
GSK-3β, FYN, and DYRK1A: Master Regulators in Neurodegenerative Pathways



Abstract
1. The Neurokinome in Drug Discovery
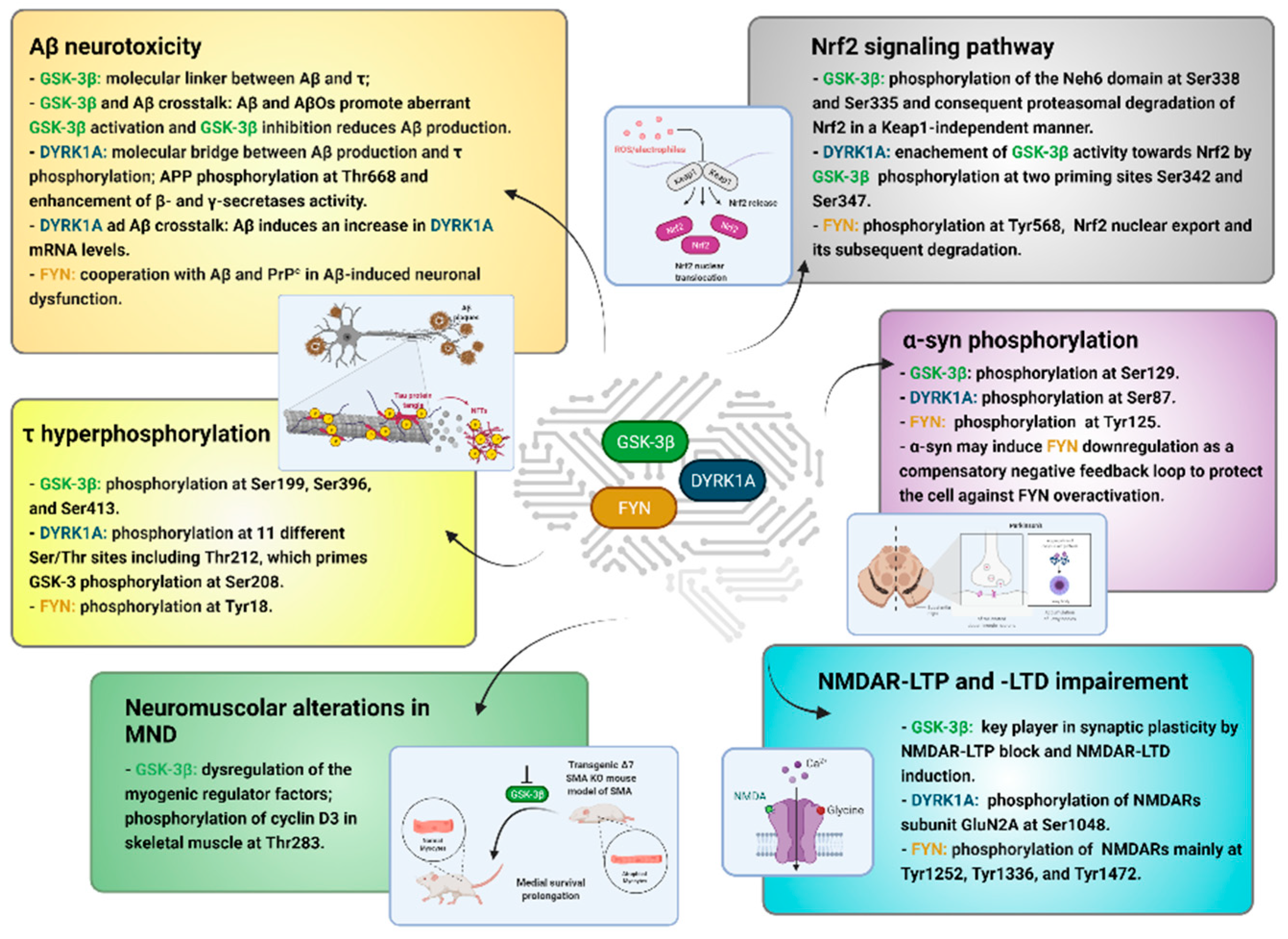
2. GSK-3β, FYN, and DYRK1A, Emerging Targets in the Neurokinome
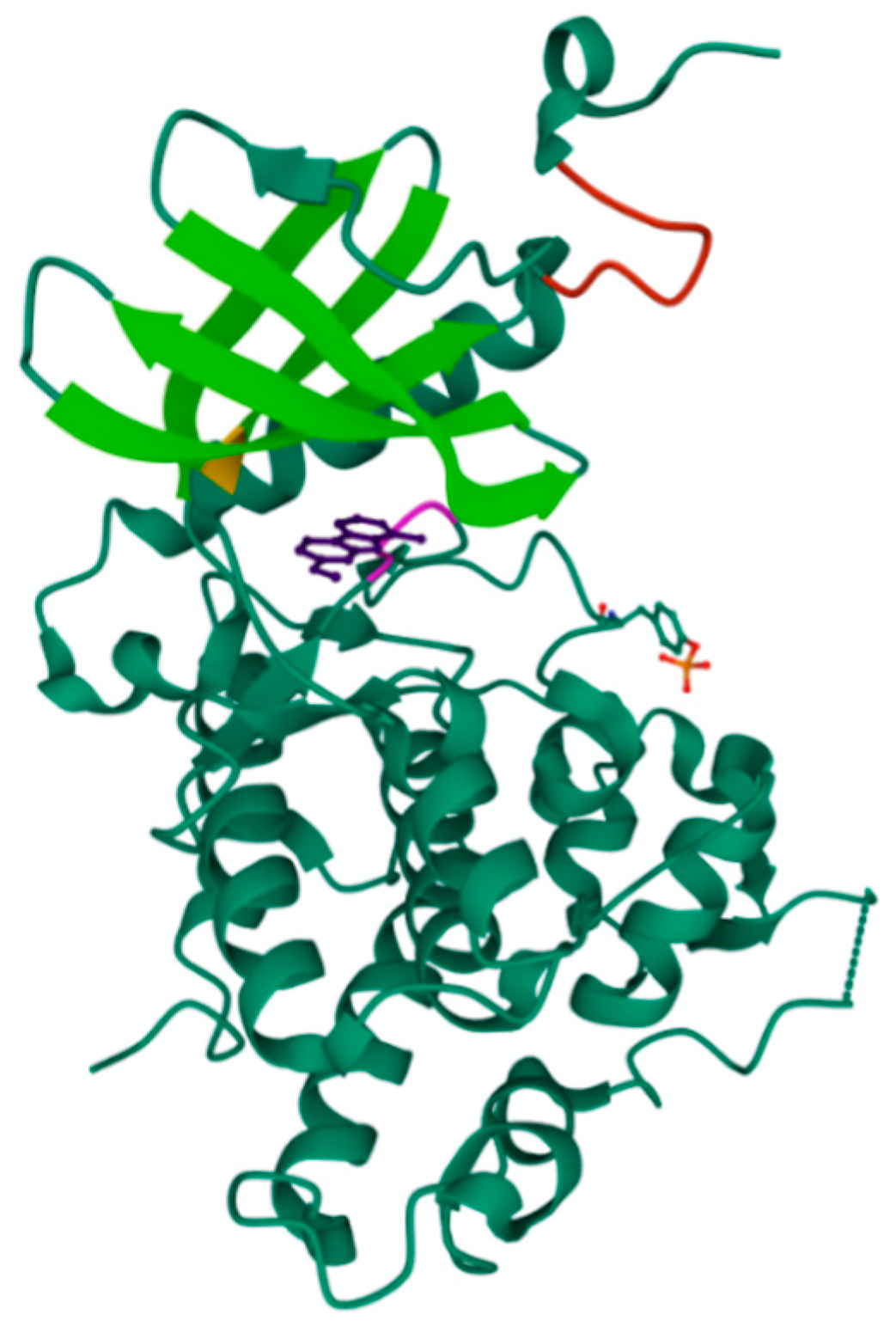
2.1. GSK-3β
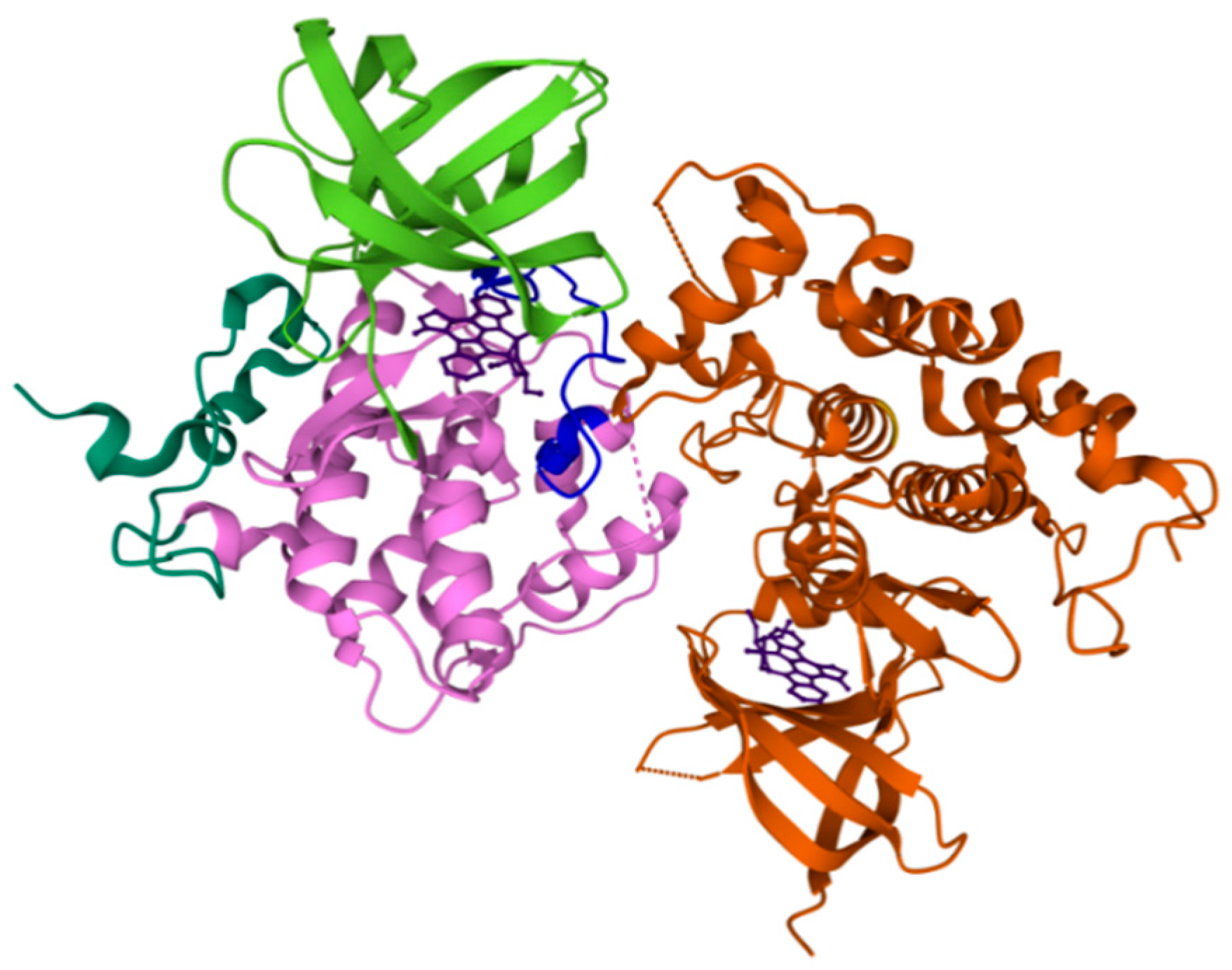
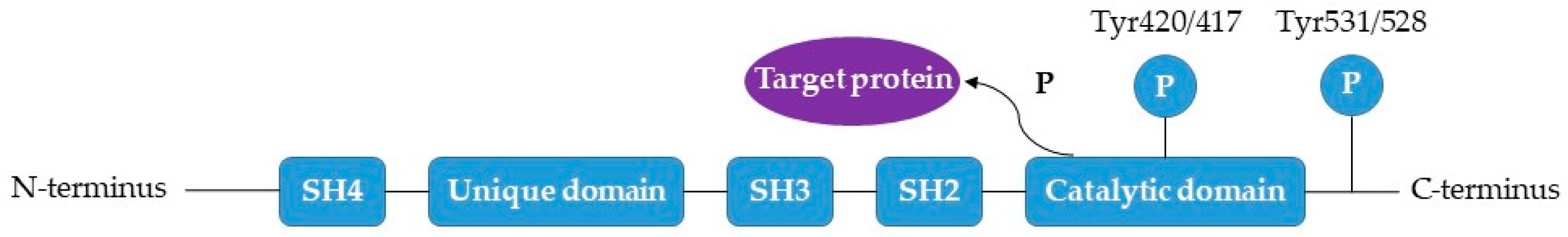
2.2. FYN
2.3. DYRK1A
3. τ Hyperphosphorylation
4. Aβ Neurotoxicity
5. Nrf2 Signaling Pathway
6. α-syn Phosphorylation
7. NMDAR-LTP and LTD Impairment
8. Neuromuscular Alterations in MND
9. GSK-3β Modulation
9.1. Covalent Inhibitors
9.2. Substrate Competitive Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Structure | pKi/IC50 Values | Purpose/Biological Activities |
|---|---|---|
 | IC50 34.3 nM |
|
 | Ki 31 nM |
|
 | IC50 0.5–2.5 µM | |
 | IC50 17 nM |
|
 | IC50 50 nM |
|
 | IC50 0.6–7 µM |
|
 | 22: IC50 184.9 µM 23: IC50 0.59 µM |
|
 | IC50 ≈ 1–4 µM |
|
 | 26: IC50 1.5 µM |
|
 | IC50 1.9 µM |
|
 | IC50 2.01–3.01 µM |
|
9.3. Non-ATP Competitive Modulators
10. FYN Inhibition
| Chemical Structure | Ki/IC50 Values | Purpose/Biological Activities |
|---|---|---|
 | IC50 2 nM |
|
 | IC50 240 nM |
|
 | IC50 4.8 μM |
|
 | IC50 0.76 μM |
|
 | Ki 2.1 μM |
|
 | Ki 2.25 μM |
|
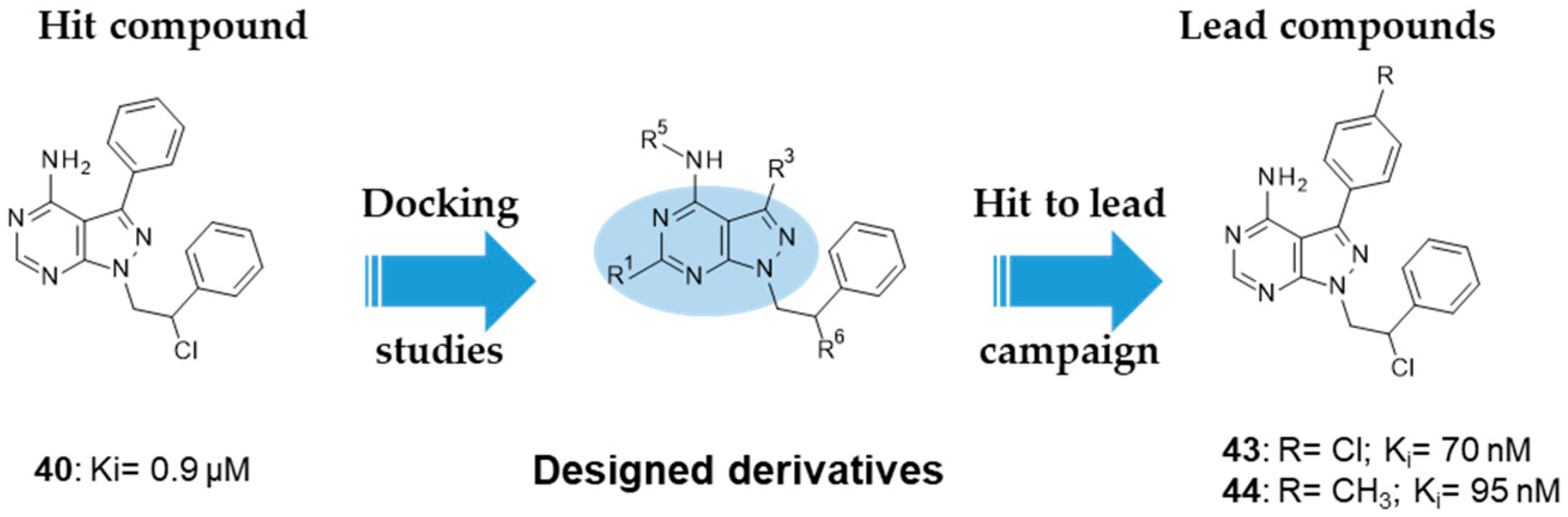
 | 40: Ki 0.9 µM 43: Ki 70 nM 44: Ki 95 nM |
|
 | IC50 60 nM |
|
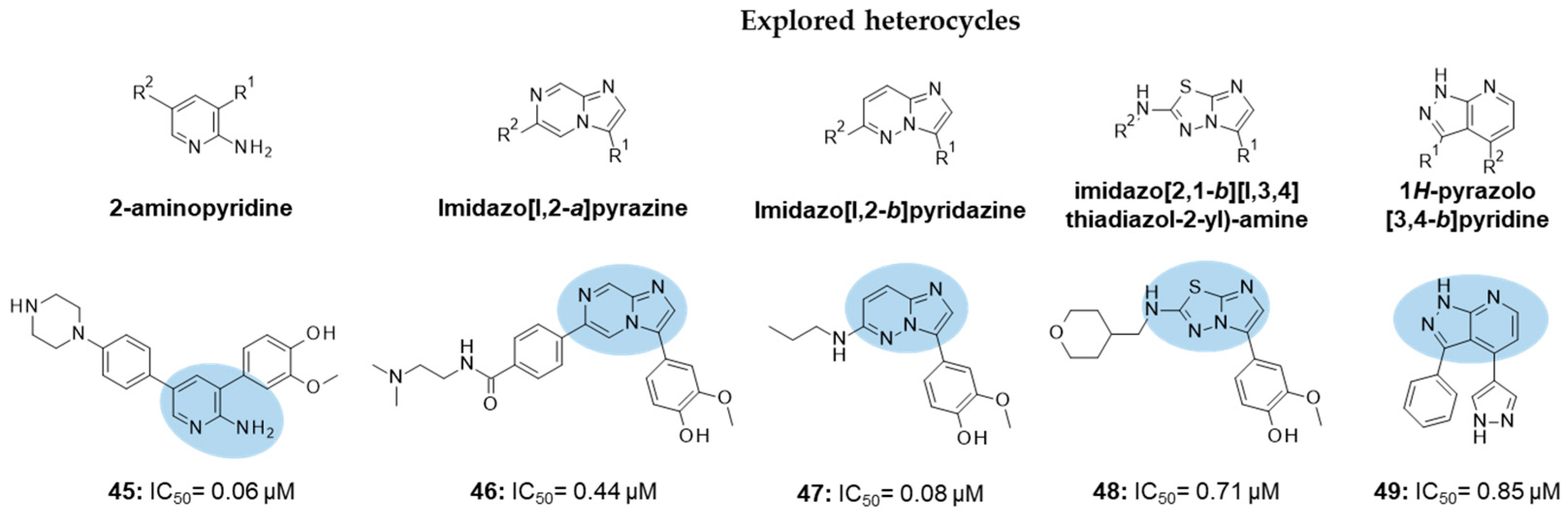
 | IC50 0.44 μM |
|
 | IC50 80 nM |
|
 | IC50 0.71 μM |
|
 | IC50 0.85 μM |
|
 | IC50 0.20 μM |
|
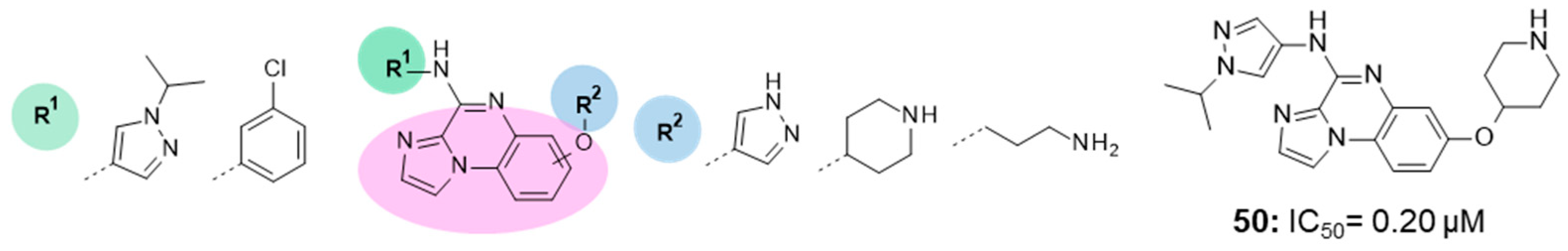
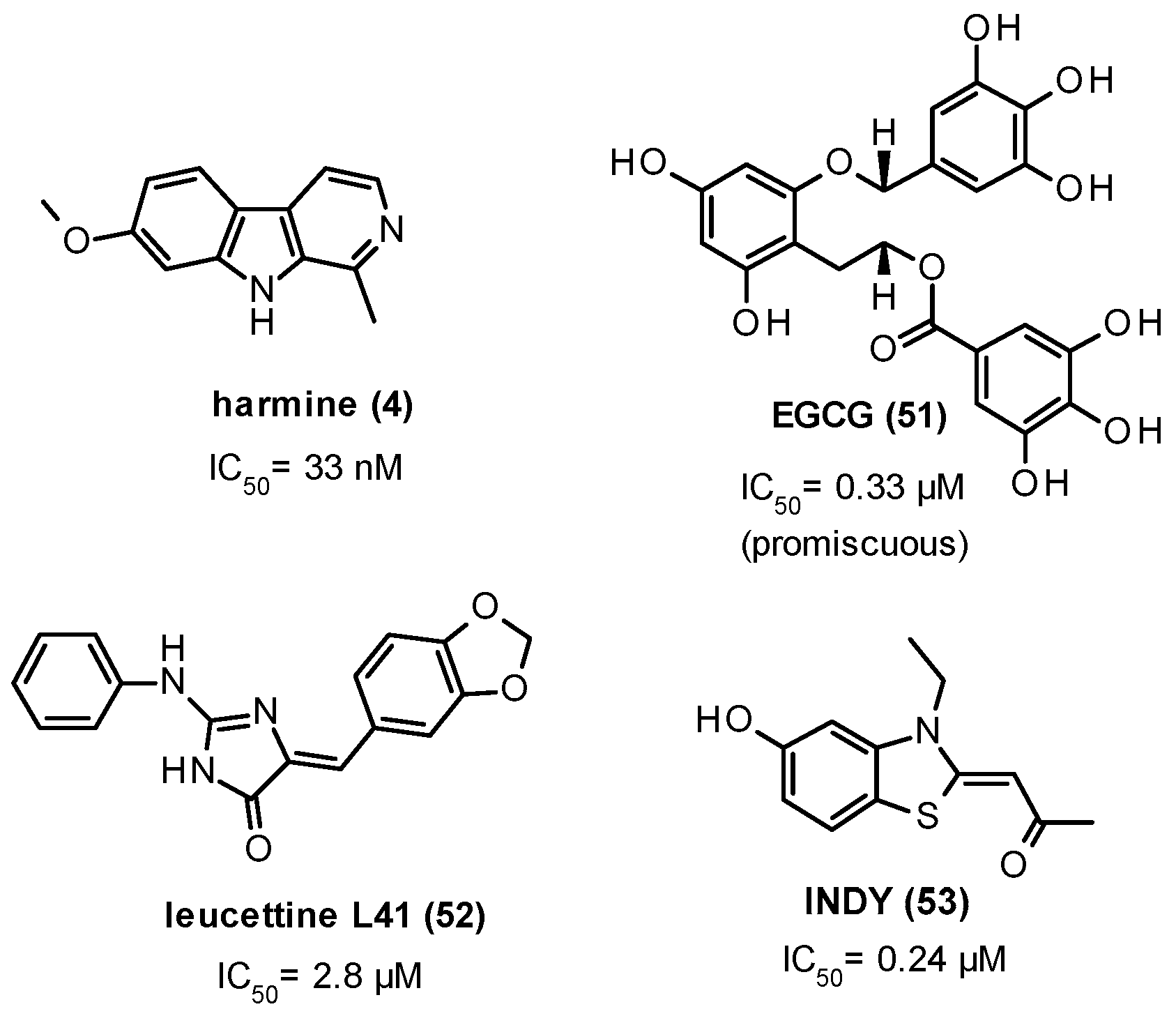
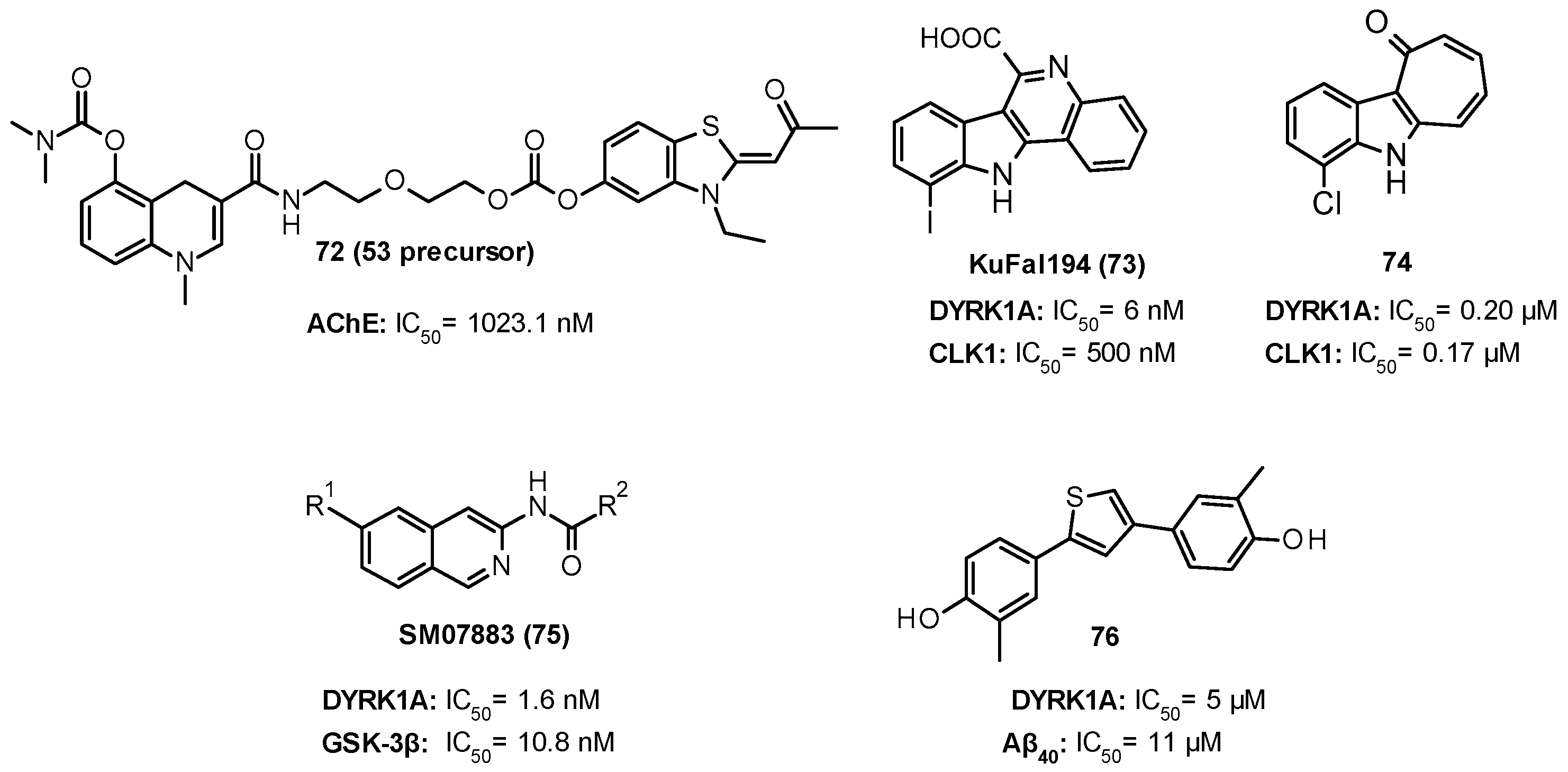
11. DYRK1A Inhibitors
| Chemical Structure | IC50 Values | Purpose/Biological Activities |
|---|---|---|
 | 181 nM |
|
 | 34 nM |
|
 | * IC50 data not available |
|
 | 6.8 nM |
|
 | 59: 0.36 nM 60: 0.22 nM |
|
 | 532 nM |
|
 | 76.9 nM |
|
12. Multi-Target Compounds with Potential CNS Application
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AChE | acetylcholinesterase |
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| APP | amyloid precursor protein |
| ARE | antioxidant response element |
| ASF | alternative splicing factor |
| Aβ | amyloid-β |
| BACE-1 | β-site APP cleaving enzyme |
| BBB | blood–brain barrier |
| CDKs | cyclin-dependent kinases |
| CEP | chefalic |
| CLKs | CDC2-like kinases |
| ClogD | calculated distribution coefficient at pH 7.4 |
| ClogP | calculated partition coefficient |
| CMCTs | canine mast cell tumors |
| CNS | central nervous system |
| DM1 | myotonic dystrophy type 1 |
| DSCR | Down syndrome critical region |
| DYRK1A | dual-specificity tyrosine phosphorylation-regulated kinase 1A |
| FDA | Food and Drug Administration |
| FTLD | frontotemporal lobar degeneration |
| GSK-3 | glycogen synthase kinase-3 |
| HD | Huntington’s disease |
| HMK | halomethylketone |
| ITDZs | 5-imino-1,2,4-thiadiazoles |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LTD | long-term depression |
| LTP | long-term potentiation |
| MAPKs | mitogen-activated protein kinases |
| MND | motor neuron disease |
| MTDLs | multi-target-directed ligands |
| MW | molecular weight |
| NF-kB | nuclear factor-kB |
| NFTs | neurofibrillary tangles |
| NMDARs | N-methyl-D-Aspartate receptors |
| PAMPA | parallel artificial membrane permeability assay |
| PD | Parkinson’s disease |
| P-gp | P-glycoprotein |
| PiD | Pick’s Disease |
| Pka | acid dissociation constant |
| PKs | protein kinases |
| PP1 | protein phosphatase 1 |
| PrPc | cellular prion protein |
| PS1 | presenilin 1 |
| PSD | postsynaptic density |
| PSP | progressive supranuclear palsy |
| PTMs | post-translational modifications |
| SCIs | substrate competitive inhibitors |
| SFKs | Src family kinases |
| SH | Src homology |
| SMA | spinal muscular atrophy |
| SMN | protein survival of motor neuron |
| TDZDs | thiadiazolidindiones |
| TK | tyrosine kinase |
| α-syn | α-synuclein |
References
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 1–23. [Google Scholar] [CrossRef]
- Wells, C.I.; Al-Ali, H.; Andrews, D.M.; Asquith, C.R.M.; Axtman, A.D.; Dikic, I.; Ebner, D.; Ettmayer, P.; Fischer, C.; Frederiksen, M.; et al. The Kinase Chemogenomic Set (KCGS): An Open Science Resource for Kinase Vulnerability Identification. Int. J. Mol. Sci. 2021, 22, 566. [Google Scholar] [CrossRef]
- Krahn, A.I.; Wells, C.; Drewry, D.H.; Beitel, L.K.; Durcan, T.M.; Axtman, A.D. Defining the Neural Kinome: Strategies and Opportunities for Small Molecule Drug Discovery to Target Neurodegenerative Diseases. ACS Chem. Neurosci. 2020, 11, 1871–1886. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood–brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.S.; Bauer, B.; Hartz, A.M.S. Modulation of P-Glycoprotein at the Blood-Brain Barrier: Opportunities to Improve Central Nervous System Pharmacotherapy. Pharmacol. Rev. 2008, 60, 196–209. [Google Scholar] [CrossRef]
- Danon, J.J.; Reekie, T.A.; Kassiou, M. Challenges and Opportunities in Central Nervous System Drug Discovery. Trends Chem. 2019, 1, 612–624. [Google Scholar] [CrossRef]
- Ford, J.M.; Hait, W.N. Pharmacologic circumvention of multidrug resistance. Cytotechnology 1993, 12, 171–212. [Google Scholar] [CrossRef]
