
Sclerostin Depletion Induces Inflammation in the Bone Marrow of Mice

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
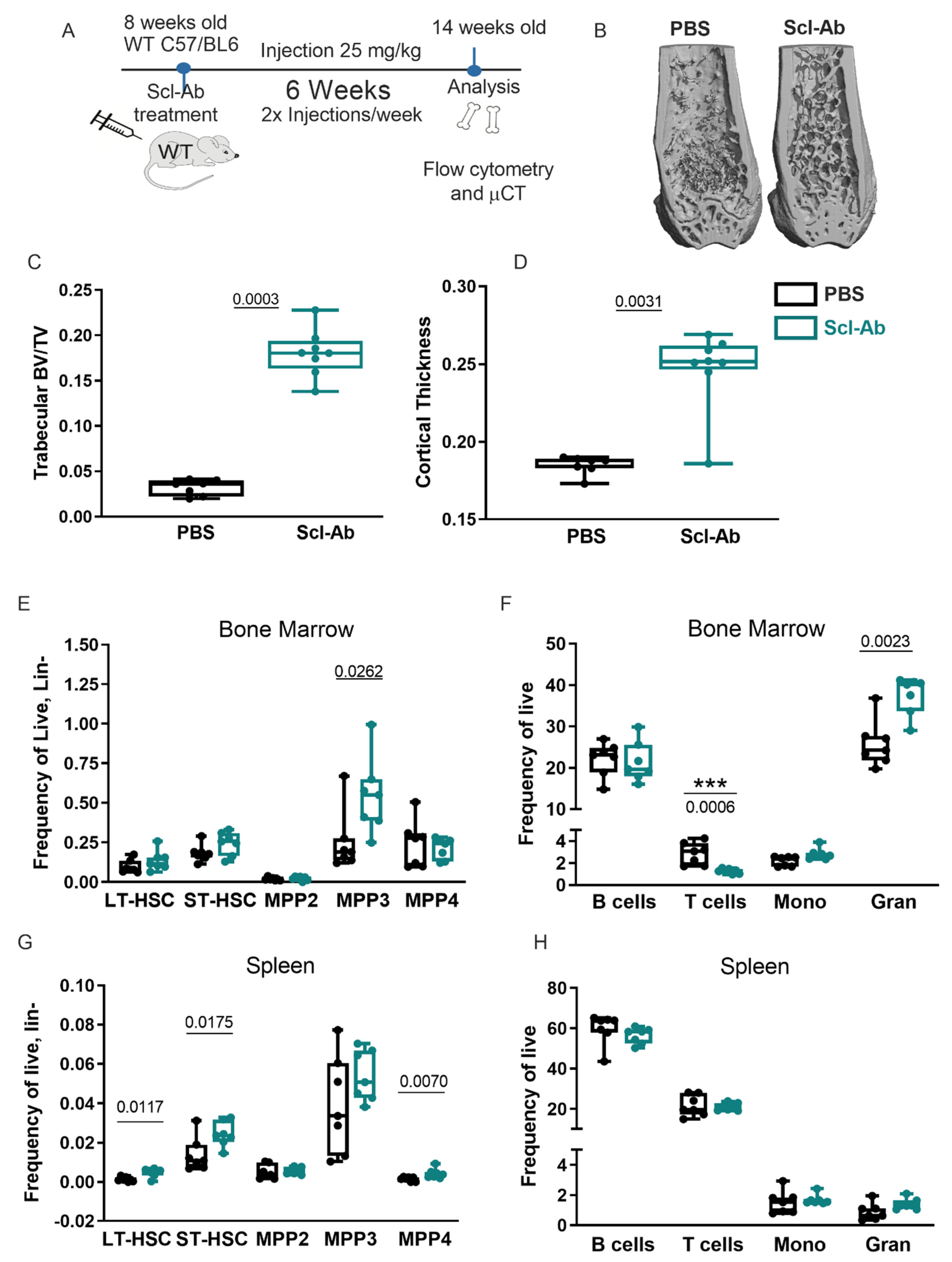
2.1. Sclerostin-Depleting Antibody Treatment Changes Hematopoietic Differentiation
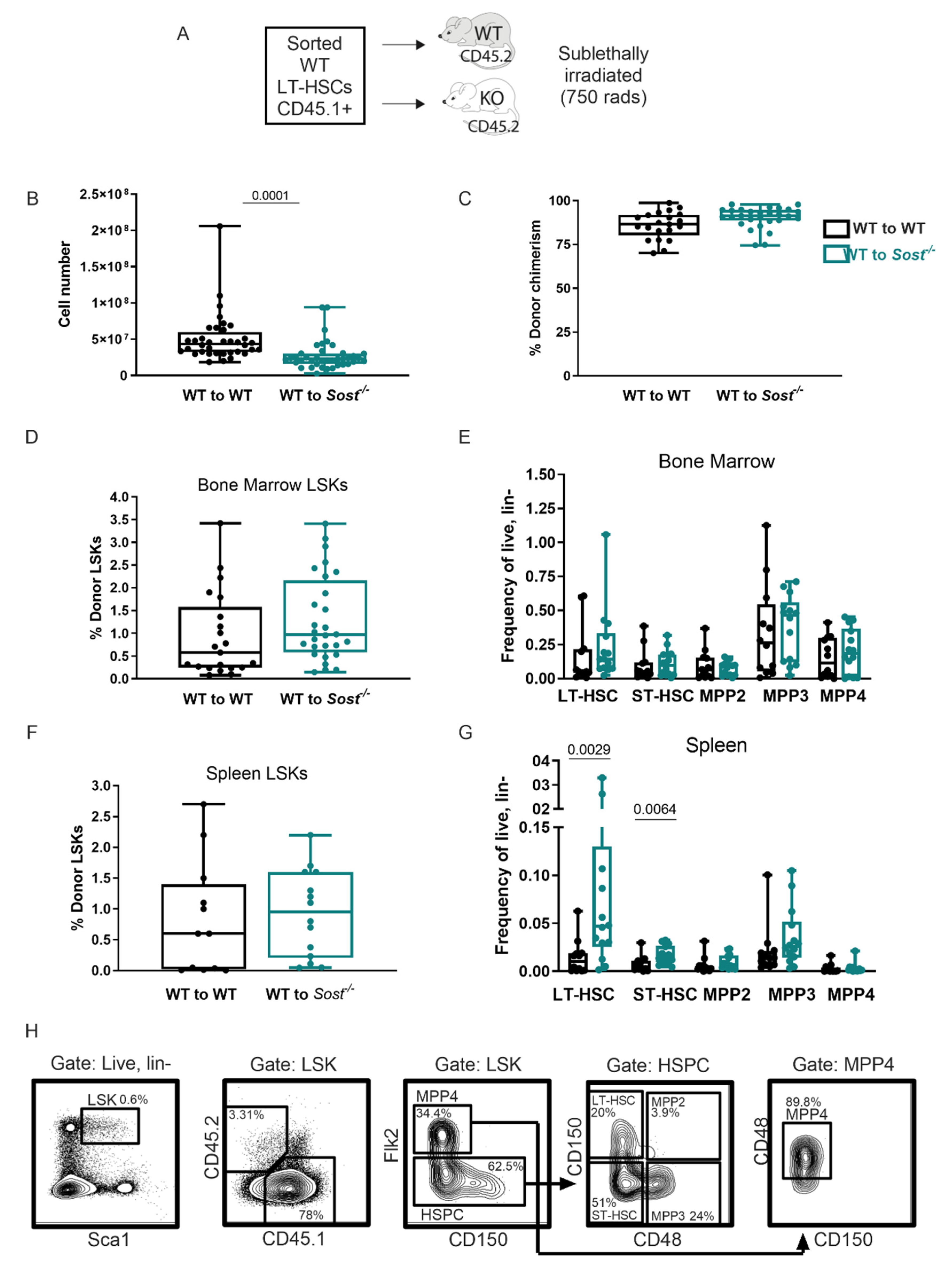
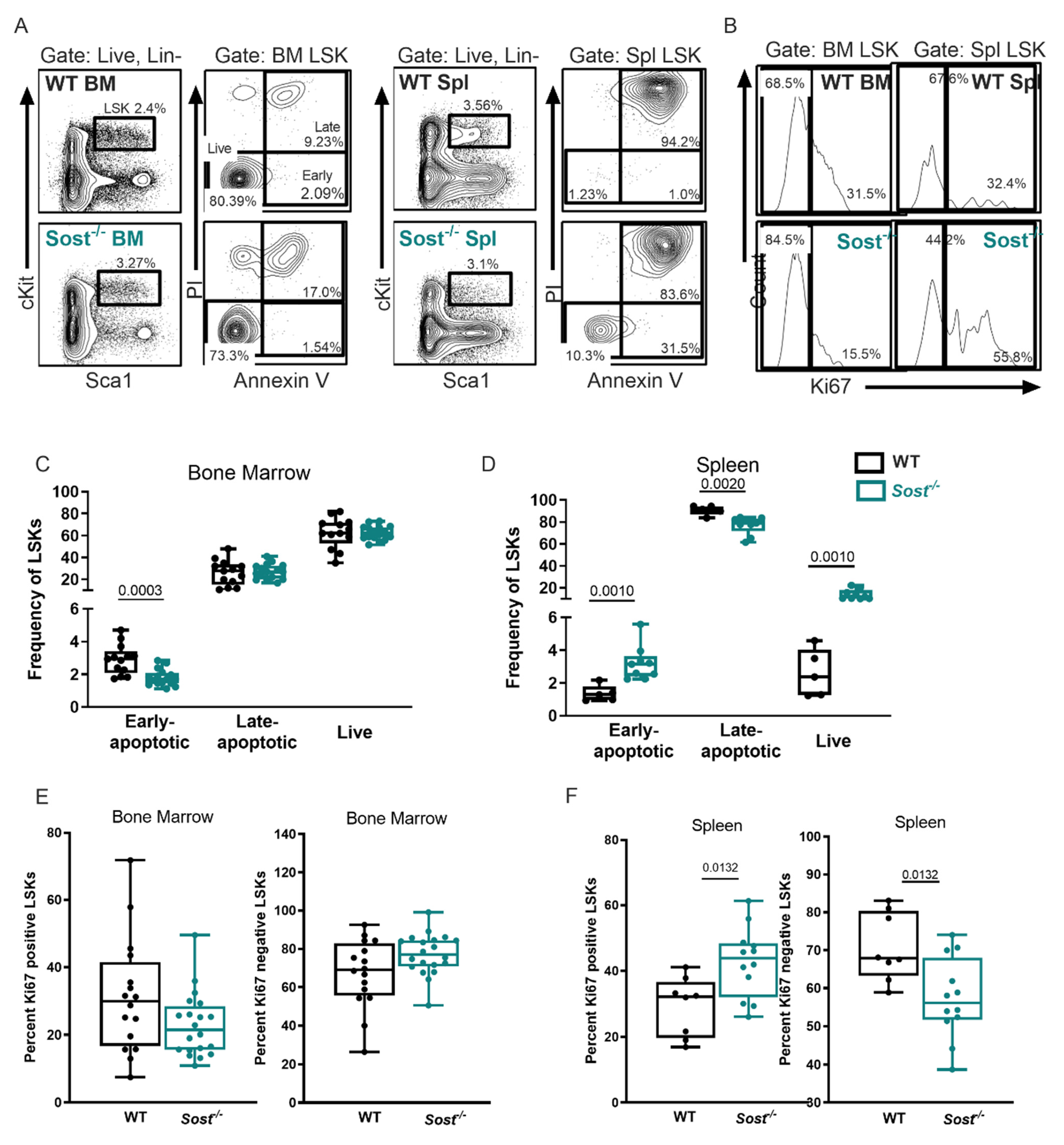
2.2. Lack of Sclerostin in the Bone Does Not Affect Hematopoietic Progenitor Distributions in the Bone Marrow
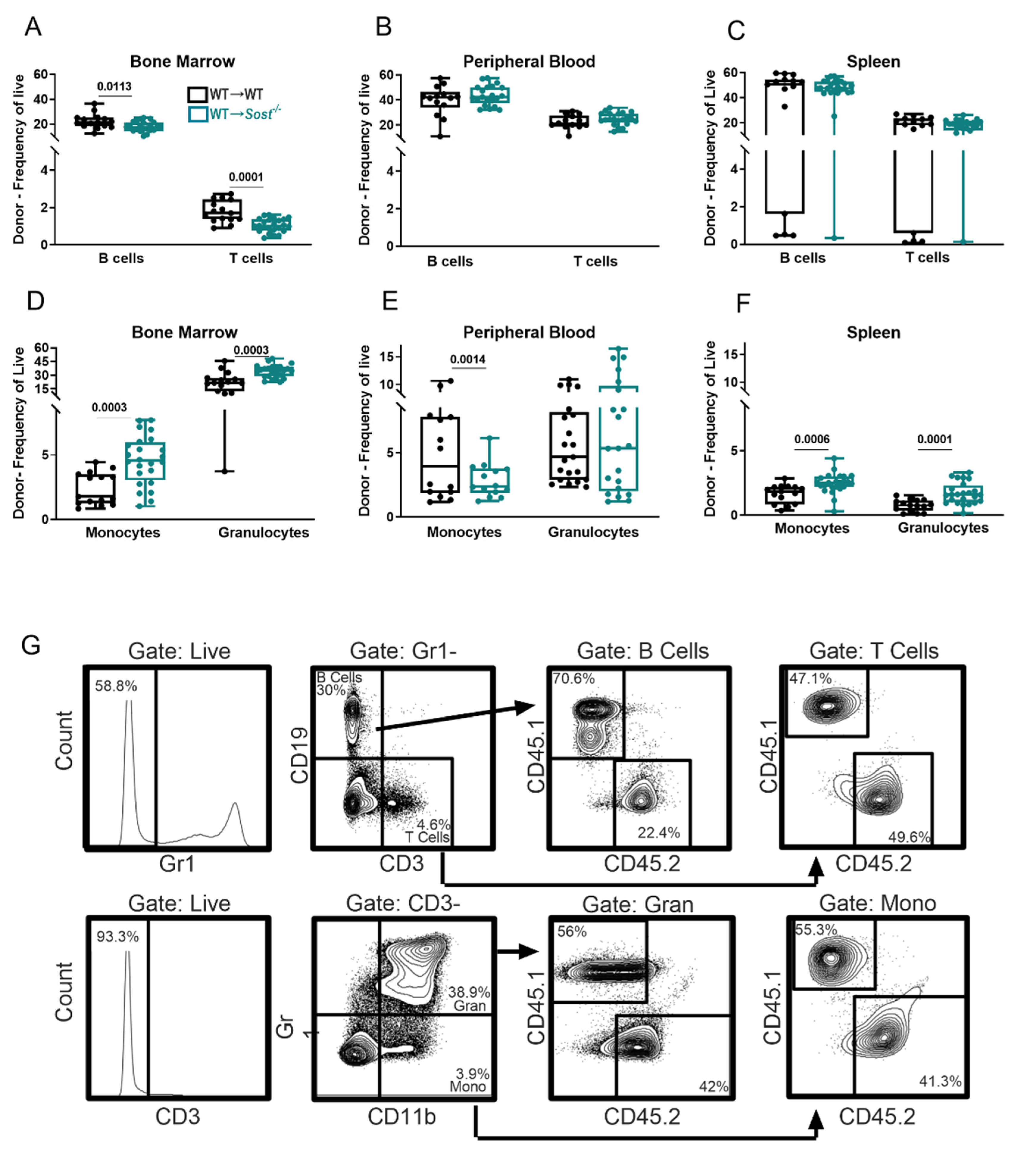
2.3. Lack of Sclerostin in the Bone Microenvironment Results in a Myeloid Bias
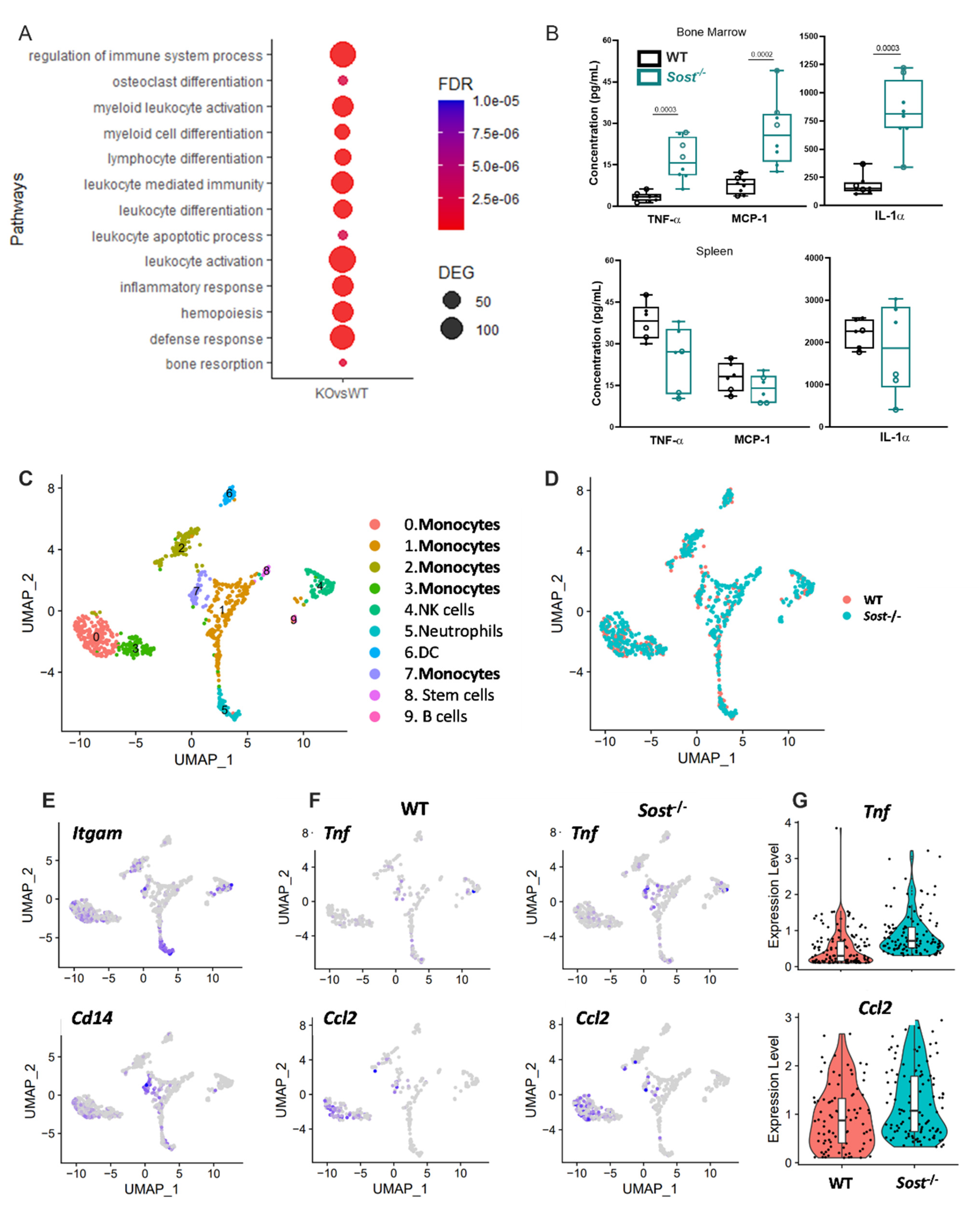
2.4. Sost−/− Mice Express High Levels of Inflammatory Cytokines in the Bone Marrow
2.5. Bone Marrow Monocytes in Sost−/− Mice Exhibit Upregulated Tnf and Ccl2 Gene Expression
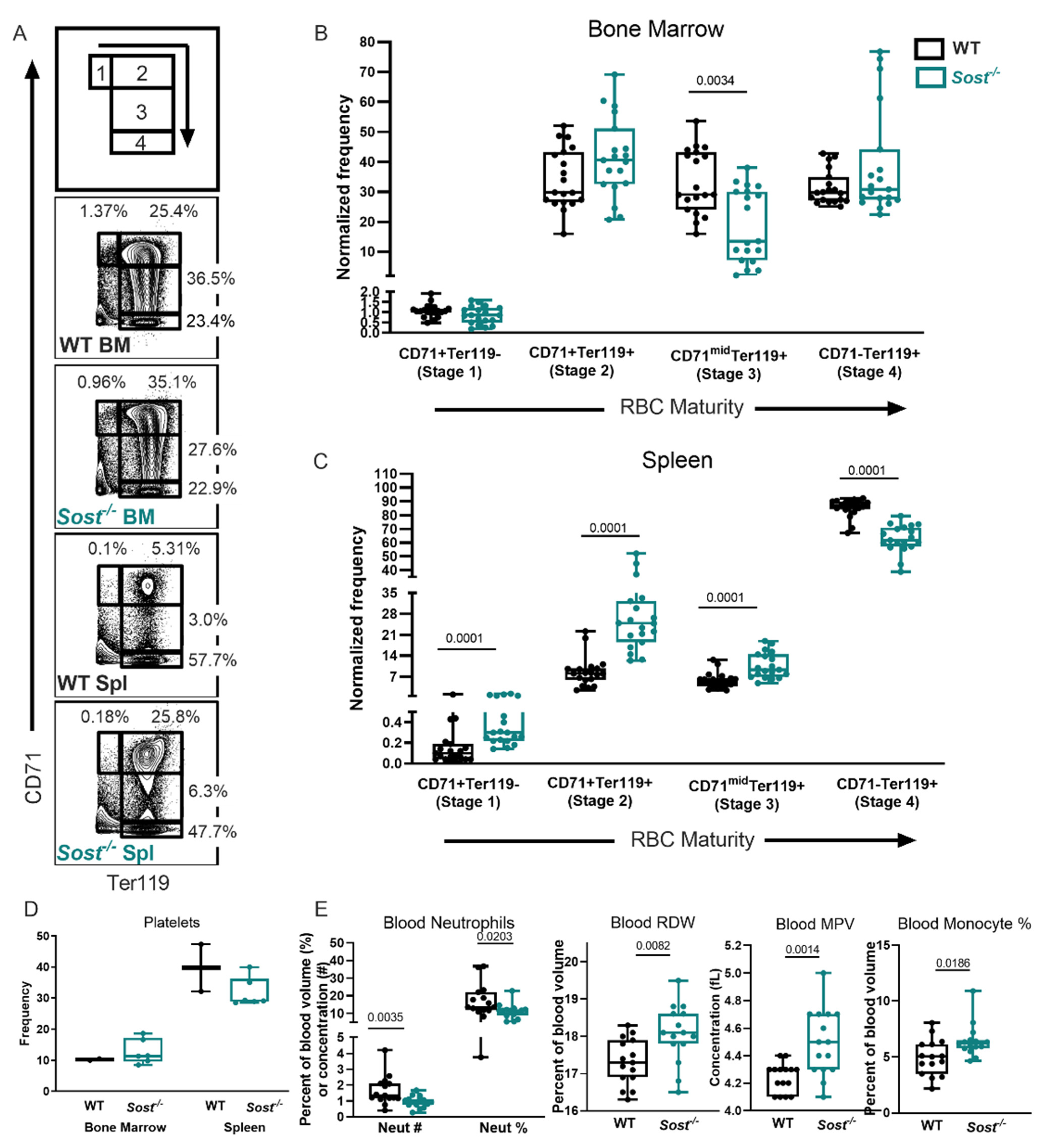
2.6. Evidence of Extramedullary Hematopoiesis in Sost−/− Mice
2.7. Erythrocyte Development Is Altered in Sost−/− Mice
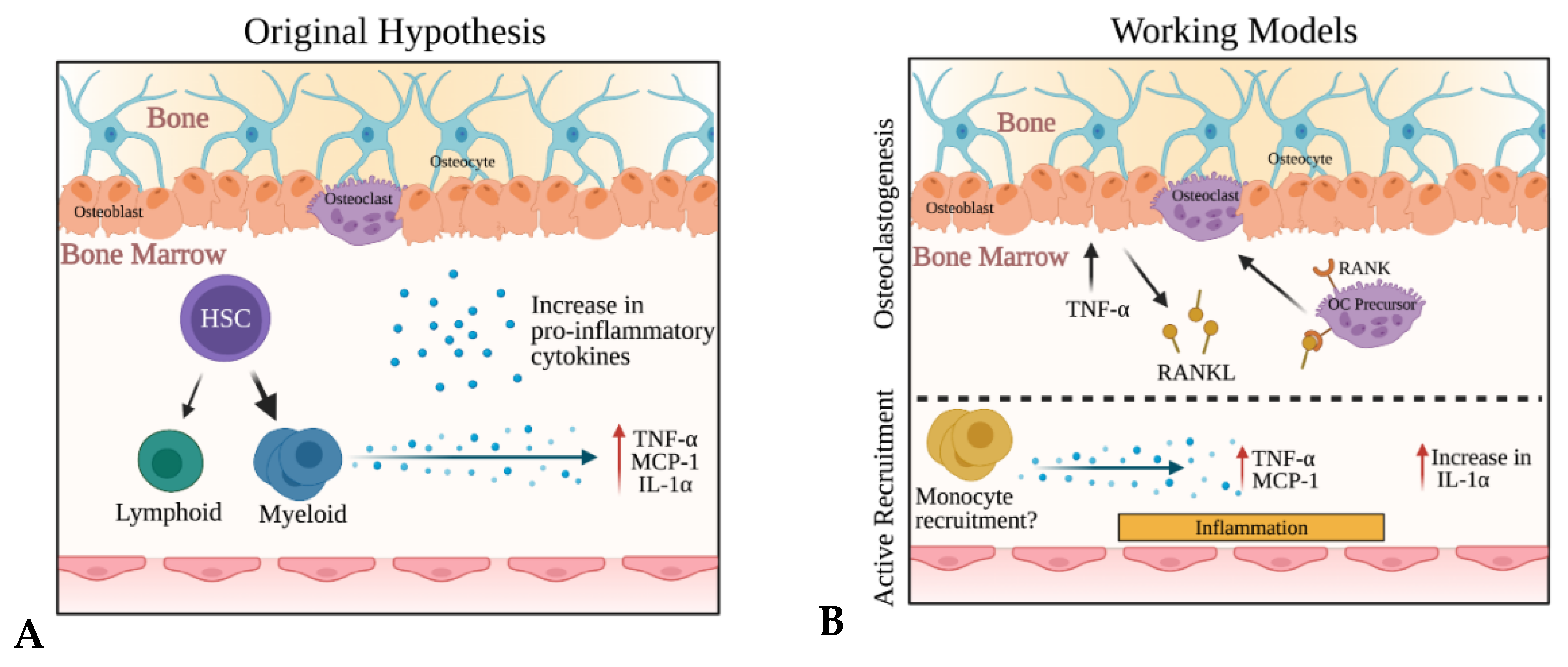
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Balemans, W.; Ebeling, M.; Patel, N.; van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; van den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef]
- Balemans, W.; Patel, N.; Ebeling, M.; van Hul, E.; Wuyts, W.; Lacza, C.; Dioszegi, M.; Dikkers, F.G.; Hildering, P.; Willems, P.J.; et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002, 39, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Piters, E.; Halleux, C.; Hu, S.; Kramer, I.; Morvan, F.; Bouwmeester, T.; Schirle, M.; Bueno-Lozano, M.; Fuentes, F.J.; et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 2011, 286, 19489–19500. