
Modeling and Structure Determination of Homo-Oligomeric Proteins: An Overview of Challenges and Current Approaches


Abstract
1. Introduction
1.1. Protein Homo-Oligomerization as an Efficient Design Principle
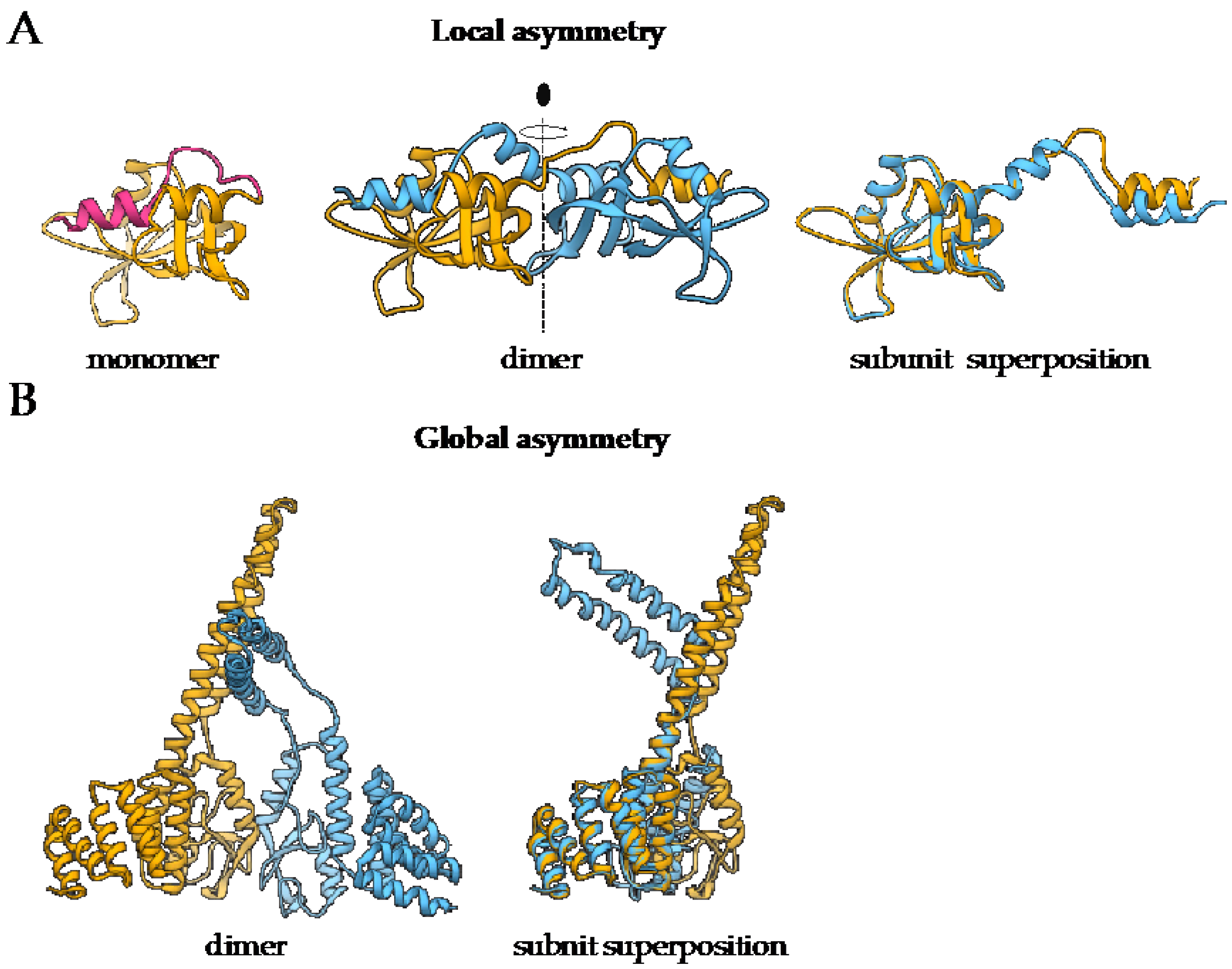
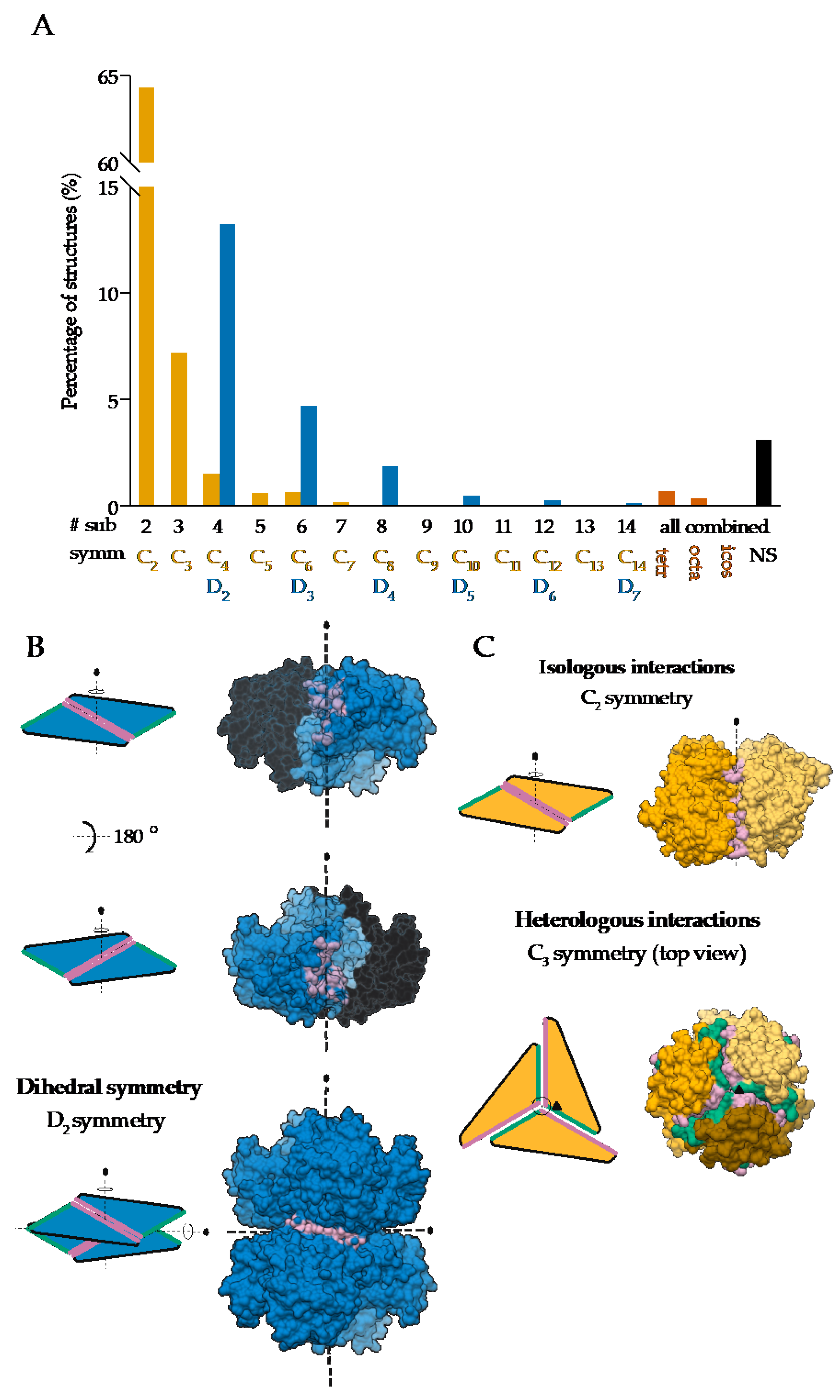
1.2. Most Protein Homo-Oligomers Are Symmetric
2. High-Resolution Structure Determination of Homo-Oligomers
2.1. X-ray Crystallography
2.1.1. Characteristics of Protein Crystals
2.1.2. Symmetry Operations
2.1.3. Approaches to Distinguish between Crystal-Only and Biologically Relevant Interaction Interfaces
2.2. Nuclear Magnetic Resonance Spectroscopy
2.3. Cryo-Electron Microscopy
3. Computational Approaches for Modeling of Homo-Oligomers
3.1. Ab Initio Docking of Protein Complexes with Cyclic Symmetries
3.2. Ab Initio Docking of Protein Complexes with Dihedral and Cubic Symmetries
3.3. Homology-Based Modeling of Homo-Oligomers
3.4. Other Computational Approaches, Used for Modeling Homo-Oligomers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levy, E.D.; Teichmann, S. Structural, evolutionary, and assembly principles of protein oligomerization. Prog. Mol. Biol. Transl. Sci. 2013, 117, 25–51. [Google Scholar] [PubMed]
- Marsh, J.A.; Rees, H.A.; Ahnert, S.E.; Teichmann, S.A. Structural and evolutionary versatility in protein complexes with uneven stoichiometry. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- Stossel, T.P. From signal to pseudopod. How cells control cytoplasmic actin assembly. J. Biol. Chem. 1989, 264, 18261–18264. [Google Scholar] [CrossRef]
- Mitchison, T.; Kirschner, M. Dynamic instability of microtubule growth. Nature 1984, 312, 237–242. [Google Scholar] [CrossRef]
- Renatus, M.; Stennicke, H.R.; Scott, F.L.; Liddington, R.C.; Salvesen, G.S. Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. USA 2001, 98, 14250–14255. [Google Scholar] [CrossRef]
- Yu, X.; Sharma, K.D.; Takahashi, T.; Iwamoto, R.; Mekada, E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol. Biol. Cell 2002, 13, 2547–2557. [Google Scholar] [CrossRef]
- Jiang, G.; den Hertog, J.; Hunter, T. Receptor-like protein tyrosine phosphatase alpha homodimerizes on the cell surface. Mol. Cell. Biol. 2000, 20, 5917–5929. [Google Scholar] [CrossRef]
- Navia, M.A.; Fitzgerald, P.M.; McKeever, B.M.; Leu, C.T.; Heimbach, J.C.; Herber, W.K.; Sigal, I.S.; Darke, P.L.; Springer, J.P. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 1989, 337, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Schamel, W.W.A.; Alarcon, B.; Höfer, T.; Minguet, S. The Allostery Model of TCR Regulation. J. Immunol. 2017, 198, 47–52. [Google Scholar] [CrossRef]
- Fushinobu, S.; Ohta, T.; Matsuzawa, H. Homotropic Activation via the Subunit Interaction and Allosteric Symmetry Revealed on Analysis of Hybrid Enzymes ofl-Lactate Dehydrogenase *. J. Biol. Chem. 1998, 273, 2971–2976. [Google Scholar] [CrossRef]
- Bergendahl, L.T.; Marsh, J.A. Functional determinants of protein assembly into homomeric complexes. Sci. Rep. 2017, 7, 4932. [Google Scholar] [CrossRef]
- Park, H.H. Domain swapping of death domain superfamily: Alternative strategy for dimerization. Int. J. Biol. Macromol. 2019, 138, 565–572. [Google Scholar] [CrossRef]
- Zegers, I.; Deswarte, J.; Wyns, L. Trimeric domain-swapped barnase. Proc. Natl. Acad. Sci. USA 1999, 96, 818–822. [Google Scholar] [CrossRef]