- Kuhnke, D.; Jedlitschky, G.; Grube, M.; Krohn, M.; Jucker, M.; Mosyagin, I.; Cascorbi, I.; Walker, L.; Kroemer, H.K.; Warzok, R.W.; et al. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer’s Amyloid-β Peptides—Implications for the Mechanisms of Aβ Clearance at the Blood–Brain Barrier. Brain Pathol. 2007, 17, 347–353. [Google Scholar] [CrossRef]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci. 2011, 3, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef]
- Caban, A.; Pisarczyk, K.; Kopacz, K.; Kapuśniak, A.; Toumi, M.; Rémuzat, C.; Kornfeld, A. Filling the gap in CNS drug development: Evaluation of the role of drug repurposing. J. Mark. Access Health Policy 2017, 5, 1299833. [Google Scholar] [CrossRef]
- García-Cárceles, J.; Caballero, E.; Gil, C.; Martínez, A. Kinase Inhibitors as Underexplored Antiviral Agents. J. Med. Chem. 2021. [Google Scholar] [CrossRef]
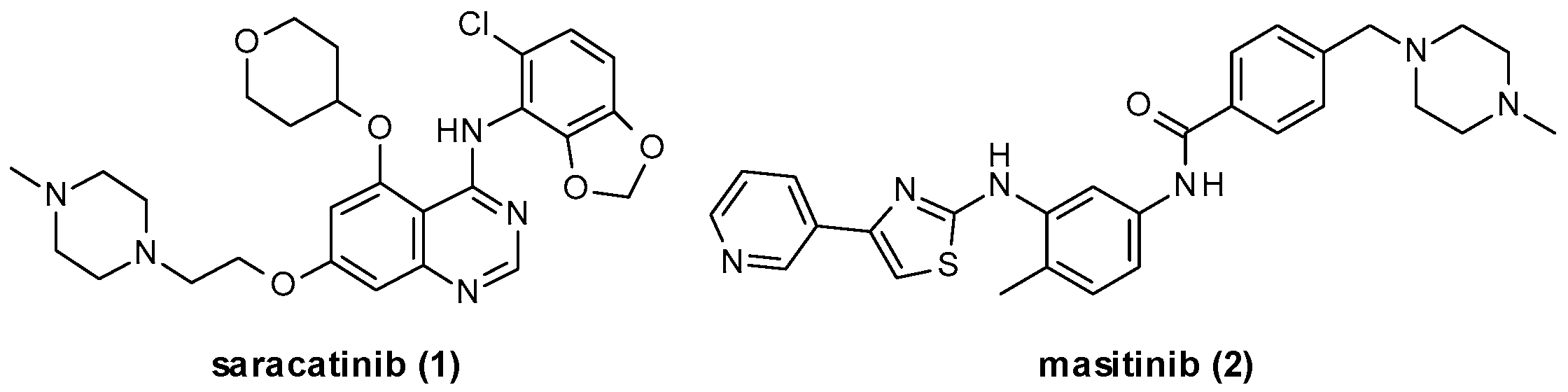
- Hennequin, L.F.; Allen, J.; Breed, J.; Curwen, J.; Fennell, M.; Green, T.P.; der Brempt, C.L.-V.; Morgentin, R.; Norman, R.A.; Olivier, A.; et al. N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a Novel, Highly Selective, Orally Available, Dual-Specific c-Src/Abl Kinase Inhibitor. J. Med. Chem. 2006, 49, 6465–6488. [Google Scholar] [CrossRef]
- Baselga, J.; Cervantes, A.; Martinelli, E.; Chirivella, I.; Hoekman, K.; Hurwitz, H.I.; Jodrell, D.I.; Hamberg, P.; Casado, E.; Elvin, P.; et al. Phase I Safety, Pharmacokinetics, and Inhibition of Src Activity Study of Saracatinib in Patients with Solid Tumors. Clin. Cancer Res. 2010, 16, 4876–4883. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef]
- Marech, I.; Patruno, R.; Zizzo, N.; Gadaleta, C.; Introna, M.; Zito, A.F.; Gadaleta, C.D.; Ranieri, G. Masitinib (AB1010), from canine tumor model to human clinical development: Where we are? Crit. Rev. Oncol. 2014, 91, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolicb, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef]
- Garuti, L.; Roberti, M.; Bottegoni, G. Multi-Kinase Inhibitors. Curr. Med. Chem. 2015, 22, 695–712. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Martinez, A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef]
- Palomo, V.; Perez, D.I.; Roca, C.; Anderson, C.; Rodriguez-Muela, N.; Perez, C.; Morales-Garcia, J.A.; Reyes, J.A.; Campillo, N.; Perez-Castillo, A.; et al. Subtly Modulating Glycogen Synthase Kinase 3 β: Allosteric Inhibitor Development and Their Potential for the Treatment of Chronic Diseases. J. Med. Chem. 2017, 60, 4983–5001. [Google Scholar] [CrossRef]
- Chen, P.C.; Gaisina, I.; El-Khodor, B.F.; Ramboz, S.; Makhortova, N.R.; Rubin, L.; Kozikowski, A.P. Identification of a Maleimide-Based Glycogen Synthase Kinase-3 (GSK-3) Inhibitor, BIP-135, That Prolongs the Median Survival Time of Δ7 SMA KO Mouse Model of Spinal Muscular Atrophy. ACS Chem. Neurosci. 2011, 3, 5–11. [Google Scholar] [CrossRef]
- Di Martino, R.M.C.; Bottegoni, G.; Seghetti, F.; Russo, D.; Penna, I.; De Simone, A.; Ottonello, G.; Bertozzi, S.M.; Armirotti, A.; Bandiera, T.; et al. Multitarget Compounds for Bipolar Disorder: From Rational Design to Preliminary Pharmacokinetic Evaluation. ChemMedChem 2020, 15, 949–954. [Google Scholar] [CrossRef]
- Nygaard, H.B. Targeting Fyn Kinase in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Julian, T.; Shaikh, M.F.; Piperi, C. Pivotal Role of Fyn Kinase in Parkinson’s Disease and Levodopa-Induced Dyskinesia: A Novel Therapeutic Target? Mol. Neurobiol. 2021, 58, 1372–1391. [Google Scholar] [CrossRef]
- Arbones, M.; Thomazeau, A.; Nakano-Kobayashi, A.; Hagiwara, M.; Delabar, J.M. DYRK1A and cognition: A lifelong relationship. Pharmacol. Ther. 2019, 194, 199–221. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Z.; Wegiel, J.; Hwang, Y.; Iqbal, K.; Grundke-Iqbal, I.; Ramakrishna, N.; Gong, C. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008, 22, 3224–3233. [Google Scholar] [CrossRef]
- Durrenberger, P.F.; Fernando, F.S.; Kashefi, S.N.; Bonnert, T.P.; Seilhean, D.; Nait-Oumesmar, B.; Schmitt, A.; Gebicke-Haerter, P.J.; Falkai, P.; Grünblatt, E.; et al. Common mechanisms in neurodegeneration and neuroinflammation: A BrainNet Europe gene expression microarray study. J. Neural Transm. 2015, 122, 1055–1068. [Google Scholar] [CrossRef]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Rippin, I.; Eldar-Finkelman, H. Mechanisms and Therapeutic Implications of GSK-3 in Treating Neurodegeneration. Cells 2021, 10, 262. [Google Scholar] [CrossRef]
- Ter Haar, E.; Coll, J.T.; A Austen, D.; Hsiao, H.M.; Swenson, L.; Jain, J. Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat. Genet. 2001, 8, 593–596. [Google Scholar] [CrossRef]
- Bertrand, J.; Thieffine, S.; Vulpetti, A.; Cristiani, C.; Valsasina, B.; Knapp, S.; Kalisz, H.; Flocco, M. Structural Characterization of the GSK-3β Active Site Using Selective and Non-selective ATP-mimetic Inhibitors. J. Mol. Biol. 2003, 333, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Palomo, V.; Soteras, I.; Perez, D.I.; Pérez, C.; Gil, C.; Campillo, N.; Martin, P. Exploring the Binding Sites of Glycogen Synthase Kinase 3. Identification and Characterization of Allosteric Modulation Cavities. J. Med. Chem. 2011, 54, 8461–8470. [Google Scholar] [CrossRef]
- Di Martino, R.M.C. Naturally Inspired Privileged Structures in Drug Discovery: Multifunctional Compounds for Alzheimer’s Disease Treatment; Alma Mater Studiorum-University of Bologna: Bologna, Italy, 2016. [Google Scholar]
