
Molecular Mechanisms of Skewed X-Chromosome Inactivation in Female Hemophilia Patients—Lessons from Wide Genome Analyses

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Patients and Methods
2.1. Female HA Patients
2.2. Genomic DNA Extraction
2.3. XCI Analysis
2.4. Whole-Exome Sequencing (WES)
2.5. Confirmation of Mutations and Segregation Studies
3. Results
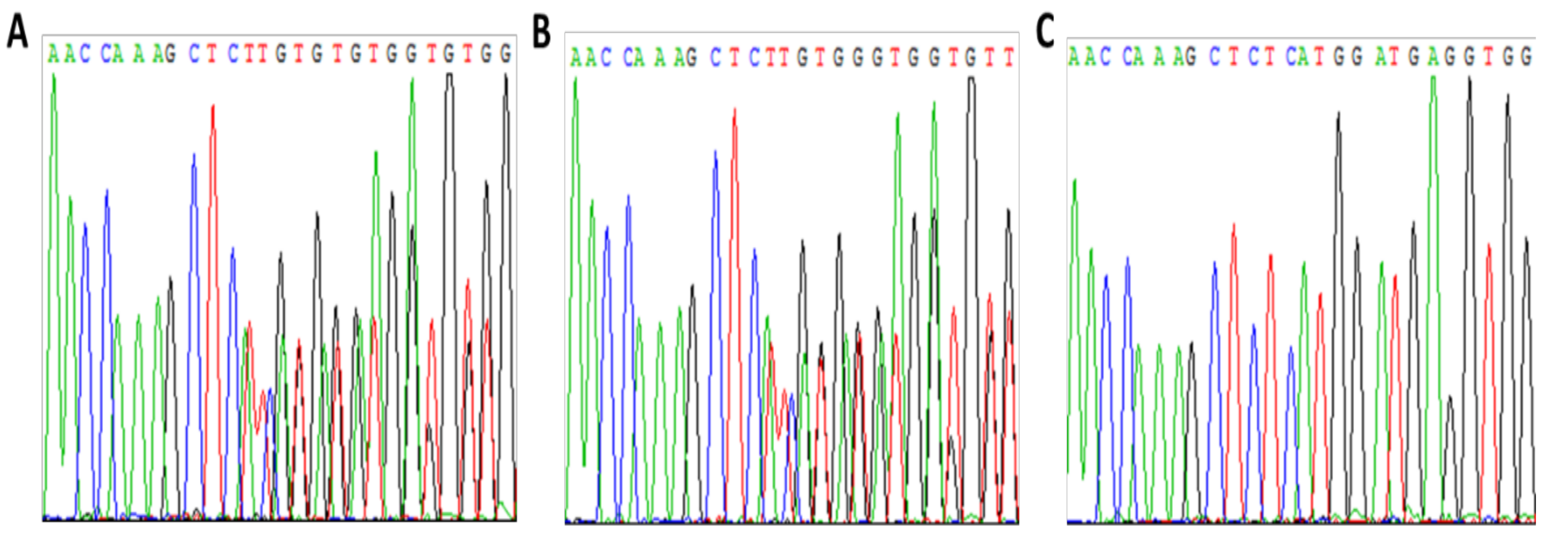
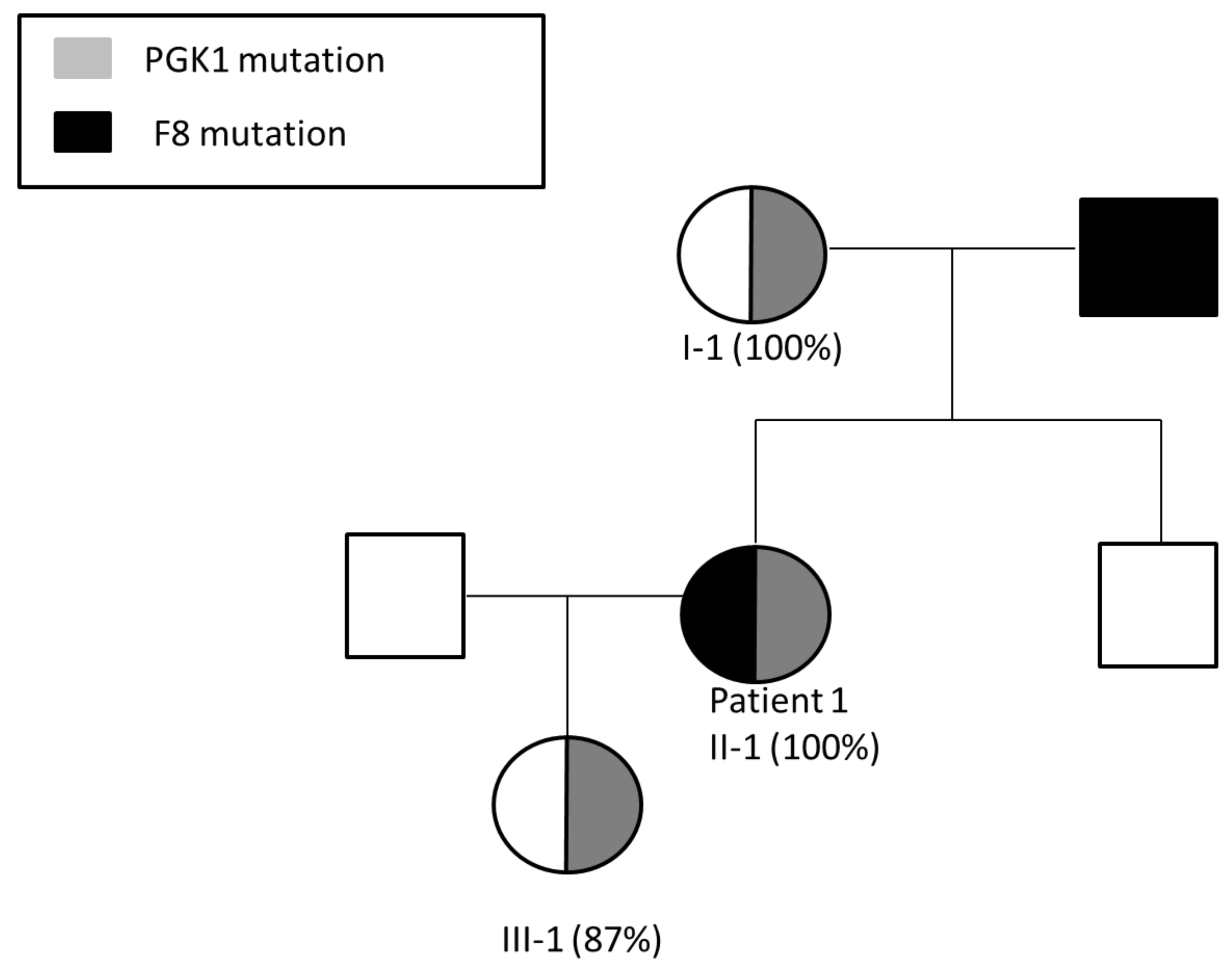
3.1. Patient 1