- Bennett, M.J.; Sawaya, M.R.; Eisenberg, D. Deposition diseases and 3D domain swapping. Structure 2006, 14, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. The evolution of multimeric protein assemblages. Mol. Biol. Evol. 2012, 29, 1353–1366. [Google Scholar] [CrossRef]
- Lynch, M. Evolutionary diversification of the multimeric states of proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E2821–E2828. [Google Scholar] [CrossRef] [PubMed]
- Hagner, K.; Setayeshgar, S.; Lynch, M. Stochastic protein multimerization, activity, and fitness. Phys. Rev. E 2018, 98, 062401. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Olson, A.J. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 105–153. [Google Scholar] [CrossRef]
- Ali, M.H.; Imperiali, B. Protein oligomerization: How and why. Bioorg. Med. Chem. 2005, 13, 5013–5020. [Google Scholar] [CrossRef]
- Perica, T.; Marsh, J.A.; Sousa, F.L.; Natan, E.; Colwell, L.J.; Ahnert, S.E.; Teichmann, S.A. The emergence of protein complexes: Quaternary structure, dynamics and allostery. Colworth Medal Lecture. Biochem. Soc. Trans. 2012, 40, 475–491. [Google Scholar] [CrossRef]
- Griffin, M.D.W.; Gerrard, J.A. The relationship between oligomeric state and protein function. Adv. Exp. Med. Biol. 2012, 747, 74–90. [Google Scholar] [PubMed]
- Bonjack, M.; Avnir, D. The near-symmetry of protein oligomers: NMR-derived structures. Sci. Rep. 2020, 10, 8367. [Google Scholar] [CrossRef]
- Bonjack-Shterengartz, M.; Avnir, D. The enigma of the near-symmetry of proteins: Domain swapping. PLoS ONE 2017, 12, e0180030. [Google Scholar] [CrossRef] [PubMed]
- Swapna, L.S.; Srikeerthana, K.; Srinivasan, N. Extent of structural asymmetry in homodimeric proteins: Prevalence and relevance. PLoS ONE 2012, 7, e36688. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Ritchie, D.W.; Levy, E.D. PDB-wide identification of biological assemblies from conserved quaternary structure geometry. Nat. Methods 2017, 15, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.E.; Olson, A.J. Icosahedral virus structures and the protein data bank. J. Biol. Chem. 2021, 296, 100554. [Google Scholar] [CrossRef]
- Lukatsky, D.B.; Zeldovich, K.B.; Shakhnovich, E.I. Statistically enhanced self-attraction of random patterns. Phys. Rev. Lett. 2006, 97, 178101. [Google Scholar] [CrossRef]
- Lukatsky, D.B.; Shakhnovich, B.E.; Mintseris, J.; Shakhnovich, E.I. Structural similarity enhances interaction propensity of proteins. J. Mol. Biol. 2007, 365, 1596–1606. [Google Scholar] [CrossRef]
- André, I.; Strauss, C.E.M.; Kaplan, D.B.; Bradley, P.; Baker, D. Emergence of symmetry in homooligomeric biological assemblies. Proc. Natl. Acad. Sci. USA 2008, 105, 16148–16152. [Google Scholar] [CrossRef]
- Schulz, G.E. The dominance of symmetry in the evolution of homo-oligomeric proteins. J. Mol. Biol. 2010, 395, 834–843. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009, 5, 15–22. [Google Scholar] [CrossRef]
- Liu, Y.; Hart, P.J.; Schlunegger, M.P.; Eisenberg, D. The crystal structure of a 3D domain-swapped dimer of RNase A at a 2.1-A resolution. Proc. Natl. Acad. Sci. USA 1998, 95, 3437–3442. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.A.; Karplus, P.A.; Dodge, R.W.; Laity, J.H.; Scheraga, H.A. Crystal structures of two mutants that have implications for the folding of bovine pancreatic ribonuclease A. Protein Sci. 1998, 7, 1255–1258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Windheim, M.; Roe, S.M.; Peggie, M.; Cohen, P.; Prodromou, C.; Pearl, L.H. Chaperoned ubiquitylation—crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol. Cell 2005, 20, 525–538. [Google Scholar] [CrossRef]