- Rampa, A.; Gobbi, S.; Di Martino, R.M.C.; Belluti, F.; Bisi, A. Dual BACE-1/GSK-3β Inhibitors to Combat Alzheimer’s Disease: A Focused Review. Curr. Top. Med. Chem. 2018, 17, 3361–3369. [Google Scholar] [CrossRef] [PubMed]
- Emedina, M.; Egarrido, J.J.; Wandosell, F.G. Modulation of GSK-3 as a Therapeutic Strategy on Tau Pathologies. Front. Mol. Neurosci. 2011, 4, 24. [Google Scholar] [CrossRef]
- Di Martino, R.M.C.; Pruccoli, L.; Bisi, A.; Gobbi, S.; Rampa, A.; Martinez, A.; Pérez, C.; Martinez-Gonzalez, L.; Paglione, M.; Di Schiavi, E.; et al. Novel Curcumin-Diethyl Fumarate Hybrid as a Dualistic GSK-3β Inhibitor/Nrf2 Inducer for the Treatment of Parkinson’s Disease. ACS Chem. Neurosci. 2020, 11, 2728–2740. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-J.; Cha, S.J.; Lee, J.-W.; Kim, H.-J.; Kim, K. Recent Advances on the Role of GSK3β in the Pathogenesis of Amyotrophic Lateral Sclerosis. Brain Sci. 2020, 10, 675. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef]
- Kypta, R.M.; Hemming, A.; Courtneidge, S.A. Identification and characterization of p59fyn (a src-like protein tyrosine kinase) in normal and polyoma virus transformed cells. EMBO J. 1988, 7, 3837–3844. [Google Scholar] [CrossRef]
- Tang, X.; Feng, Y.; Ye, K. Src-family tyrosine kinase fyn phosphorylates phosphatidylinositol 3-kinase enhancer-activating Akt, preventing its apoptotic cleavage and promoting cell survival. Cell Death Differ. 2006, 14, 368–377. [Google Scholar] [CrossRef]
- Sperber, B.R.; Boyle-Walsh, É.A.; Engleka, M.J.; Gadue, P.; Peterson, A.C.; Stein, P.L.; Scherer, S.S.; McMorris, F.A. A Unique Role for Fyn in CNS Myelination. J. Neurosci. 2001, 21, 2039–2047. [Google Scholar] [CrossRef]
- Goldsmith, J.F.; Hall, C.G.; Atkinson, T. Identification of an alternatively spliced isoform of the fyn tyrosine kinase. Biochem. Biophys. Res. Commun. 2002, 298, 501–504. [Google Scholar] [CrossRef]
- Schenone, S.; Brullo, C.; Musumeci, F.M.; Biava, M.; Falchi, F.; Botta, M. Fyn kinase in brain diseases and cancer: The search for inhibitors. Curr. Med. Chem. 2011, 18, 2921–2942. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Alzheimer’s Disease II. Topics in Medicinal Chemistry; Wolfe, M.S., Ed.; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Lapillo, M.; Granchi, C.; Caciolla, J.; Mouawad, N.; Caligiuri, I.; Rizzolio, F.; Langer, T.; Minutolo, F.; Tuccinardi, T. Binding investigation and preliminary optimisation of the 3-amino-1,2,4-triazin-5(2H)-one core for the development of new Fyn inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Himpel, S.; Panzer, P.; Eirmbter, K.; Czajkowska, H.; Sayed, M.; Packman, L.C.; Blundell, T.; Kentrup, H.; Grötzinger, J.; Joost, H.-G.; et al. Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. Biochem. J. 2001, 359, 497–505. [Google Scholar] [CrossRef]
- Soundararajan, M.; Roos, A.K.; Savitsky, P.; Filippakopoulos, P.; Kettenbach, A.; Olsen, J.; Gerber, S.A.; Eswaran, J.; Knapp, S.; Elkins, J.M. Structures of Down Syndrome Kinases, DYRKs, Reveal Mechanisms of Kinase Activation and Substrate Recognition. Structure 2013, 21, 986–996. [Google Scholar] [CrossRef]
- Hämmerle, B.; Ulin, E.; Guimera, J.; Becker, W.; Guillemot, F.; Tejedor, F.J. Transient expression of Mnb/Dyrk1a couples cell cycle exit and differentiation of neuronal precursors by inducing p27KIP1 expression and suppressing NOTCH signaling. Development 2011, 138, 2543–2554. [Google Scholar] [CrossRef]
- Park, J.; Oh, Y.; Yoo, L.; Jung, M.-S.; Song, W.-J.; Lee, S.-H.; Seo, H.; Chung, K.C. Dyrk1A Phosphorylates p53 and Inhibits Proliferation of Embryonic Neuronal Cells. J. Biol. Chem. 2010, 285, 31895–31906. [Google Scholar] [CrossRef]
- Ogawa, Y.; Nonaka, Y.; Goto, T.; Ohnishi, E.; Hiramatsu, T.; Kii, I.; Yoshida, M.; Ikura, T.; Onogi, H.; Shibuya, H.; et al. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat. Commun. 2010, 1, 86. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Ung, P.M.-U.; Wang, P.; Wang, H.; Li, H.; Andrews, M.K.; Stewart, A.F.; Schlessinger, A.; DeVita, R.J. Novel selective thiadiazine DYRK1A inhibitor lead scaffold with human pancreatic β-cell proliferation activity. Eur. J. Med. Chem. 2018, 157, 1005–1016. [Google Scholar] [CrossRef]
- Treiber, D.K.; Shah, N.P. Ins and Outs of Kinase DFG Motifs. Chem. Biol. 2013, 20, 745–746. [Google Scholar] [CrossRef]
- Guo, X.; Williams, J.G.; Schug, T.T.; Li, X. DYRK1A and DYRK3 Promote Cell Survival through Phosphorylation and Activation of SIRT1. J. Biol. Chem. 2010, 285, 13223–13232. [Google Scholar] [CrossRef]
- Abbassi, R.; Johns, T.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Adarkwah, C.C.; Jegan, N.; Heinzel-Gutenbrunner, M.; Kühne, F.; Siebert, U.; Popert, U.; Donner-Banzhoff, N.; Kürwitz, S. Time-to-event versus ten-year-absolute-risk in cardiovascular risk prevention—Does it make a difference? Results from the Optimizing-Risk-Communication (OptRisk) randomized-controlled trial. BMC Med. Inform. Decis. Mak. 2016, 16, 1–12. [Google Scholar] [CrossRef]
