of the SiNPs/DOX group was two-fold smaller than that of the free DOX group and four-fold smaller than that of the PBS group. | [38] | |
| DOX | Mesoporous silica nanoparticles/Peptide-BSA-LA | -/In the presence of metalloproteinases | After 20 days of the experiment, the control tumor was 6.7 times larger than the encapsulated drug-treated tumor. | [39] | |
| DOX and Mn2+-chelated chlorin e6 (Ce6(Mn)) | CaCO3/PEG | -/Highly sensitive to reduced pH | Tumor growth on mice treated by carriers with the loaded drug was greatly inhibited after combined photodynamic & chemotherapy, demonstrating the superior synergistic antitumor effect by those two kinds of therapies. | [40] | |
| DOX | CaCO3/poly(acrylic acid) | -/pH-sensitive | Carriers with the loaded drug showed significantly higher tumor suppression than free drugs due to the enhanced permeability and retention effect and the pH responsiveness of NPs. | [41] | |
| BACE1 siRNA | -/Rabies viral glycoprotein (RVG) exosomes | Targeting was achieved by engineering the dendritic cells to express Lamp2b/- | Three days after administration and a significant protein knockdown in both siRNA-RVG-9R-treated and siRNA-RVG exosome-treated mice was observed, resulting from a significant decrease in BACE1 mRNA levels. | [42] | |
| KRASG12D siRNA | -/Exosomes derived from normal fibroblast-like mesenchymal cells | -/- | Diminished pancreas desmoplasia, enhanced cancer cell apoptosis, suppressed cancer cell proliferation, reduced phospho-ERK, phospho-AKT, and Kras levels are noted in KTC tumors, as well as diminished oncogenic KrasG12D expression with iExosomes treatment. | [43] | |
| Nucleic acid | Anti-EGFR siRNA | rPAA-Chol polymer/Cationic lipid | -/- | All mice treated with siRNA formulations did not show significantly increased IFN-α and IL-6 levels compared with the 5% glucose group. These results demonstrated that LP/siRNA NPs can silence specific gene without arising innate immune responses in vivo. Carriers promising delivery systems for siRNA application in cancer treatment. | [44] |
| Let-7 | -/Exosomes released by HEK293 cells | GE11 at membrane/- | Exosomes delivered let-7a potently inhibited the expression of HMGA2 mRNA in A549 lung adenocarcinoma cells. Exosomes delivered let-7a inhibits tumor development via previously unidentified or uncharacterized genes in HCC70 breast cancer cells. | [45] | |
| Fluorescently labeled FAM-siRNA | Calcium phosphate/PEGylated carboxymethyl chitosan | -/pH-depending release | On day 14, the tumor volume in mice treated with the sihTERT nanoparticles was around 43.5% of the average volume of the PBS group. This indicated that sihTERT, the delivery of which was mediated by NPPEG-CMCS/CaP, was responsible for tumor growth suppression. | [46] | |
| Cas9/sgRNA | -/Exosomes released by SKOV-3 cells | -/- | Taken together, CRISPR/Cas9-loaded exosomes administered intravenously or intratumorally could deliver PARP-1 to tumor sites, causing anticancer effects. The tumor volume decreases 2.7 times after 20 days. | [47] | |
| PHD2 | -/Poly[DMAEMA-b-(BMA-co-PAA-co-DMAEMA)] | -/pH-depending release | PHD2-NPs increased both the number and size of vessels within the scaffolds. PHD2-NPs increased the vascular volume by 300% and increased the mean vascular thickness by 137%. | [48] | |
| Anti-luciferase gene | -/PEG-CPB-PEI (PCPP) | -/Phenylboronic acid is targeting the SA-terminated sugar chains on cancer cells | In vivo studies demonstrated that PBA-based nanoparticles effectively accumulated in tumors and inhibited tumor growth and metastasis in the 4T1 orthotopic mammary tumor model after intravenous administration. | [49] | |