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Delgado-Calle, J.; Condon, K.W.; Maycas, M.; Zhang, H.; Carlesso, N.; Taketo, M.M.; Burr, D.B.; Plotkin, L.I.; Bellido, T. Osteocytes mediate the anabolic actions of canonical Wnt/beta-catenin signaling in bone. Proc. Natl. Acad. Sci. USA 2015, 112, E478–E486. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Romosozumab: First global approval. Drugs 2019, 79, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Donham, C.; Manilay, J.O. The effects of sclerostin on the immune system. Curr. Osteoporos. Rep. 2020, 18, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Donham, C.; Chicana, B.; Millan, A.J.; Manilay, J.O. Wnt antagonists in hematopoietic and immune cell fate: Implications for osteoporosis therapies. Curr. Osteoporos. Rep. 2019, 17, 49–58. [Google Scholar]
- Blank, U.; Karlsson, G.; Karlsson, S. Signaling pathways governing stem-cell fate. Blood 2008, 111, 492–503. [Google Scholar] [CrossRef]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef]
- Kirstetter, P.; Anderson, K.; Porse, B.T.; Jacobsen, S.E.; Nerlov, C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat. Immunol. 2006, 7, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Scheller, M.; Huelsken, J.; Rosenbauer, F.; Taketo, M.M.; Birchmeier, W.; Tenen, D.G.; Leutz, A. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat. Immunol. 2006, 7, 1037–1047. [Google Scholar] [CrossRef]