- Nakano-Kobayashi, A.; Awaya, T.; Kii, I.; Sumida, Y.; Okuno, Y.; Yoshida, S.; Sumida, T.; Inoue, H.; Hosoya, T.; Hagiwara, M. Prenatal neurogenesis induction therapy normalizes brain structure and function in Down syndrome mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10268–10273. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Schade, N.; Opitz, A.; Hilbrich, I.; Stieler, J.; Vogel, T.; Neukel, V.; Oberstadt, M.; Totzke, F.; Schächtele, C.; et al. Novel Protein Kinase Inhibitors Related to Tau Pathology Modulate Tau Protein-Self Interaction Using a Luciferase Complementation Assay. Molecules 2018, 23, 2335. [Google Scholar] [CrossRef]
- Kimura, T.; Ishiguro, K.; Hisanaga, S.-I. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Jana, P.; Chakrabarti, S.K.; Hamid, M.R.A. Curcumin Downregulates GSK3 and Cdk5 in Scopolamine-Induced Alzheimer’s Disease Rats Abrogating Aβ40/42 and Tau Hyperphosphorylation. J. Alzheimer’s Dis. Rep. 2019, 3, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, R.M.C.; De Simone, A.; Andrisano, V.; Bisignano, P.; Bisi, A.; Gobbi, S.; Rampa, A.; Fato, R.; Bergamini, C.; Perez, D.I.; et al. Versatility of the Curcumin Scaffold: Discovery of Potent and Balanced Dual BACE-1 and GSK-3β Inhibitors. J. Med. Chem. 2016, 59, 531–544. [Google Scholar] [CrossRef]
- Billingsley, M.L.; Kincaid, R.L. Regulated phosphorylation and dephosphorylation of tau protein: Effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem. J. 1997, 323, 577–591. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, T.; Zhou, C.; Chohan, M.; Gu, X.; Wegiel, J.; Zhou, J.; Hwang, Y.-W.; Iqbal, K.; Grundke-Iqbal, I.; et al. Increased Dosage of Dyrk1A Alters Alternative Splicing Factor (ASF)-regulated Alternative Splicing of Tau in Down Syndrome. J. Biol. Chem. 2008, 283, 28660–28669. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Ayton, S.; Bush, A.I.; Adlard, P.A. GSK-3 in Neurodegenerative Diseases. Int. J. Alzheimer’s Dis. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Decker, H.; Lo, K.; Unger, S.M.; Ferreira, S.T.; Silverman, M.A. Amyloid- Peptide Oligomers Disrupt Axonal Transport through an NMDA Receptor-Dependent Mechanism That Is Mediated by Glycogen Synthase Kinase 3 in Primary Cultured Hippocampal Neurons. J. Neurosci. 2010, 30, 9166–9171. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Gong, C.-X.; Hwang, Y.-W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2010, 278, 236–245. [Google Scholar] [CrossRef]
- Stotani, S.; Giordanetto, F.; Medda, F. DYRK1A inhibition as potential treatment for Alzheimer’s disease. Future Med. Chem. 2016, 8, 681–696. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Park, S.Y.; Jung, M.-S.; Yoon, S.-H.; Kwen, M.-Y.; Lee, S.-Y.; Choi, S.-H.; Radnaabazar, C.; Kim, M.-K.; Kim, H.; et al. Dyrk1A-mediated phosphorylation of Presenilin 1: A functional link between Down syndrome and Alzheimer’s disease. J. Neurochem. 2010, 115, 574–584. [Google Scholar] [CrossRef]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H.; et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between β-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2006, 16, 15–23. [Google Scholar] [CrossRef]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Lesné, S.E. The Complex PrPc-Fyn Couples Human Oligomeric A with Pathological Tau Changes in Alzheimer’s Disease. J. Neurosci. 2012, 32, 16857–16871. [Google Scholar] [CrossRef]
- Bs, A.C.K.; Bs, S.V.S.; Haas, L.T.; Yang, J.; Bs, M.A.K.; Bs, A.T.J.; Robinson, S.; Gunther, E.C.; Van Dyck, C.H.; Nygaard, H.B.; et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. 2015, 77, 953–971. [Google Scholar] [CrossRef]
- Liu, Z.; Li, P.; Wu, J.; Wang, Y.; Li, P.; Hou, X.; Zhang, Q.; Wei, N.; Zhao, Z.; Liang, H.; et al. Alzheimer’s Disease—Challenges for the Future; Zerr, I., Ed.; IntechOpen: London, UK, 2015. [Google Scholar]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer’s Disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef]
- Gameiro, I.; Michalska, P.; Tenti, G.; Cores, Á.; Buendia, I.; Rojo, A.I.; Georgakopoulos, N.D.; Hernández-Guijo, J.M.; Ramos, M.T.; Wells, G.; et al. Discovery of the first dual GSK3β inhibitor/Nrf2 inducer. A new multitarget therapeutic strategy for Alzheimer’s disease. Sci. Rep. 2017, 7, 45701. [Google Scholar] [CrossRef]
- Robertson, H. Mechanisms of Repression of the Transcription Factor NRF2 by KEAP1-and B-TrCP-Dependent Ubiquitin Ligases and How the Dysregulation of NRF2 Contributes to Lung Cancer Progression; University of Dundee: Dundee, UK, 2019. [Google Scholar]
- Boo, Y.C. Natural Nrf2 Modulators for Skin Protection. Antioxidants 2020, 9, 812. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2—An update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef]
- Jain, A.K.; Jaiswal, A.K. Retraction: Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J. Biol. Chem. 2017, 292, 2051. [Google Scholar] [CrossRef]
- Kanninen, K.; White, A.R.; Koistinaho, J.; Malm, T. Targeting Glycogen Synthase Kinase-3βfor Therapeutic Benefit against Oxidative Stress in Alzheimer’s Disease: Involvement of the Nrf2-ARE Pathway. Int. J. Alzheimer’s Dis. 2011, 2011, 1–9. [Google Scholar] [CrossRef]
- Culbreth, M.; Aschner, M. GSK-3β, a double-edged sword in Nrf2 regulation: Implications for neurological dysfunction and disease. F1000Research 2018, 7, 1043. [Google Scholar] [CrossRef]