- Ortíz, C.; Botti, H.; Buschiazzo, A.; Comini, M.A. Glucose-6-phosphate dehydrogenase from the human pathogen Trypanosoma cruzi evolved unique structural features to support efficient product formation. J. Mol. Biol. 2019, 431, 2143–2162. [Google Scholar] [CrossRef]
- Kerfeld, C.A.; Sawaya, M.R.; Brahmandam, V.; Cascio, D.; Ho, K.K.; Trevithick-Sutton, C.C.; Krogmann, D.W.; Yeates, T.O. The crystal structure of a cyanobacterial water-soluble carotenoid binding protein. Structure 2003, 11, 55–65. [Google Scholar] [CrossRef]
- Mera, P.E.; St Maurice, M.; Rayment, I.; Escalante-Semerena, J.C. Structural and functional analyses of the human-type corrinoid adenosyltransferase (PduO) from Lactobacillus reuteri. Biochemistry 2007, 46, 13829–13836. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Yip, K.M.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Atomic-resolution protein structure determination by cryo-EM. Nature 2020, 587, 157–161. [Google Scholar] [CrossRef]
- Powell, H.R. X-ray data processing. Biosci. Rep. 2017, 37, BSR20170227. [Google Scholar] [CrossRef]
- Wlodawer, A.; Minor, W.; Dauter, Z.; Jaskolski, M. Protein crystallography for aspiring crystallographers or how to avoid pitfalls and traps in macromolecular structure determination. FEBS J. 2013, 280, 5705–5736. [Google Scholar] [CrossRef]
- Vonck, J.; Mills, D.J. Advances in high-resolution cryo-EM of oligomeric enzymes. Curr. Opin. Struct. Biol. 2017, 46, 48–54. [Google Scholar] [CrossRef]
- Wlodawer, A.; Minor, W.; Dauter, Z.; Jaskolski, M. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J. 2008, 275, 1–21. [Google Scholar] [CrossRef]
- Kantardjieff, K.A.; Rupp, B. Matthews coefficient probabilities: Improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci. 2003, 12, 1865–1871. [Google Scholar] [CrossRef]
- Eyal, E.; Gerzon, S.; Potapov, V.; Edelman, M.; Sobolev, V. The limit of accuracy of protein modeling: Influence of crystal packing on protein structure. J. Mol. Biol. 2005, 351, 431–442. [Google Scholar] [CrossRef]
- Juers, D.H.; Matthews, B.W. Reversible lattice repacking illustrates the temperature dependence of macromolecular interactions. J. Mol. Biol. 2001, 311, 851–862. [Google Scholar] [CrossRef]
- Dafforn, T.R. So how do you know you have a macromolecular complex? Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. What macromolecular crowding can do to a protein. Int. J. Mol. Sci. 2014, 15, 23090–23140. [Google Scholar] [CrossRef] [PubMed]
- Banatao, D.R.; Cascio, D.; Crowley, C.S.; Fleissner, M.R.; Tienson, H.L.; Yeates, T.O. An approach to crystallizing proteins by synthetic symmetrization. Proc. Natl. Acad. Sci. USA 2006, 103, 16230–16235. [Google Scholar] [CrossRef] [PubMed]
- Chesterman, C.; Arnold, E. Co-crystallization with diabodies: A case study for the introduction of synthetic symmetry. Structure 2021, 29, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Chantler, C.; Bunker, B.; Boscherini, F. International Tables for Crystallography, X-ray Absorption Spectroscopy and Related Techniques; Wiley: Hoboken, NJ, USA; ISBN 9781119433941. in press.