- Staal, F.J.; Chhatta, A.; Mikkers, H. Caught in a Wnt storm: Complexities of Wnt signaling in hematopoiesis. Exp. Hematol. 2016, 44, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Till, J.E.; Mc, C.E. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat. Res. 1961, 14, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.L.; Weissman, I.L. Flk-2 is a marker in hematopoietic stem cell differentiation: A simple method to isolate long-term stem cells. Proc. Natl. Acad. Sci. USA 2001, 98, 14541–14546. [Google Scholar] [CrossRef]
- Wagers, A.J. Stem cell grand SLAM. Cell 2005, 121, 967–970. [Google Scholar] [CrossRef][Green Version]
- Mann, M.; Mehta, A.; de Boer, C.G.; Kowalczyk, M.S.; Lee, K.; Haldeman, P.; Rogel, N.; Knecht, A.R.; Farouq, D.; Regev, A. Heterogeneous responses of hematopoietic stem cells to inflammatory stimuli are altered with age. Cell Rep. 2018, 25, 2992–3005. [Google Scholar] [CrossRef]
- Pietras, E.M.; Reynaud, D.; Kang, Y.A.; Carlin, D.; Calero-Nieto, F.J.; Leavitt, A.D.; Stuart, J.M.; Gottgens, B.; Passegue, E. Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell 2015, 17, 35–46. [Google Scholar] [CrossRef]
- Morrison, S.J.; Wandycz, A.M.; Akashi, K.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef]
- Sudo, K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef]
- De Haan, G.; van Zant, G. Dynamic changes in mouse hematopoietic stem cell numbers during aging. Blood 1999, 93, 3294–3301. [Google Scholar] [CrossRef]
- Denkinger, M.D.; Leins, H.; Schirmbeck, R.; Florian, M.C.; Geiger, H. HSC aging and senescent immune remodeling. Trends Immunol. 2015, 36, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar]