- Jain, A.; Jaiswal, A.K. GSK-3β Acts Upstream of Fyn Kinase in Regulation of Nuclear Export and Degradation of NF-E2 Related Factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Chen, L.; Periquet, M.; Wang, X.; Negro, A.; McLean, P.; Hyman, B.T.; Feany, M.B. Tyrosine and serine phosphorylation of α-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Investig. 2009, 119, 3257–3265. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Credle, J.J.; George, J.L.; Wills, J.; Duka, V.; Shah, K.; Lee, Y.-C.; Rodriguez, O.; Simkins, T.; Winter, M.; Moechars, D.; et al. GSK-3β dysregulation contributes to parkinson’s-like pathophysiology with associated region-specific phosphorylation and accumulation of tau and α-synuclein. Cell Death Differ. 2014, 22, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Yamashita, H.; Takahashi, T.; Nakamura, S. Activated Fyn Phosphorylates alpha-Synuclein at Tyrosine Residue 125. Biochem. Biophys. Res. Commun. 2001, 280, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Sung, J.Y.; Lee, H.J.; Rhim, H.; Hasegawa, M.; Iwatsubo, T.; Min, D.S.; Kim, J.; Paik, S.R.; Chung, K.C. Dyrk1A Phosphorylates α-Synuclein and Enhances Intracellular Inclusion Formation. J. Biol. Chem. 2006, 281, 33250–33257. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, P.; Feng, J.; Wu, M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol. Sci. 2016, 37, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Bradley, C.A.; Peineau, S.; Taghibiglou, C.; Nicolas, C.S.; Whitcomb, D.J.; Bortolotto, Z.A.; Kaang, B.-K.; Cho, K.; Wang, Y.T.; Collingridge, G.L. A pivotal role of GSK-3 in synaptic plasticity. Front. Mol. Neurosci. 2012, 5, 13. [Google Scholar] [CrossRef]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.C.; Saule, E.; Bouschet, T.; et al. LTP Inhibits LTD in the Hippocampus via Regulation of GSK3β. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef]
- Georgievska, B.; Sandin, J.; Doherty, J.; Mörtberg, A.; Neelissen, J.; Andersson, A.; Gruber, S.; Nilsson, Y.; Schött, P.; I Arvidsson, P.; et al. AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J. Neurochem. 2013, 125, 446–456. [Google Scholar] [CrossRef]
- Grant, S.; O’Dell, T.; Karl, K.; Stein, P.; Soriano, P.; Kandel, E. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 1992, 258, 1903–1910. [Google Scholar] [CrossRef]
- Trepanier, C.H.; Jackson, M.F.; Macdonald, J.F. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J. 2011, 279, 12–19. [Google Scholar] [CrossRef]
- Zhang, W.-B.; Ross, P.J.; Tu, Y.; Wang, Y.; Beggs, S.; Sengar, A.S.; Ellis, J.; Salter, M.W. Fyn Kinase regulates GluN2B subunit-dominant NMDA receptors in human induced pluripotent stem cell-derived neurons. Sci. Rep. 2016, 6, 23837. [Google Scholar] [CrossRef]
- Grau, C.; Arató, K.; Fernã¡ndez-Fernã¡ndez, J.M.; Valderrama, A.; Sindreu, C.; Fillat, C.; Ferrer, I.; De La Luna, S.; Altafaj, X. DYRK1A-mediated phosphorylation of GluN2A at Ser1048 regulates the surface expression and channel activity of GluN1/GluN2A receptors. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Hoolachan, J.M.; Sutton, E.R.; Bowerman, M. Teaching an old drug new tricks: Repositioning strategies for spinal muscular atrophy. Future Neurol. 2019, 14, FNL25. [Google Scholar] [CrossRef]
- Jones, K.; Wei, C.; Iakova, P.; Bugiardini, E.; Schneider-Gold, C.; Meola, G.; Woodgett, J.; Killian, J.; Timchenko, N.A.; Timchenko, L.T. GSK3β mediates muscle pathology in myotonic dystrophy. J. Clin. Investig. 2012, 122, 4461–4472. [Google Scholar] [CrossRef]
- Andreev, S.; Pantsar, T.; Tesch, R.; Kahlke, N.; El-Gokha, A.; Ansideri, F.; Grätz, L.; Romasco, J.; Sita, G.; Geibel, C.; et al. Addressing a Trapped High-Energy Water: Design and Synthesis of Highly Potent Pyrimidoindole-Based Glycogen Synthase Kinase-3β Inhibitors. J. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Tsui, H.; Zeng, Q.; Chen, K.; Zhang, K. 7.10—Inhibiting Kinases in the CNS, in Comprehensive Medicinal Chemistry III; Chackalamannil, S., Rotella, D., Ward, S.E., Eds.; Elsevier: Oxford, UK, 2017; pp. 408–446. [Google Scholar]
- Palomo, V.; Martinez, A. Glycogen synthase kinase 3 (GSK-3) inhibitors: A patent update (2014–2015). Expert Opin. Ther. Pat. 2016, 27, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Conde, S.; Perez, D.I.; Martínez, A.; Perez, C.; Moreno, F.J. Thienyl and Phenyl α-Halomethyl Ketones: New Inhibitors of Glycogen Synthase Kinase (GSK-3β) from a Library of Compound Searching. J. Med. Chem. 2003, 46, 4631–4633. [Google Scholar] [CrossRef]
- Perez, D.I.; Conde, S.; Pérez, C.; Gil, C.; Simón, D.; Wandosell, F.; Moreno, F.J.; Gelpí, J.L.; Luque, F.J.; Martínez, A. Thienylhalomethylketones: Irreversible glycogen synthase kinase 3 inhibitors as useful pharmacological tools. Bioorg. Med. Chem. 2009, 17, 6914–6925. [Google Scholar] [CrossRef]
- Perez, D.I.; Palomo, V.; Pérez, C.; Gil, C.; Dans, P.D.; Luque, F.J.; Conde, S.; Martinez, A. Switching Reversibility to Irreversibility in Glycogen Synthase Kinase 3 Inhibitors: Clues for Specific Design of New Compounds. J. Med. Chem. 2011, 54, 4042–4056. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, H.; Pan, B.; He, F.; Pan, Z. Design and synthesis of (aza)indolyl maleimide-based covalent inhibitors of glycogen synthase kinase 3β. Org. Biomol. Chem. 2018, 16, 4127–4140. [Google Scholar] [CrossRef]