| RFP siRNA | -/Hyaluronic acid-graft-poly(dimethylaminoethyl methacrylate) (HPD) conjugate | -/Biodegradability of HA | The tumor site after 5 days was significantly reduced for the mice treated with siRNA carriers. The high tumor targetability of siRNA carriers resulted in the suppression of tumor growth, owing to its cytotoxicity against cancer cells. | [50] | |
| FAM-siNC, cy3-siNC, siLuc, and siBcl2 | CaP/Disulfide cross-linked HA | -/Biodegradability of HA | The mass of tumors treated with loaded CaP/HA containers was only 25% of the tumor mass in the PBS group, and the tumor inhibition rate was about 80%. | [51] | |
| Catalase BDNF | Exosomes | -/- -/radiolabeling | Exosomes loaded with catalase efficiently accumulate in neurons and microglial cells in the brain and produce a potent neuroprotective effect. Exosomes can penetrate the vascular barrier but can make no conclusions regarding their ability to penetrate the blood–CSF barrier. | [52,53] | |
| Insulin | Polymethacrylic acid–polyethylene glycol–chitosan-based hydrogel microparticles | -/pH-sensitive | Within 2 h of receiving the encapsulated dose, a lowering of blood glucose level was observed in the diabetic animals. | [54] | |
| Growth-factor | Immunoactive TLR-3 poly(I:C) | PLGA/Thiolated silica | Toll-like receptor 3/Immunostimulation effect | The therapeutic efficacy of different formulations was evaluated in the arthritic mouse. An intravenous injection of nanoparticles into mice led to a particle accumulation mainly in the lung and in the liver. The expression of IFN-α/β, TNF-α, IL-6, and IP-10 in hepatocytes, NPCs, and LSECs was significantly increased. | [55] |
| Other | Dexamethasone sodium phosphate | Calcium phosphate gel nanoparticles/Sialic acid-modified PEGylated lipid bilayer | E-selectin-receptors-mediated endocytosis/pH-sensitive treatment of acute kidney injury | The capsulated drug significantly improved the renal function, decreased the level of pro-inflammatory factors, and adjusted the oxidative stress factors and apoptotic proteins compared to free Dsp solution in pharmacodynamic studies. Moreover, few negative effects on blood glucose and bone mineral density were observed. | [56] |
| Adapalene | PLA-PEG NP blended with low molecular weight PLA or PCL; PLGA NP/- | Receptor β (RARβ)/- | Treatment with adapalene-loaded nanoparticles was able to elicit a biological response as quickly as 4 h post injection in reporter mice, and these effects were sustained for a minimum of 24 h. | [57] |
| Exposure Limits | Correction Factor, CA | ||
|---|---|---|---|
| Exposure Duration, t | Definition | Wavelength, λ | Definition |
| 1 ns–100 ns | 200∙CA, J∙m−2 | 400–700 nm | 1.0 |
| 100 ns–10 s | 11∙CA∙t0.25, kJ∙m−2 | 700–1050 nm | 100.002(λ/1–700) |
| 10 s–30 ks | 2.0∙CA, kW∙m−2 | 1050–1400 nm | 5.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voronin, D.V.; Abalymov, A.A.; Svenskaya, Y.I.; Lomova, M.V. Key Points in Remote-Controlled Drug Delivery: From the Carrier Design to Clinical Trials. Int. J. Mol. Sci. 2021, 22, 9149. https://doi.org/10.3390/ijms22179149
Voronin DV, Abalymov AA, Svenskaya YI, Lomova MV. Key Points in Remote-Controlled Drug Delivery: From the Carrier Design to Clinical Trials. International Journal of Molecular Sciences. 2021; 22(17):9149. https://doi.org/10.3390/ijms22179149
Chicago/Turabian StyleVoronin, Denis V., Anatolii A. Abalymov, Yulia I. Svenskaya, and Maria V. Lomova. 2021. "Key Points in Remote-Controlled Drug Delivery: From the Carrier Design to Clinical Trials" International Journal of Molecular Sciences 22, no. 17: 9149. https://doi.org/10.3390/ijms22179149
APA StyleVoronin, D. V., Abalymov, A. A., Svenskaya, Y. I., & Lomova, M. V. (2021). Key Points in Remote-Controlled Drug Delivery: From the Carrier Design to Clinical Trials. International Journal of Molecular Sciences, 22(17), 9149. https://doi.org/10.3390/ijms22179149