- Boulais, P.E.; Frenette, P.S. Making sense of hematopoietic stem cell niches. Blood 2015, 125, 2621–2629. [Google Scholar] [CrossRef]
- Czechowicz, A.; Kraft, D.; Weissman, I.L.; Bhattacharya, D. Efficient transplantation via antibody-based clearance of hematopoietic stem cell niches. Science 2007, 318, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.R.; Kottmann, A.H.; Kuroda, M.; Taniuchi, I.; Littman, D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393, 595–599. [Google Scholar] [CrossRef]
- Nagasawa, T.; Hirota, S.; Tachibana, K.; Takakura, N.; Nishikawa, S.; Kitamura, Y.; Yoshida, N.; Kikutani, H.; Kishimoto, T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996, 382, 635–638. [Google Scholar] [CrossRef]
- McDermott, D.H.; Gao, J.L.; Murphy, P.M. Chromothriptic cure of WHIM syndrome: Implications for bone marrow transplantation. Rare Dis. 2015, 3, e1073430. [Google Scholar] [CrossRef]
- Kimura, S.; Roberts, A.W.; Metcalf, D.; Alexander, W.S. Hematopoietic stem cell deficiencies in mice lacking c-Mpl, the receptor for thrombopoietin. Proc. Natl. Acad. Sci. USA 1998, 95, 1195–1200. [Google Scholar] [CrossRef] [PubMed]
- Kaushansky, K.; Broudy, V.C.; Lin, N.; Jorgensen, M.J.; McCarty, J.; Fox, N.; Zucker-Franklin, D.; Lofton-Day, C. Thrombopoietin, the Mp1 ligand, is essential for full megakaryocyte development. Proc. Natl. Acad. Sci. USA 1995, 92, 3234–3238. [Google Scholar] [CrossRef] [PubMed]
- Cain, C.J.; Rueda, R.; McLelland, B.; Collette, N.M.; Loots, G.G.; Manilay, J.O. Absence of sclerostin adversely affects B-cell survival. J. Bone Miner. Res. 2012, 27, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ominsky, M.S.; Niu, Q.T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef]