- Martinez, A.; Alonso, M.; Castro, A.; Pérez, A.C.; Moreno, F.J. First Non-ATP Competitive Glycogen Synthase Kinase 3 β (GSK-3β) Inhibitors: Thiadiazolidinones (TDZD) as Potential Drugs for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2002, 45, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Gil, C.; Perez, D.I. Glycogen Synthase Kinase 3 Inhibitors in the Next Horizon for Alzheimer’s Disease Treatment. Int. J. Alzheimer’s Dis. 2011, 2011, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andrés, M.V.; Gómez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; León, T. Treatment of Alzheimer’s Disease with the GSK-3 Inhibitor Tideglusib: A Pilot Study. J. Alzheimer’s Dis. 2012, 33, 205–215. [Google Scholar] [CrossRef]
- Tolosa, E.; Litvan, I.; Höglinger, G.; Burn, D.; Lees, A.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; Del Ser, T. TAUROS Investigators A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disord. 2014, 29, 470–478. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.-J.; Calero, M.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; et al. A Phase II Trial of Tideglusib in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Palomo, V.; Perez, D.I.; Pérez, C.; Morales-Garcia, J.A.; Soteras, I.; Alonso-Gil, S.; Encinas, A.; Castro, A.; Campillo, N.; Pérez-Castillo, A.; et al. 5-Imino-1,2,4-Thiadiazoles: First Small Molecules As Substrate Competitive Inhibitors of Glycogen Synthase Kinase 3. J. Med. Chem. 2012, 55, 1645–1661. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Li, Q.X. Discovery of Selective, Substrate-Competitive, and Passive Membrane Permeable Glycogen Synthase Kinase-3β Inhibitors: Synthesis, Biological Evaluation, and Molecular Modeling of New C-Glycosylflavones. ACS Chem. Neurosci. 2018, 9, 1166–1183. [Google Scholar] [CrossRef]
- Amemiya, T.; Koike, R.; Fuchigami, S.; Ikeguchi, M.; Kidera, A. Classification and Annotation of the Relationship between Protein Structural Change and Ligand Binding. J. Mol. Biol. 2011, 408, 568–584. [Google Scholar] [CrossRef]
- Avrahami, L.; Licht-Murava, A.; Eisenstein, M.; Eldar-Finkelman, H. GSK-3 inhibition: Achieving moderate efficacy with high selectivity. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Rippin, I.; Khazanov, N.; Ben Joseph, S.; Kudinov, T.; Berent, E.; Ruiz, S.M.A.; Marciano, D.; Levy, L.; Gruzman, A.; Senderowitz, H.; et al. Discovery and Design of Novel Small Molecule GSK-3 Inhibitors Targeting the Substrate Binding Site. Int. J. Mol. Sci. 2020, 21, 8709. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Kudrimoti, S.; Prasanna, S.; Odde, S.; Doerksen, R.J.; Pennaka, H.K.; Choo, Y.M.; Rao, K.V.; Tekwani, B.L.; Madgula, V.; et al. Structure−Activity Relationship and Mechanism of Action Studies of Manzamine Analogues for the Control of Neuroinflammation and Cerebral Infections. J. Med. Chem. 2010, 53, 61–76. [Google Scholar] [CrossRef]
- Hamann, M.; Alonso, D.; Martín-Aparicio, E.; Fuertes, A.; Pérez-Puerto, M.J.; Castro, A.; Morales, S.; Navarro, M.L.; Del Monte-Millán, M.; Medina, M.; et al. Glycogen Synthase Kinase-3 (GSK-3) Inhibitory Activity and Structure–Activity Relationship (SAR) Studies of the Manzamine Alkaloids. Potential for Alzheimer’s Disease. J. Nat. Prod. 2007, 70, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Bidon-Chanal, A.; Fuertes, A.; Alonso, D.; Pérez, D.I.; Martínez, A.; Luque, F.J.; Medina, M. Evidence for a new binding mode to GSK-3: Allosteric regulation by the marine compound palinurin. Eur. J. Med. Chem. 2013, 60, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Martinez Gil, A.; Gil Ayuso-Gontán, C.; Palomo Ruiz, V.; Perez Martín, C.; Pérez Fernández, D.I.; Reyes Rodríguez, J.A. Heterocyclic GSK-3 Allosteric Modulators. ES Patent WO2013045736, 4 April 2013. [Google Scholar]
- Beurel, E.; Kaidanovich-Beilin, O.; Yeh, W.-I.; Song, L.; Palomo, V.; Michalek, S.M.; Woodgett, J.R.; Harrington, L.E.; Eldar-Finkelman, H.; Martinez, A.; et al. Regulation of Th1 Cells and Experimental Autoimmune Encephalomyelitis by Glycogen Synthase Kinase-3. J. Immunol. 2013, 190, 5000–5011. [Google Scholar] [CrossRef] [PubMed]
- Franklin, A.V.; King, M.K.; Palomo, V.; Martinez, A.; McMahon, L.L.; Jope, R.S. Glycogen Synthase Kinase-3 Inhibitors Reverse Deficits in Long-term Potentiation and Cognition in Fragile X Mice. Biol. Psychiatry 2014, 75, 198–206. [Google Scholar] [CrossRef]
- Davidson, D.; Viallet, J.; Veillette, A. Unique catalytic properties dictate the enhanced function of p59fynT, the hemopoietic cell-specific isoform of the Fyn tyrosine protein kinase, in T cells. Mol. Cell. Biol. 1994, 14, 4554–4564. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yadikar, H.; Torres, I.; Aiello, G.; Kurup, M.; Yang, Z.; Lin, F.; Kobeissy, F.; Yost, R.; Wang, K.K. Screening of tau protein kinase inhibitors in a tauopathy-relevant cell-based model of tau hyperphosphorylation and oligomerization. PLoS ONE 2020, 15, e0224952. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.J.; Fesharaki-Zadeh, A.; Takahashi, H.; Nies, S.H.; Smith, L.M.; Luo, A.; Chyung, A.; Chiasseu, M.; Strittmatter, S.M. Fyn kinase inhibition reduces protein aggregation, increases synapse density and improves memory in transgenic and traumatic Tauopathy. Acta Neuropathol. Commun. 2020, 8, 1–21. [Google Scholar] [CrossRef]
- Nygaard, H.B.; Wagner, A.; Bowen, G.S.; Good, S.P.; MacAvoy, M.G.; A Strittmatter, K.; Kaufman, A.C.; Rosenberg, B.J.; Sekine-Konno, T.; Varma, P.; et al. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 35. [Google Scholar] [CrossRef]