- International Tables for Crystallography. International Tables for Crystallography, 2012.
- Dauter, Z.; Jaskolski, M. How to read (and understand) Volume A of International Tables for Crystallography: An introduction for nonspecialists. J. Appl. Crystallogr. 2010, 43, 1150–1171. [Google Scholar] [CrossRef]
- Chruszcz, M.; Potrzebowski, W.; Zimmerman, M.D.; Grabowski, M.; Zheng, H.; Lasota, P.; Minor, W. Analysis of solvent content and oligomeric states in protein crystals--does symmetry matter? Protein Sci. 2008, 17, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Jouravel, N.; Sablin, E.; Togashi, M.; Baxter, J.D.; Webb, P.; Fletterick, R.J. Molecular basis for dimer formation of TRbeta variant D355R. Proteins 2009, 75, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Capitani, G.; Duarte, J.M.; Baskaran, K.; Bliven, S.; Somody, J.C. Understanding the fabric of protein crystals: Computational classification of biological interfaces and crystal contacts. Bioinformatics 2016, 32, 481–489. [Google Scholar] [CrossRef]
- Elez, K.; Bonvin, A.M.J.J.; Vangone, A. Biological vs. Crystallographic Protein Interfaces: An Overview of Computational Approaches for Their Classification. Crystals 2020, 10, 114. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Krissinel, E. Crystal contacts as nature’s docking solutions. J. Comput. Chem. 2010, 31, 133–143. [Google Scholar] [CrossRef]
- Shrake, A.; Rupley, J.A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 1973, 79, 351–371. [Google Scholar] [CrossRef]
- Yueh, C.; Hall, D.R.; Xia, B.; Padhorny, D.; Kozakov, D.; Vajda, S. ClusPro-DC: Dimer Classification by the Cluspro Server for Protein-Protein Docking. J. Mol. Biol. 2017, 429, 372–381. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.M.; Srebniak, A.; Schärer, M.A.; Capitani, G. Protein interface classification by evolutionary analysis. BMC Bioinformatics 2012, 13, 334. [Google Scholar] [CrossRef]
- Liu, S.; Li, Q.; Lai, L. A combinatorial score to distinguish biological and nonbiological protein-protein interfaces. Proteins 2006, 64, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, Z.; Li, J. Use B-factor related features for accurate classification between protein binding interfaces and crystal packing contacts. BMC Bioinform. 2014, 15 (Suppl. 16), S3. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Kinoshita, K.; Ito, N.; Nakamura, H. PreBI: Prediction of biological interfaces of proteins in crystals. Nucleic Acids Res. 2006, 34, W320–W324. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsuchiya, Y.; Nakamura, H.; Kinoshita, K. Discrimination between biological interfaces and crystal-packing contacts. Adv. Appl. Bioinform. Chem. 2008, 1, 99–113. [Google Scholar] [CrossRef]
- Fukasawa, Y.; Tomii, K. Accurate Classification of Biological and non-Biological Interfaces in Protein Crystal Structures using Subtle Covariation Signals. Sci. Rep. 2019, 9, 12603. [Google Scholar] [CrossRef]
- Elez, K.; Bonvin, A.M.J.J.; Vangone, A. Distinguishing crystallographic from biological interfaces in protein complexes: Role of intermolecular contacts and energetics for classification. BMC Bioinform. 2018, 19, 438. [Google Scholar] [CrossRef]
- Jiménez-García, B.; Elez, K.; Koukos, P.I.; Bonvin, A.M.; Vangone, A. PRODIGY-crystal: A web-tool for classification of biological interfaces in protein complexes. Bioinformatics 2019, 35, 4821–4823. [Google Scholar] [CrossRef]
- Baskaran, K.; Duarte, J.M.; Biyani, N.; Bliven, S.; Capitani, G. A PDB-wide, evolution-based assessment of protein-protein interfaces. BMC Struct. Biol. 2014, 14, 22. [Google Scholar] [CrossRef]
- Hu, J.; Liu, H.-F.; Sun, J.; Wang, J.; Liu, R. Integrating co-evolutionary signals and other properties of residue pairs to distinguish biological interfaces from crystal contacts. Protein Sci. 2018, 27, 1723–1735. [Google Scholar] [CrossRef]
- Luo, M.; Tanner, J.J. Structural basis of substrate recognition by aldehyde dehydrogenase 7A1. Biochemistry 2015, 54, 5513–5522. [Google Scholar] [CrossRef] [PubMed]
- Dhatwalia, R.; Singh, H.; Oppenheimer, M.; Karr, D.B.; Nix, J.C.; Sobrado, P.; Tanner, J.J. Crystal structures and small-angle x-ray scattering analysis of UDP-galactopyranose mutase from the pathogenic fungus Aspergillus fumigatus. J. Biol. Chem. 2012, 287, 9041–9051. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.H.; Mobli, M.; Gooley, P.R.; King, G.F.; Mackay, J.P. Macromolecular NMR spectroscopy for the non-spectroscopist. FEBS J. 2011, 278, 687–703. [Google Scholar] [CrossRef]
- Yu, H. Extending the size limit of protein nuclear magnetic resonance. Proc. Natl. Acad. Sci. USA 1999, 96, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Sgourakis, N.G.; Lange, O.F.; DiMaio, F.; André, I.; Fitzkee, N.C.; Rossi, P.; Montelione, G.T.; Bax, A.; Baker, D. Determination of the structures of symmetric protein oligomers from NMR chemical shifts and residual dipolar couplings. J. Am. Chem. Soc. 2011, 133, 6288–6298. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.P.; McElroy, C.A.; Amero, C.D. Solution NMR of large molecules and assemblies. Biochemistry 2007, 46, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Tjandra, N. The use of residual dipolar coupling in studying proteins by NMR. Top. Curr. Chem. 2012, 326, 47–67. [Google Scholar]