- Asada, N.; Takeishi, S.; Frenette, P.S. Complexity of bone marrow hematopoietic stem cell niche. Int. J. Hematol. 2017, 106, 45–54. [Google Scholar] [CrossRef]
- Szade, K.; Gulati, G.S.; Chan, C.K.F.; Kao, K.S.; Miyanishi, M.; Marjon, K.D.; Sinha, R.; George, B.M.; Chen, J.Y.; Weissman, I.L. Where hematopoietic stem cells live: The bone marrow niche. Antioxid. Redox Signal. 2018, 29, 191–204. [Google Scholar] [CrossRef]
- Cain, C.J.; Manilay, J.O. Hematopoietic stem cell fate decisions are regulated by Wnt antagonists: Comparisons and current controversies. Exp. Hematol. 2013, 41, 3–16. [Google Scholar] [CrossRef]
- Collette, N.M.; Genetos, D.C.; Murugesh, D.; Harland, R.M.; Loots, G.G. Genetic evidence that SOST inhibits WNT signaling in the limb. Dev. Biol. 2010, 342, 169–179. [Google Scholar] [CrossRef]
- Williams, D.K.; Parham, S.G.; Schryver, E.; Akel, N.S.; Shelton, R.S.; Webber, J.; Swain, F.L.; Schmidt, J.; Suva, L.J.; Gaddy, D. Sclerostin antibody treatment stimulates bone formation to normalize bone mass in male down syndrome mice. JBMR Plus 2018, 2, 47–54. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.-M.; Will, B. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Schepers, K.; Hsiao, E.C.; Garg, T.; Scott, M.J.; Passegue, E. Activated Gs signaling in osteoblastic cells alters the hematopoietic stem cell niche in mice. Blood 2012, 120, 3425–3435. [Google Scholar] [CrossRef] [PubMed]