- Van Dyck, C.H.; Nygaard, H.B.; Chen, K.; Donohue, M.C.; Raman, R.; Rissman, R.A.; Brewer, J.B.; Koeppe, R.A.; Chow, T.W.; Rafii, M.S.; et al. Effect of AZD0530 on Cerebral Metabolic Decline in Alzheimer Disease. JAMA Neurol. 2019, 76, 1219. [Google Scholar] [CrossRef]
- King’s College London. NCT03661125, SRC Inhibition as a Potential Target for Parkinson’s Disease Psychosis (SCRIPT); King’s College London: London, UK, 2019. [Google Scholar]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.-P.; et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimer’s Res. Ther. 2011, 3, 16. [Google Scholar] [CrossRef]
- Poli, G.; Tuccinardi, T.; Rizzolio, F.; Caligiuri, I.; Botta, L.; Granchi, C.; Ortore, G.; Minutolo, F.; Schenone, S.; Martinelli, A. Identification of New Fyn Kinase Inhibitors Using a FLAP-Based Approach. J. Chem. Inf. Model. 2013, 53, 2538–2547. [Google Scholar] [CrossRef]
- Tintori, C.; La Sala, G.; Vignaroli, G.; Botta, L.; Fallacara, A.L.; Falchi, F.; Radi, M.; Zamperini, C.; Dreassi, E.; Iacono, L.D.; et al. Studies on the ATP Binding Site of Fyn Kinase for the Identification of New Inhibitors and Their Evaluation as Potential Agents against Tauopathies and Tumors. J. Med. Chem. 2015, 58, 4590–4609. [Google Scholar] [CrossRef]
- Lau, W.C. Methods, Compositions and Uses of Novel Fyn Kinase Inhibitors. U.S. Patent WO2017/044623, 16 March 2017. [Google Scholar]
- Paraselli, B.R.; Nangunoori, S.K.; Appala, V.R.; Kanthasamy, A.G.; Anatharam, V.; Guntupalli, P. Novel fyn Kinase Inhibitors. U.S. Patent WO2017/037604, 9 March 2017. [Google Scholar]
- Folch, J.; Petrov, D.; Ettcheto, M.; Pedrós, I.; Abad, S.; Beas-Zarate, C.; Lazarowski, A.; Marin, M.; Olloquequi, J.; Auladell, C.; et al. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev. Neurother. 2015, 15, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Frost, D.; Meechoovet, B.; Wang, T.; Gately, S.; Giorgetti, M.; Shcherbakova, I.; Dunckley, T. β-Carboline Compounds, Including Harmine, Inhibit DYRK1A and Tau Phosphorylation at Multiple Alzheimer’s Disease-Related Sites. PLoS ONE 2011, 6, e19264. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, J.; Mclauchlan, H.; Klevernic, I.; Arthur, S.; Alessi, D.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Gompel, M.; Leost, M.; Joffe, E.D.B.D.K.; Puricelli, L.; Franco, L.H.; Palermo, J.; Meijer, L. Meridianins, a new family of protein kinase inhibitors isolated from the Ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. [Google Scholar] [CrossRef] [PubMed]
- Debdab, M.; Carreaux, F.; Renault, S.; Soundararajan, M.; Fedorov, O.; Filippakopoulos, P.; Lozach, O.; Babault, L.; Tahtouh, T.; Baratte, B.; et al. Leucettines, a Class of Potent Inhibitors of cdc2-Like Kinases and Dual Specificity, Tyrosine Phosphorylation Regulated Kinases Derived from the Marine Sponge Leucettamine B: Modulation of Alternative Pre-RNA Splicing. J. Med. Chem. 2011, 54, 4172–4186. [Google Scholar] [CrossRef]
- Pathak, A.; Rohilla, A.; Gupta, T.; Akhtar, J.; Haider, R.; Sharma, K.; Haider, K.; Yar, M.S. DYRK1A kinase inhibition with emphasis on neurodegeneration: A comprehensive evolution story-cum-perspective. Eur. J. Med. Chem. 2018, 158, 559–592. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Fruit, C.; Hérault, Y.; Meijer, L.; Besson, T. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017, 27, 1183–1199. [Google Scholar] [CrossRef]
- Smith, B.; Medda, F.; Gokhale, V.; Dunckley, T.; Hulme, C. Recent Advances in the Design, Synthesis, and Biological Evaluation of Selective DYRK1A Inhibitors: A New Avenue for a Disease Modifying Treatment of Alzheimer’s? ACS Chem. Neurosci. 2012, 3, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.; Soppa, U.; Tejedor, F.J. DYRK1A: A potential drug target for multiple Down syndrome neuropathologies. CNS Neurol. Disord. Drug Targets 2014, 13, 26–33. [Google Scholar] [CrossRef]
- Göckler, N.; Jofre, G.; Papadopoulos, C.; Soppa, U.; Tejedor, F.J.; Becker, W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 2009, 276, 6324–6337. [Google Scholar] [CrossRef]
- Kim, H.; Sablin, S.O.; Ramsay, R. Inhibition of Monoamine Oxidase A by β-Carboline Derivatives. Arch. Biochem. Biophys. 1997, 337, 137–142. [Google Scholar] [CrossRef]
- Rüben, K.; Wurzlbauer, A.; Walte, A.; Sippl, W.; Bracher, F.; Becker, W. Selectivity Profiling and Biological Activity of Novel β-Carbolines as Potent and Selective DYRK1 Kinase Inhibitors. PLoS ONE 2015, 10, e0132453. [Google Scholar] [CrossRef]
- Bain, J.; Mclauchlan, H.; Elliott, M.; Cohen, P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef]
- De la Torre, R.; de Sola, S.; Hernandez, G.; Farré, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A.; et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar] [CrossRef]
- Franco, L.H.; Joffé, E.B.D.K.; Puricelli, L.; Tatián, M.; Seldes, A.M.; Palermo, J.A. Indole Alkaloids from the TunicateAplidiummeridianum. J. Nat. Prod. 1998, 61, 1130–1132. [Google Scholar] [CrossRef]
- Yadav, R.R.; Sharma, S.; Joshi, P.; Wani, A.; Vishwakarma, R.A.; Kumar, A.; Bharate, S.B. Meridianin derivatives as potent Dyrk1A inhibitors and neuroprotective agents. Bioorg. Med. Chem. Lett. 2015, 25, 2948–2952. [Google Scholar] [CrossRef]