- Wang, J.; Zuo, X.; Yu, P.; Byeon, I.-J.L.; Jung, J.; Wang, X.; Dyba, M.; Seifert, S.; Schwieters, C.D.; Qin, J.; et al. Determination of multicomponent protein structures in solution using global orientation and shape restraints. J. Am. Chem. Soc. 2009, 131, 10507–10515. [Google Scholar] [CrossRef]
- Yu, X.; Jin, L.; Zhou, Z.H. 3.88 A structure of cytoplasmic polyhedrosis virus by cryo-electron microscopy. Nature 2008, 453, 415–419. [Google Scholar] [CrossRef]
- van Heel, M. Multivariate statistical classification of noisy images (randomly oriented biological macromolecules). Ultramicroscopy 1984, 13, 165–183. [Google Scholar] [CrossRef]
- Costa, A.; Patwardhan, A. A novel mirror-symmetry analysis approach for the study of macromolecular assemblies imaged by electron microscopy. J. Mol. Biol. 2008, 378, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Reboul, C.F.; Kiesewetter, S.; Elmlund, D.; Elmlund, H. Point-group symmetry detection in three-dimensional charge density of biomolecules. Bioinformatics 2020, 36, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Moro, S.; Gotte, G. Elucidation of the ribonuclease A aggregation process mediated by 3D domain swapping: A computational approach reveals possible new multimeric structures. Biopolymers 2008, 89, 26–39. [Google Scholar] [CrossRef]
- Pierce, B.; Tong, W.; Weng, Z. M-ZDOCK: A grid-based approach for Cn symmetric multimer docking. Bioinformatics 2005, 21, 1472–1478. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Geometry-based flexible and symmetric protein docking. Proteins 2005, 60, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Comeau, S.R.; Camacho, C.J. Predicting oligomeric assemblies: N-mers a primer. J. Struct. Biol. 2005, 150, 233–244. [Google Scholar] [CrossRef]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins 2013, 81, 2159–2166. [Google Scholar] [CrossRef]
- Desta, I.T.; Porter, K.A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and Its Limits in Rigid Body Protein-Protein Docking. Structure 2020, 28, 1071–1081. [Google Scholar] [CrossRef]
- Tovchigrechko, A.; Vakser, I.A. Development and testing of an automated approach to protein docking. Proteins 2005, 60, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Tovchigrechko, A.; Vakser, I.A. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006, 34, W310–W314. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Velankar, S.; Kryshtafovych, A.; Huang, S.-Y.; Schneidman-Duhovny, D.; Sali, A.; Segura, J.; Fernandez-Fuentes, N.; Viswanath, S.; Elber, R.; et al. Prediction of homoprotein and heteroprotein complexes by protein docking and template-based modeling: A CASP-CAPRI experiment. Proteins 2016, 84 (Suppl. 1), 323–348. [Google Scholar] [CrossRef]
- Berchanski, A.; Eisenstein, M. Construction of molecular assemblies via docking: Modeling of tetramers with D2 symmetry. Proteins 2003, 53, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Katchalski-Katzir, E.; Shariv, I.; Eisenstein, M.; Friesem, A.A.; Aflalo, C.; Vakser, I.A. Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. USA 1992, 89, 2195–2199. [Google Scholar] [CrossRef]
- Berchanski, A.; Segal, D.; Eisenstein, M. Modeling oligomers with Cn or Dn symmetry: Application to CAPRI target 10. Proteins 2005, 60, 202–206. [Google Scholar] [CrossRef]
- Ritchie, D.W.; Grudinin, S. Spherical polar Fourier assembly of protein complexes with arbitrary point group symmetry. J. Appl. Crystallogr. 2016, 49, 158–167. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Zou, X. An iterative knowledge-based scoring function for protein-protein recognition. Proteins Struct. Funct. Bioinform. 2008, 72, 557–579. [Google Scholar] [CrossRef]
- Yan, Y.; Wen, Z.; Wang, X.; Huang, S.-Y. Addressing recent docking challenges: A hybrid strategy to integrate template-based and free protein-protein docking. Proteins 2017, 85, 497–512. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; Huang, S.-Y. HSYMDOCK: A docking web server for predicting the structure of protein homo-oligomers with Cn or Dn symmetry. Nucleic Acids Res. 2018, 46, W423–W431. [Google Scholar] [CrossRef]
- Park, T.; Baek, M.; Lee, H.; Seok, C. GalaxyTongDock: Symmetric and asymmetric ab initio protein-protein docking web server with improved energy parameters. J. Comput. Chem. 2019, 40, 2413–2417. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Karaca, E.; Melquiond, A.S.J.; de Vries, S.J.; Kastritis, P.L.; Bonvin, A.M.J.J. Building macromolecular assemblies by information-driven docking: Introducing the HADDOCK multibody docking server. Mol. Cell. Proteom. 2010, 9, 1784–1794. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- André, I.; Bradley, P.; Wang, C.; Baker, D. Prediction of the structure of symmetrical protein assemblies. Proc. Natl. Acad. Sci. USA 2007, 104, 17656–17661. [Google Scholar] [CrossRef] [PubMed]