- Oda, A.; Tezuka, T.; Ueno, Y.; Hosoda, S.; Amemiya, Y.; Notsu, C.; Kasahara, T.; Nishiyama, C.; Goitsuka, R. Niche-induced extramedullary hematopoiesis in the spleen is regulated by the transcription factor Tlx1. Sci. Rep. 2018, 8, 8308. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H. Homeostatic and pathogenic extramedullary hematopoiesis. J. Blood Med. 2010, 1, 13. [Google Scholar] [CrossRef]
- Zaretsky, A.G.; Engiles, J.B.; Hunter, C.A. Infection-induced changes in hematopoiesis. J. Immunol. 2014, 192, 27–33. [Google Scholar] [CrossRef]
- MacNamara, K.C.; Jones, M.; Martin, O.; Winslow, G.M. Transient activation of hematopoietic stem and progenitor cells by IFNγ during acute bacterial infection. PLoS ONE 2011, 6, e28669. [Google Scholar] [CrossRef]
- Price, E.A.; Schrier, S.L. Unexplained aspects of anemia of inflammation. Adv. Hematol. 2010, 2010, 508739. [Google Scholar] [CrossRef] [PubMed]
- Gravano, D.M.; Al-Kuhlani, M.; Davini, D.; Sanders, P.D.; Manilay, J.O.; Hoyer, K.K. CD8(+) T cells drive autoimmune hematopoietic stem cell dysfunction and bone marrow failure. J. Autoimmun. 2016, 75, 58–67. [Google Scholar] [CrossRef]
- Evans, T.C.; Jehle, D. The red blood cell distribution width. J. Emerg. Med. 1991, 9, 71–74. [Google Scholar] [CrossRef]
- Amgen Inc. Available online: https://www.evenityhcp.com/ (accessed on 10 November 2020).
- Amgen Inc. 2019. Available online: https://www.amgen.com/media/news-releases/2019/01/evenity-romosozumab-receives-approval-in-japan-for-the-treatment-of-osteoporosis-in-patients-at-high-risk-of-fracture/ (accessed on 10 November 2020).