- Giraud, F.; Alves, G.; Debiton, E.; Nauton, L.; Théry, V.; Durieu, E.; Ferandin, Y.; Lozach, O.; Meijer, L.; Anizon, F.; et al. Synthesis, Protein Kinase Inhibitory Potencies, and in Vitro Antiproliferative Activities of Meridianin Derivatives. J. Med. Chem. 2011, 54, 4474–4489. [Google Scholar] [CrossRef]
- Kii, I.; Sumida, Y.; Goto, T.; Sonamoto, R.; Okuno, Y.; Yoshida, S.; Kato-Sumida, T.; Koike, Y.; Abe, M.; Nonaka, Y.; et al. Selective inhibition of the kinase DYRK1A by targeting its folding process. Nat. Commun. 2016, 7, 11391. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, K.-S.; Kim, A.-K.; Choi, M.; Choi, K.; Kang, M.; Chi, S.-W.; Lee, M.-S.; Lee, J.-S.; Lee, S.-Y.; et al. A chemical with proven clinical safety restores Down syndrome-related phenotypes via DYRK1A inhibition. Dis. Model. Mech. 2016, 9, 839–848. [Google Scholar] [CrossRef]
- Chaikuad, A.; Diharce, J.; Schröder, M.; Foucourt, A.; Leblond, B.; Casagrande, A.-S.; Désiré, L.; Bonnet, P.; Knapp, S.; Besson, T. An Unusual Binding Model of the Methyl 9-Anilinothiazolo[5,4-f] quinazoline-2-carbimidates (EHT 1610 and EHT 5372) Confers High Selectivity for Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases. J. Med. Chem. 2016, 59, 10315–10321. [Google Scholar] [CrossRef]
- Coutadeur, S.; Benyamine, H.; Delalonde, L.; de Oliveira, C.; Leblond, B.; Foucourt, A.; Besson, T.; Casagrande, A.-S.; Taverne, T.; Girard, A.; et al. A novel DYRK1A (Dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: Effect on Tau and amyloid pathologies in vitro. J. Neurochem. 2015, 133, 440–451. [Google Scholar] [CrossRef]
- Czarna, A.; Wang, J.; Zelencova, D.; Liu, Y.; Deng, X.; Choi, H.G.; Zhang, T.; Zhou, W.; Chang, J.W.; Kildalsen, H.; et al. Novel Scaffolds for Dual Specificity Tyrosine-Phosphorylation-Regulated Kinase (DYRK1A) Inhibitors. J. Med. Chem. 2018, 61, 7560–7572. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A. Multitarget Drug Discovery and Polypharmacology. ChemMedChem 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Bottegoni, G.; Favia, A.D.; Recanatini, M.; Cavalli, A. The role of fragment-based and computational methods in polypharmacology. Drug Discov. Today 2012, 17, 23–24. [Google Scholar] [CrossRef] [PubMed]
- De Simone, A.; Tumiatti, V.; Andrisano, V.; Milelli, A. Glycogen Synthase Kinase 3β: A New Gold Rush in Anti-Alzheimer’s Disease Multitarget Drug Discovery? J. Med. Chem. 2021, 64, 26–41. [Google Scholar] [CrossRef]
- Redenti, S.; Marcovich, I.; De Vita, T.; Pérez, C.; DE Zorzi, R.; Demitri, N.; Perez, D.I.; Bottegoni, G.; Bisignano, P.; Bissaro, M.; et al. A Triazolotriazine-Based Dual GSK-3β/CK-1δ Ligand as a Potential Neuroprotective Agent Presenting Two Different Mechanisms of Enzymatic Inhibition. ChemMedChem 2019, 14, 310–314. [Google Scholar] [CrossRef]
- Barré, A.; Azzouz, R.; Gembus, V.; Papamicaël, C.; Levacher, V. Design, Synthesis, and In Vitro Biological Activities of a Bio-Oxidizable Prodrug to Deliver Both ChEs and DYRK1A Inhibitors for AD Therapy. Molecules 2019, 24, 1264. [Google Scholar] [CrossRef]
- Mehta, M.; Adem, A.; Sabbagh, M. New Acetylcholinesterase Inhibitors for Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lechner, C.; Flaßhoff, M.; Falke, H.; Preu, L.; Loaëc, N.; Meijer, L.; Knapp, S.; Chaikuad, A.; Kunick, C. [b]-Annulated Halogen-Substituted Indoles as Potential DYRK1A Inhibitors. Molecules 2019, 24, 4090. [Google Scholar] [CrossRef] [PubMed]
- Melchior, B.; Mittapalli, G.K.; Lai, C.; Duong-Polk, K.; Stewart, J.; Güner, B.; Hofilena, B.; Tjitro, A.; Anderson, S.D.; Herman, D.S.; et al. Tau pathology reduction with SM07883, a novel, potent, and selective oral DYRK1A inhibitor: A potential therapeutic for Alzheimer’s disease. Aging Cell 2019, 18, e13000. [Google Scholar] [CrossRef] [PubMed]
- Lubrook, G. ACTRN12619000327189, A phase 1, open-label study evaluating the safety, tolerability, and pharmacokinetics of a single ascending dose of SM07883, a novel DYRK1A inhibitor, following oral administration to healthy subjects. Australian New Zealand Clinical Trials Registry, 2019.
- Mariano, M.; Schmitt, C.; Miralinaghi, P.; Catto, M.; Hartmann, R.W.; Carotti, A.; Engel, M. First Selective Dual Inhibitors of Tau Phosphorylation and Beta-Amyloid Aggregation, Two Major Pathogenic Mechanisms in Alzheimer’s Disease. ACS Chem. Neurosci. 2014, 5, 1198–1202. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demuro, S.; Di Martino, R.M.C.; Ortega, J.A.; Cavalli, A. GSK-3β, FYN, and DYRK1A: Master Regulators in Neurodegenerative Pathways. Int. J. Mol. Sci. 2021, 22, 9098. https://doi.org/10.3390/ijms22169098
Demuro S, Di Martino RMC, Ortega JA, Cavalli A. GSK-3β, FYN, and DYRK1A: Master Regulators in Neurodegenerative Pathways. International Journal of Molecular Sciences. 2021; 22(16):9098. https://doi.org/10.3390/ijms22169098
Chicago/Turabian StyleDemuro, Stefania, Rita M. C. Di Martino, Jose A. Ortega, and Andrea Cavalli. 2021. "GSK-3β, FYN, and DYRK1A: Master Regulators in Neurodegenerative Pathways" International Journal of Molecular Sciences 22, no. 16: 9098. https://doi.org/10.3390/ijms22169098
APA StyleDemuro, S., Di Martino, R. M. C., Ortega, J. A., & Cavalli, A. (2021). GSK-3β, FYN, and DYRK1A: Master Regulators in Neurodegenerative Pathways. International Journal of Molecular Sciences, 22(16), 9098. https://doi.org/10.3390/ijms22169098