- Lyskov, S.; Chou, F.-C.; Conchúir, S.Ó.; Der, B.S.; Drew, K.; Kuroda, D.; Xu, J.; Weitzner, B.D.; Douglas Renfrew, P.; Sripakdeevong, P.; et al. Serverification of Molecular Modeling Applications: The Rosetta Online Server That Includes Everyone (ROSIE). PLoS ONE 2013, 8, e63906. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; André, I.; Shen, Y.; Wu, Y.; Lemak, A.; Bansal, S.; Arrowsmith, C.H.; Szyperski, T.; Baker, D. Simultaneous prediction of protein folding and docking at high resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 18978–18983. [Google Scholar] [CrossRef]
- Roy Burman, S.S.; Yovanno, R.A.; Gray, J.J. Flexible Backbone Assembly and Refinement of Symmetrical Homomeric Complexes. Structure 2019, 27, 1041–1051.e8. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Lee, H.; Park, H.; Ko, J.; Seok, C. GalaxyGemini: A web server for protein homo-oligomer structure prediction based on similarity. Bioinformatics 2013, 29, 1078–1080. [Google Scholar] [CrossRef]
- Baek, M.; Park, T.; Heo, L.; Park, C.; Seok, C. GalaxyHomomer: A web server for protein homo-oligomer structure prediction from a monomer sequence or structure. Nucleic Acids Res. 2017, 45, W320–W324. [Google Scholar] [CrossRef]
- Baek, M.; Park, T.; Heo, L.; Seok, C. Modeling Protein Homo-Oligomer Structures with GalaxyHomomer Web Server. In Protein Structure Prediction; Kihara, D., Ed.; Springer US: New York, NY, 2020; pp. 127–137. ISBN 9781071607084. [Google Scholar]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.A.; Padhorny, D.; Desta, I.; Ignatov, M.; Beglov, D.; Kotelnikov, S.; Sun, Z.; Alekseenko, A.; Anishchenko, I.; Cong, Q.; et al. Template-based modeling by ClusPro in CASP13 and the potential for using co-evolutionary information in docking. Proteins 2019, 87, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, F.; Leaver-Fay, A.; Bradley, P.; Baker, D.; André, I. Modeling symmetric macromolecular structures in Rosetta3. PLoS ONE 2011, 6, e20450. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; DiMaio, F.; Wang, R.Y.-R.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-resolution comparative modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Hourai, Y.; Weng, Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS ONE 2011, 6, e24657. [Google Scholar] [CrossRef] [PubMed]
- Mintseris, J.; Pierce, B.; Wiehe, K.; Anderson, R.; Chen, R.; Weng, Z. Integrating statistical pair potentials into protein complex prediction. Proteins 2007, 69, 511–520. [Google Scholar] [CrossRef]
- Vreven, T.; Schweppe, D.K.; Chavez, J.D.; Weisbrod, C.R.; Shibata, S.; Zheng, C.; Bruce, J.E.; Weng, Z. Integrating Cross-Linking Experiments with Ab Initio Protein–Protein Docking. J. Mol. Biol. 2018, 430, 1814–1828. [Google Scholar] [CrossRef]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient Unbound Docking of Rigid Molecules. In Proceedings of the Algorithms in Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2002; pp. 185–200. [Google Scholar]
- Gaber, A.; Kim, S.J.; Kaake, R.M.; Benčina, M.; Krogan, N.; Šali, A.; Pavšič, M.; Lenarčič, B. EpCAM homo-oligomerization is not the basis for its role in cell-cell adhesion. Sci. Rep. 2018, 8, 13269. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.A.; Sali, A. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016, 44, W424–W429. [Google Scholar] [CrossRef]
- Xia, B.; Vajda, S.; Kozakov, D. Accounting for pairwise distance restraints in FFT-based protein–protein docking. Bioinformatics 2016, 32, 3342–3344. [Google Scholar] [CrossRef]
- Gaber, A.; Gunčar, G.; Pavšič, M. Proper evaluation of chemical cross-linking-based spatial restraints improves the precision of modeling homo-oligomeric protein complexes. BMC Bioinform. 2019, 20, 464. [Google Scholar] [CrossRef]
- Xia, B.; Mamonov, A.; Leysen, S.; Allen, K.N.; Strelkov, S.V.; Paschalidis, I.C.; Vajda, S.; Kozakov, D. Accounting for observed small angle X-ray scattering profile in the protein-protein docking server cluspro. J. Comput. Chem. 2015, 36, 1568–1572. [Google Scholar] [CrossRef]
- Ignatov, M.; Kazennov, A.; Kozakov, D. ClusPro FMFT-SAXS: Ultra-fast Filtering Using Small-Angle X-ray Scattering Data in Protein Docking. J. Mol. Biol. 2018, 430, 2249–2255. [Google Scholar] [CrossRef]
- Yan, Y.; Huang, S.-Y. CHDOCK: A hierarchical docking approach for modeling Cn symmetric homo-oligomeric complexes. Biophys. Rep. 2019, 5, 65–72. [Google Scholar] [CrossRef][Green Version]
- Yan, Y.; Huang, S.-Y. Protein-Protein Docking with Improved Shape Complementarity. In Proceedings of the Intelligent Computing Theories and Application, Wuhan, China, 15–18 August 2018; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 600–605. [Google Scholar]
- van Dijk, A.D.J.; Boelens, R.; Bonvin, A.M.J.J. Data-driven docking for the study of biomolecular complexes. FEBS J. 2005, 272, 293–312. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, A.D.J.; Fushman, D.; Bonvin, A.M.J.J. Various strategies of using residual dipolar couplings in NMR-driven protein docking: Application to Lys48-linked di-ubiquitin and validation against 15N-relaxation data. Proteins 2005, 60, 367–381. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, A.D.J.; Kaptein, R.; Boelens, R.; Bonvin, A.M.J.J. Combining NMR relaxation with chemical shift perturbation data to drive protein-protein docking. J. Biomol. NMR 2006, 34, 237–244. [Google Scholar] [CrossRef][Green Version]