- Fuentes, E.; Fuentes, M.; Alarcon, M.; Palomo, I. Immune system dysfunction in the elderly. An. Acad. Bras. Cienc. 2017, 89, 285–299. [Google Scholar] [CrossRef]
- Kaveh, S.; Hosseinifard, H.; Ghadimi, N.; Vojdanian, M.; Aryankhesal, A. Efficacy and safety of Romosozumab in treatment for low bone mineral density: A systematic review and meta-analysis. Clin. Rheumatol. 2020, 39, 3261–3276. [Google Scholar] [CrossRef]
- Nathan, C.F. Secretory products of macrophages. J. Clin. Investig. 1987, 79, 319–326. [Google Scholar] [CrossRef]
- Mundy, G.R.; Boyce, B.; Hughes, D.; Wright, K.; Bonewald, L.; Dallas, S.; Harris, S.; Ghosh-Choudhury, N.; Chen, D.; Dunstan, C.; et al. The effects of cytokines and growth factors on osteoblastic cells. Bone 1995, 17, 71S–75S. [Google Scholar] [CrossRef]
- Rahimi, P.; Wang, C.Y.; Stashenko, P.; Lee, S.K.; Lorenzo, J.A.; Graves, D.T. Monocyte chemoattractant protein-1 expression and monocyte recruitment in osseous inflammation in the mouse. Endocrinology 1995, 136, 2752–2759. [Google Scholar] [CrossRef] [PubMed]
- Wehmeyer, C.; Frank, S.; Beckmann, D.; Bottcher, M.; Cromme, C.; Konig, U.; Fennen, M.; Held, A.; Paruzel, P.; Hartmann, C.; et al. Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction. Sci. Transl. Med. 2016, 8, 330ra35. [Google Scholar] [CrossRef] [PubMed]
- Courbon, G.; Lamarque, R.; Gerbaix, M.; Caire, R.; Linossier, M.T.; Laroche, N.; Thomas, M.; Thomas, T.; Vico, L.; Marotte, H. Early sclerostin expression explains bone formation inhibition before arthritis onset in the rat adjuvant-induced arthritis model. Sci. Rep. 2018, 8, 3492. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.; Hwang, H.R.; Park, H.J.; Kwon, A.; Qadir, A.S.; Ko, S.H.; Woo, K.M.; Ryoo, H.M.; Kim, G.S.; Baek, J.H. TNF-alpha upregulates sclerostin expression in obese mice fed a high-fat diet. J. Cell Physiol. 2014, 229, 640–650. [Google Scholar] [CrossRef]
- Marenzana, M.; Vugler, A.; Moore, A.; Robinson, M. Effect of sclerostin-neutralising antibody on periarticular and systemic bone in a murine model of rheumatoid arthritis: A microCT study. Arthritis Res. Ther. 2013, 15, R125. [Google Scholar] [CrossRef]
- Metzger, C.E.; Narayanan, A.; Zawieja, D.C.; Bloomfield, S.A. Inflammatory bowel disease in a rodent model alters osteocyte protein levels controlling bone turnover. J. Bone Miner. Res. 2017, 32, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Karmarkar, D.; Iwakura, Y.; Rock, K.L. Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J. Immunol. 2010, 184, 4470–4478. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.; Carmi, Y.; Guttman, O.; Braiman, A.; Cohen, I.; Voronov, E.; White, M.R.; Dinarello, C.A.; Apte, R.N. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol. 2011, 187, 4835–4843. [Google Scholar] [CrossRef]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Dominguez, A.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef]
- Kovtonyuk, L.V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front. Immunol. 2016, 7, 502. [Google Scholar] [CrossRef]
- Ho, Y.H.; Mendez-Ferrer, S. Microenvironmental contributions to hematopoietic stem cell aging. Haematologica 2020, 105, 38–46. [Google Scholar] [CrossRef]
- Jiang, N.; Li, Y.; Shu, T.; Wang, J. Cytokines and inflammation in adipogenesis: An updated review. Front. Med. 2019, 13, 314–329. [Google Scholar] [CrossRef]
- Fairfield, H.; Falank, C.; Harris, E.; Demambro, V.; McDonald, M.; Pettitt, J.A.; Mohanty, S.T.; Croucher, P.; Kramer, I.; Kneissel, M. The skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis. J. Cell. Physiol. 2018, 233, 1156–1167. [Google Scholar] [CrossRef]
- Horowitz, M.C.; Berry, R.; Holtrup, B.; Sebo, Z.; Nelson, T.; Fretz, J.A.; Lindskog, D.; Kaplan, J.L.; Ables, G.; Rodeheffer, M.S. Bone marrow adipocytes. Adipocyte 2017, 6, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.E.; Witte, P.L.; Knight, K.L. Bone marrow fat and the decline of B lymphopoiesis in rabbits. Dev. Comp. Immunol. 2016, 58, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Yee, C.S.; Manilay, J.O.; Chang, J.C.; Hum, N.R.; Murugesh, D.K.; Bajwa, J.; Mendez, M.E.; Economides, A.E.; Horan, D.J.; Robling, A.G.; et al. Conditional deletion of sost in MSC-derived lineages identifies specific cell-type contributions to bone mass and B-cell development. J. Bone Miner. Res. 2018, 33, 1748–1759. [Google Scholar] [CrossRef]
- Aurrand-Lions, M.; Mancini, S.J. Murine bone marrow niches from hematopoietic stem cells to B cells. Int. J. Mol. Sci. 2018, 19, 2353. [Google Scholar] [CrossRef]
- Turner, R.T.; Martin, S.A.; Iwaniec, U.T. Metabolic coupling between bone marrow adipose tissue and hematopoiesis. Curr. Osteoporos. Rep. 2018, 16, 95–104. [Google Scholar] [CrossRef]
- Liu, J.J.; Hou, S.C.; Shen, C.K. Erythroid gene suppression by NF-kappa B. J. Biol. Chem. 2003, 278, 19534–19540. [Google Scholar] [CrossRef] [PubMed]
- Rusten, L.S.; Jacobsen, S.E. Tumor necrosis factor (TNF)-alpha directly inhibits human erythropoiesis in vitro: Role of p55 and p75 TNF receptors. Blood 1995, 85, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Theurl, I.; Hilgendorf, I.; Nairz, M.; Tymoszuk, P.; Haschka, D.; Asshoff, M.; He, S.; Gerhardt, L.M.; Holderried, T.A.; Seifert, M. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat. Med. 2016, 22, 945–951. [Google Scholar] [CrossRef]
- England, J.; Down, M. Red-cell-volume distribution curves and the measurement of anisocytosis. Lancet 1974, 303, 701–703. [Google Scholar] [CrossRef]
- Bessman, J.D.; Gilmer JR, P.R.; Gardner, F.H. Improved classification of anemias by MCV and RDW. Am. J. Clin. Pathol. 1983, 80, 322–326. [Google Scholar] [CrossRef]
- Van Wijngaarden, J.; Doets, E.; Szczecińska, A.; Souverein, O.; Duffy, M.E.; Dullemeijer, C.; Cavelaars, A.; Pietruszka, B.; van’t Veer, P.; Brzozowska, A. Vitamin B12, folate, homocysteine, and bone health in adults and elderly people: A systematic review with meta-analyses. J. Nutr. Metab. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Bessman, J.D. The inverse relation between platelet volume and platelet number: Abnormalities in hematologic disease and evidence that platelet size does not correlate with platelet age. J. Lab. Clin. Med. 1983, 101, 295–307. [Google Scholar]
- Kissova, J.; Bulikova, A.; Ovesna, P.; Bourkova, L.; Penka, M. Increased mean platelet volume and immature platelet fraction as potential predictors of thrombotic complications in BCR/ABL-negative myeloproliferative neoplasms. Int. J. Hematol. 2014, 100, 429–436. [Google Scholar] [CrossRef]
- Raszeja-Specht, A.; Skibowska, A.; Kabata, J.; Hellmann, A. Platelet defects in chronic myeloproliferative disorders. Acta Haematol. Pol. 1994, 25, 253. [Google Scholar]
- Yan, K.; Ding, B.; Huang, J.; Dai, Y.; Xiong, S.; Zhai, Z. Normal platelet counts mask abnormal thrombopoiesis in patients with chronic myeloid leukemia. Oncol. Lett. 2015, 10, 2390–2394. [Google Scholar] [CrossRef]
- Lucijanic, M.; Mitrovic, Z.; Cicic, D.; Prka, Z.; Pejsa, V.; Livun, A.; Stoos-Veic, T.; Romic, Z.; Zivkovic, M.; Lucijanic, I. Increased mean platelet volume (MPV) is an independent predictor of inferior survival in patients with primary and secondary myelofibrosis. Int. J. Hematol. 2018, 107, 166–172. [Google Scholar] [CrossRef]
- Masutani, R.; Ikemoto, T.; Maki, A.; Tanada, H.; Iwatani, Y.; Watanabe, M.; Takubo, T.; Okada, Y. Mean platelet component and mean platelet volume as useful screening markers for myelodysplastic syndrome. Health Sci. Rep. 2018, 1, e50. [Google Scholar] [CrossRef]
- Sohawon, D.; Lau, K.K.; Lau, T.; Bowden, D.K. Extra-medullary haematopoiesis: A pictorial review of its typical and atypical locations. J. Med. Imaging Radiat. Oncol. 2012, 56, 538–544. [Google Scholar] [CrossRef]
- Holdsworth, G.; Greenslade, K.; Jose, J.; Stencel, Z.; Kirby, H.; Moore, A.; Ke, H.Z.; Robinson, M.K. Dampening of the bone formation response following repeat dosing with sclerostin antibody in mice is associated with up-regulation of Wnt antagonists. Bone 2018, 107, 93–103. [Google Scholar] [CrossRef]
- Ominsky, M.S.; Boyce, R.W.; Li, X.; Ke, H.Z. Effects of sclerostin antibodies in animal models of osteoporosis. Bone 2017, 96, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Lang, A.-G.; Buchner, A. G* Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef]
- Kedlaya, R.; Veera, S.; Horan, D.J.; Moss, R.E.; Ayturk, U.M.; Jacobsen, C.M.; Bowen, M.E.; Paszty, C.; Warman, M.L.; Robling, A.G. Sclerostin inhibition reverses skeletal fragility in an Lrp5-deficient mouse model of OPPG syndrome. Sci. Transl. Med. 2013, 5, 211ra158. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bouxsein, M.L.; Boyd, S.K.; Christiansen, B.A.; Guldberg, R.E.; Jepsen, K.J.; Müller, R. Guidelines for assessment of bone microstructure in rodents using micro–computed tomography. J. Bone Miner. Res. 2010, 25, 1468–1486. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Duarte, D.; Hawkins, E.D.; Akinduro, O.; Ang, H.; de Filippo, K.; Kong, I.Y.; Haltalli, M.; Ruivo, N.; Straszkowski, L.; Vervoort, S.J.; et al. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell 2018, 22, 64–77. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef]
- Pinho, S.; Marchand, T.; Yang, E.; Wei, Q.; Nerlov, C.; Frenette, P.S. Lineage-biased hematopoietic stem cells are regulated by distinct niches. Dev. Cell 2018, 44, 634–641. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donham, C.; Chicana, B.; Robling, A.G.; Mohamed, A.; Elizaldi, S.; Chi, M.; Freeman, B.; Millan, A.; Murugesh, D.K.; Hum, N.R.; et al. Sclerostin Depletion Induces Inflammation in the Bone Marrow of Mice. Int. J. Mol. Sci. 2021, 22, 9111. https://doi.org/10.3390/ijms22179111
Donham C, Chicana B, Robling AG, Mohamed A, Elizaldi S, Chi M, Freeman B, Millan A, Murugesh DK, Hum NR, et al. Sclerostin Depletion Induces Inflammation in the Bone Marrow of Mice. International Journal of Molecular Sciences. 2021; 22(17):9111. https://doi.org/10.3390/ijms22179111
Chicago/Turabian StyleDonham, Cristine, Betsabel Chicana, Alexander G. Robling, Asmaa Mohamed, Sonny Elizaldi, Michael Chi, Brian Freeman, Alberto Millan, Deepa K. Murugesh, Nicholas R. Hum, and et al. 2021. "Sclerostin Depletion Induces Inflammation in the Bone Marrow of Mice" International Journal of Molecular Sciences 22, no. 17: 9111. https://doi.org/10.3390/ijms22179111
APA StyleDonham, C., Chicana, B., Robling, A. G., Mohamed, A., Elizaldi, S., Chi, M., Freeman, B., Millan, A., Murugesh, D. K., Hum, N. R., Sebastian, A., Loots, G. G., & Manilay, J. O. (2021). Sclerostin Depletion Induces Inflammation in the Bone Marrow of Mice. International Journal of Molecular Sciences, 22(17), 9111. https://doi.org/10.3390/ijms22179111