- Schmitz, C.; Bonvin, A.M.J.J. Protein–protein HADDocking using exclusively pseudocontact shifts. J. Biomol. NMR 2011, 50, 263–266. [Google Scholar] [CrossRef] [PubMed]
- de Vries, S.J.; Bonvin, A.M.J.J. CPORT: A consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS ONE 2011, 6, e17695. [Google Scholar] [CrossRef] [PubMed]
- Karaca, E.; Bonvin, A.M.J.J. On the usefulness of ion-mobility mass spectrometry and SAXS data in scoring docking decoys. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 683–694. [Google Scholar] [CrossRef] [PubMed]
- van Zundert, G.C.P.; Melquiond, A.S.J.; Bonvin, A.M.J.J. Integrative Modeling of Biomolecular Complexes: HADDOCKing with Cryo-Electron Microscopy Data. Structure 2015, 23, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Trellet, M.; van Zundert, G.; Bonvin, A.M.J.J. Protein–Protein Modeling Using Cryo-EM Restraints. In Structural Bioinformatics: Methods and Protocols; Gáspári, Z., Ed.; Springer US: New York, NY, 2020; pp. 145–162. ISBN 9781071602706. [Google Scholar]
- Shen, Y.; Lange, O.; Delaglio, F.; Rossi, P.; Aramini, J.M.; Liu, G.; Eletsky, A.; Wu, Y.; Singarapu, K.K.; Lemak, A.; et al. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. USA 2008, 105, 4685–4690. [Google Scholar] [CrossRef] [PubMed]
- Kahraman, A.; Herzog, F.; Leitner, A.; Rosenberger, G.; Aebersold, R.; Malmström, L. Cross-link guided molecular modeling with ROSETTA. PLoS ONE 2013, 8, e73411. [Google Scholar] [CrossRef]
- Sønderby, P.; Rinnan, Å.; Madsen, J.J.; Harris, P.; Bukrinski, J.T.; Peters, G.H.J. Small-Angle X-ray Scattering Data in Combination with RosettaDock Improves the Docking Energy Landscape. J. Chem. Inf. Model. 2017, 57, 2463–2475. [Google Scholar] [CrossRef]
- Ovchinnikov, S.; Kamisetty, H.; Baker, D. Robust and accurate prediction of residue–residue interactions across protein interfaces using evolutionary information. Elife 2014, 3, e02030. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Söding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Park, H.; Seok, C. GalaxyTBM: Template-based modeling by building a reliable core and refining unreliable local regions. BMC Bioinform. 2012, 13, 198. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, D.; Park, H.; Coutsias, E.A.; Seok, C. Protein loop modeling by using fragment assembly and analytical loop closure. Proteins 2010, 78, 3428–3436. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Seok, C. Refinement of unreliable local regions in template-based protein models. Proteins 2012, 80, 1974–1986. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Lee, G.R.; Heo, L.; Seok, C. Protein loop modeling using a new hybrid energy function and its application to modeling in inaccurate structural environments. PLoS ONE 2014, 9, e113811. [Google Scholar]
- Heo, L.; Lee, H.; Seok, C. GalaxyRefineComplex: Refinement of protein-protein complex model structures driven by interface repacking. Sci. Rep. 2016, 6, 32153. [Google Scholar] [CrossRef]
- Park, H.; Kim, D.E.; Ovchinnikov, S.; Baker, D.; DiMaio, F. Automatic structure prediction of oligomeric assemblies using Robetta in CASP12. Proteins 2018, 86 Suppl 1, 283–291. [Google Scholar] [CrossRef]
- Lensink, M.F.; Brysbaert, G.; Nadzirin, N.; Velankar, S.; Chaleil, R.A.G.; Gerguri, T.; Bates, P.A.; Laine, E.; Carbone, A.; Grudinin, S.; et al. Blind prediction of homo- and hetero-protein complexes: The CASP13-CAPRI experiment. Proteins 2019, 87, 1200–1221. [Google Scholar] [CrossRef]
- Torchala, M.; Moal, I.H.; Chaleil, R.A.G.; Fernandez-Recio, J.; Bates, P.A. SwarmDock: A server for flexible protein–protein docking. Bioinform. 2013, 29, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Dapkunas, J.; Timinskas, A.; Olechnovic, K.; Margelevicius, M.; Diciunas, R.; Venclovas, C. The PPI3D web server for searching, analyzing and modeling protein-protein interactions in the context of 3D structures. Bioinformatics 2017, 33, 935–937. [Google Scholar] [CrossRef] [PubMed]
- Dapkūnas, J.; Venclovas, Č. Template-Based Modeling of Protein Complexes Using the PPI3D Web Server. Methods Mol. Biol. 2020, 2165, 139–155. [Google Scholar] [PubMed]
- Esquivel-Rodriguez, J.; Filos-Gonzalez, V.; Li, B.; Kihara, D. Pairwise and multimeric protein-protein docking using the LZerD program suite. Methods Mol. Biol. 2014, 1137, 209–234. [Google Scholar]
- Christoffer, C.; Chen, S.; Bharadwaj, V.; Aderinwale, T.; Kumar, V.; Hormati, M.; Kihara, D. LZerD webserver for pairwise and multiple protein–protein docking. Nucleic Acids Res. 2021, 49, W359–W365. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Zou, X. MDockPP: A hierarchical approach for protein-protein docking and its application to CAPRI rounds 15–19. Proteins Struct. Funct. Bioinform. 2010, 78, 3096–3103. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Rossi, A.D.; Blundell, T.L. ProtCHOIR: A tool for proteome-scale generation of homo-oligomers. Brief. Bioinform. 2021. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 15, 1–7. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Guzenko, D.; Lafita, A.; Monastyrskyy, B.; Kryshtafovych, A.; Duarte, J.M. Assessment of protein assembly prediction in CASP13. Proteins 2019, 87, 1190–1199. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Software | Symmetry Types | Additional Information That Can Be Used to Guide the Modeling | Website | References |
|---|---|---|---|---|
| Ab Initio Docking of Protein Complexes with Cyclic Symmetries | ||||
| M-ZDOCK | C2–24, user-defined | https://zdock.umassmed.edu/m-zdock/ (webserver) (accessed on 20 August 2021) | [86] | |
| SymmDock | C2–100, user-defined | interacting residues, distance restraints | http://bioinfo3d.cs.tau.ac.il/SymmDock/ (webserver) (accessed on 20 August 2021) | [87,88] |
| ClusPro | C2 and C3, user-defined | interacting and non-interacting residues, distance restraints (can be grouped), SAXS based restraints | https://cluspro.bu.edu/ (webserver) (accessed on 20 August 2021) | [62,89,90,91] |
| GRAMM-X | C2–8, user-defined | interacting residues | http://vakser.compbio.ku.edu/resources/gramm/grammx/ (webserver) (accessed on 20 August 2021) | [92,93,94] |
| Ab initioDocking of Protein Complexes with Dihedral and Cubic Symmetries | ||||
| MOLFIT | cyclic and dihedral, user-defined | http://www.weizmann.ac.il/Chemical_Research_Support//molfit/home.html (accessed on 20 August 2021) | [95,96,97] | |
| SAM | any, user-defined | http://sam.loria.fr/ (accessed on 20 August 2021) | [98] | |
| HSYMDOCK | cyclic and dihedral, user-defined orpredicted | interacting residues | http://huanglab.phys.hust.edu.cn/hsymdock/ (webserver) (accessed on 20 August 2021) | [99,100,101] |
| GalaxyTongDock | cyclic and dihedral, (up to 12 subunits), user-defined | interacting and non-interacting residues | http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=TONGDOCK_INTRO (webserver) (accessed on 20 August 2021) | [102] |
| HADDOCK | cyclic and dihedral, up to 20 subunits and up to 10 segment pairs for each symmetry, user-defined | a variety of experimental restraints | https://wenmr.science.uu.nl/haddock2.4/ (webserver) (accessed on 20 August 2021) | [103,104,105] |
| Homology-Based Modeling of Homo-Oligomers | ||||
| Rosetta SymDock | cyclic, dihedral, icosahedral, helical, (only cyclic and dihedral with up to 10 subunits on the webserver), user-defined | https://rosie.graylab.jhu.edu/symmetric_docking/submit (webserver) (accessed on 20 August 2021) | [106,107] | |
| Rosetta Fold-and-dock | cyclic, dihedral, icosahedral, helical, user-defined | a variety of experimental restraints | https://www.rosettacommons.org/ (accessed on 20 August 2021) | [108] |
| Rosetta SymDock2 | cyclic, dihedral, icosahedral, helical, user-defined | a variety of experimental restraints | https://www.rosettacommons.org/ (accessed on 20 August 2021) | [109] |
| SWISS-MODEL | symmetry is inferred from the templates | https://swissmodel.expasy.org/ (webserver) (accessed on 20 August 2021) | [110,111,112] | |
| GalaxyGemini | symmetry is inferred from the templates | http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=GEMINI (webserver) (accessed on 20 August 2021) | [113] | |
| GalaxyHomomer | symmetry is inferred from the templates (user-defined), Cn can also be modeled | http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=HOMOMER (webserver) (accessed on 20 August 2021) | [114,115] | |
| HDOCK | cyclic and dihedral, user-defined | the binding site, distance restraints, SAXS based restraints | http://hdock.phys.hust.edu.cn/ (webserver) (accessed on 20 August 2021) | [116] |
| ClusPro TBM | the user defines stoichiometry, not symmetry | https://tbm.cluspro.org/template_based/index.php (webserver) (accessed on 20 August 2021) | [117] | |
| Rosetta CM | cyclic, dihedral, icosahedral, helical, user-defined | a variety of experimental restraints | https://www.rosettacommons.org/ (accessed on 20 August 2021) | [118,119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaber, A.; Pavšič, M. Modeling and Structure Determination of Homo-Oligomeric Proteins: An Overview of Challenges and Current Approaches. Int. J. Mol. Sci. 2021, 22, 9081. https://doi.org/10.3390/ijms22169081
Gaber A, Pavšič M. Modeling and Structure Determination of Homo-Oligomeric Proteins: An Overview of Challenges and Current Approaches. International Journal of Molecular Sciences. 2021; 22(16):9081. https://doi.org/10.3390/ijms22169081
Chicago/Turabian StyleGaber, Aljaž, and Miha Pavšič. 2021. "Modeling and Structure Determination of Homo-Oligomeric Proteins: An Overview of Challenges and Current Approaches" International Journal of Molecular Sciences 22, no. 16: 9081. https://doi.org/10.3390/ijms22169081
APA StyleGaber, A., & Pavšič, M. (2021). Modeling and Structure Determination of Homo-Oligomeric Proteins: An Overview of Challenges and Current Approaches. International Journal of Molecular Sciences, 22(16), 9081. https://doi.org/10.3390/ijms22169081