Pregnancy Planning for Patient 1
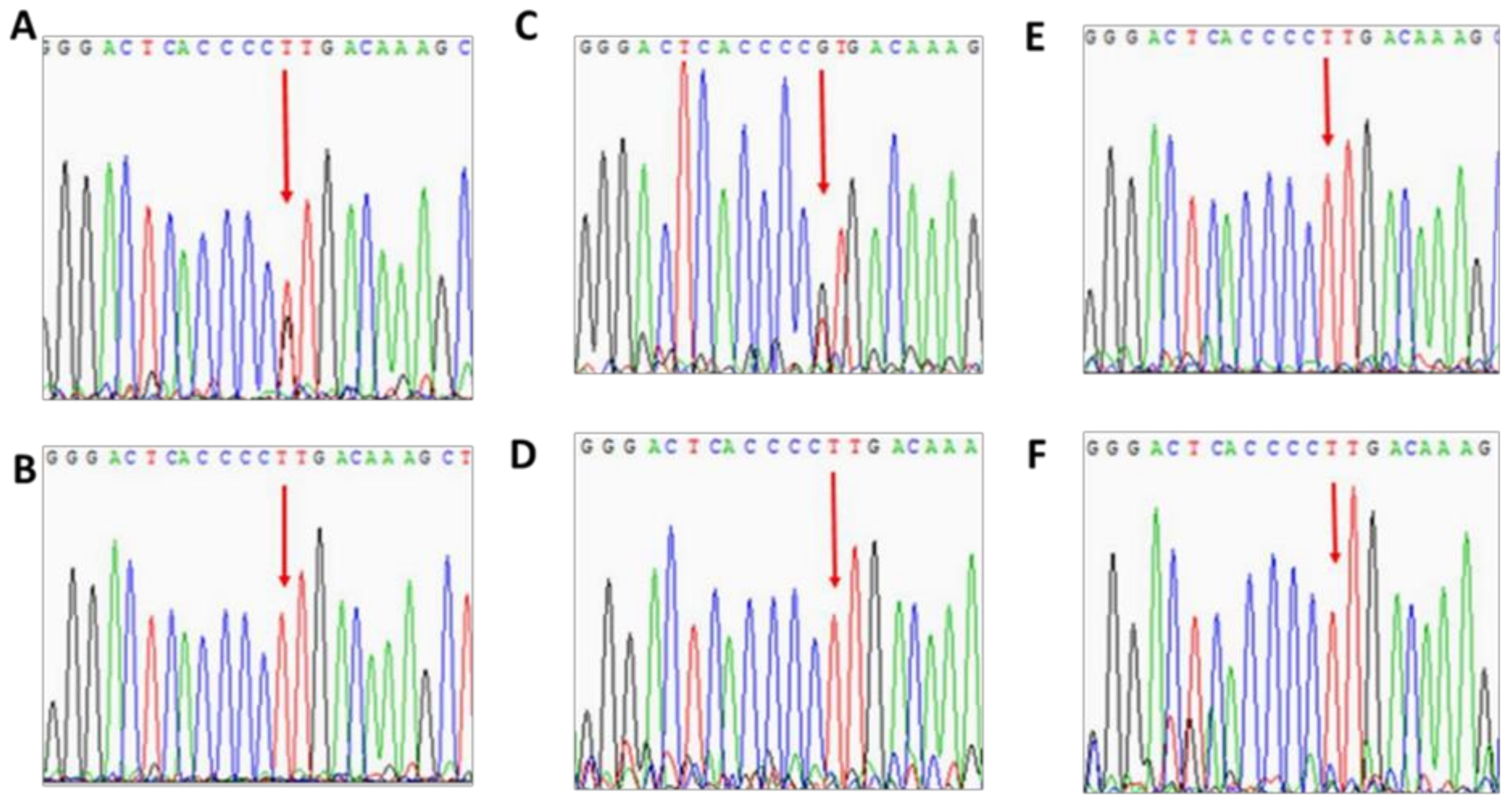
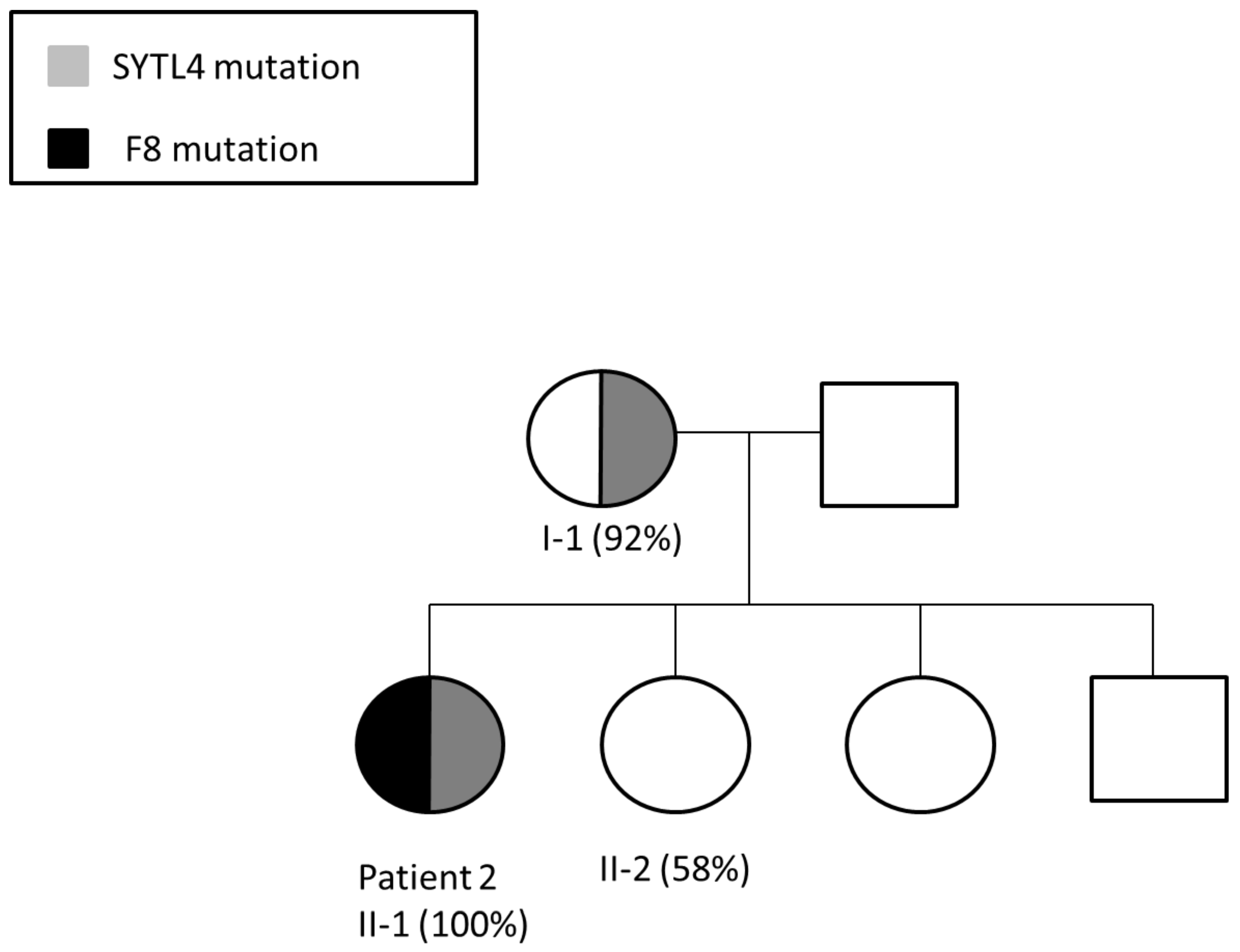
3.2. Patient 2
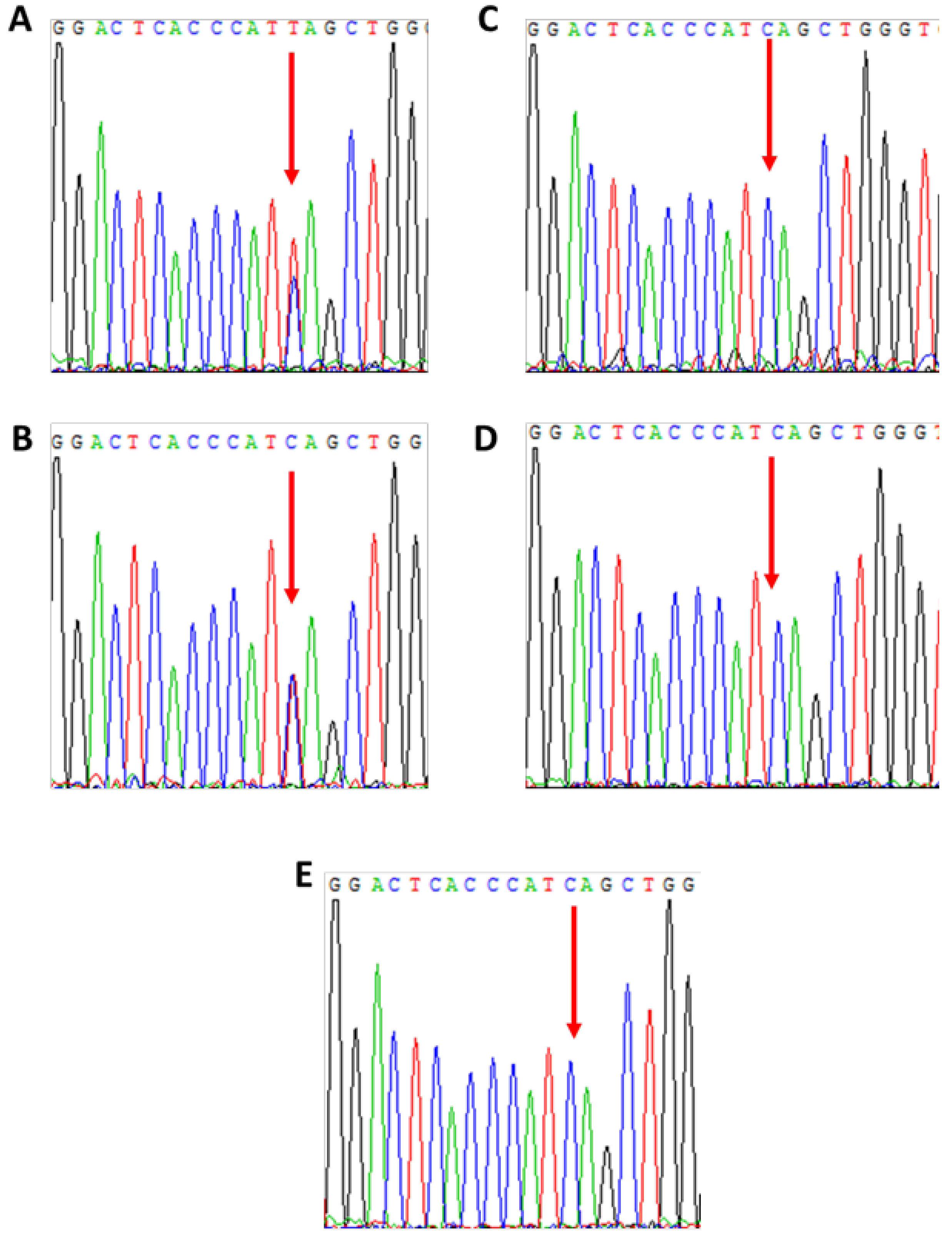
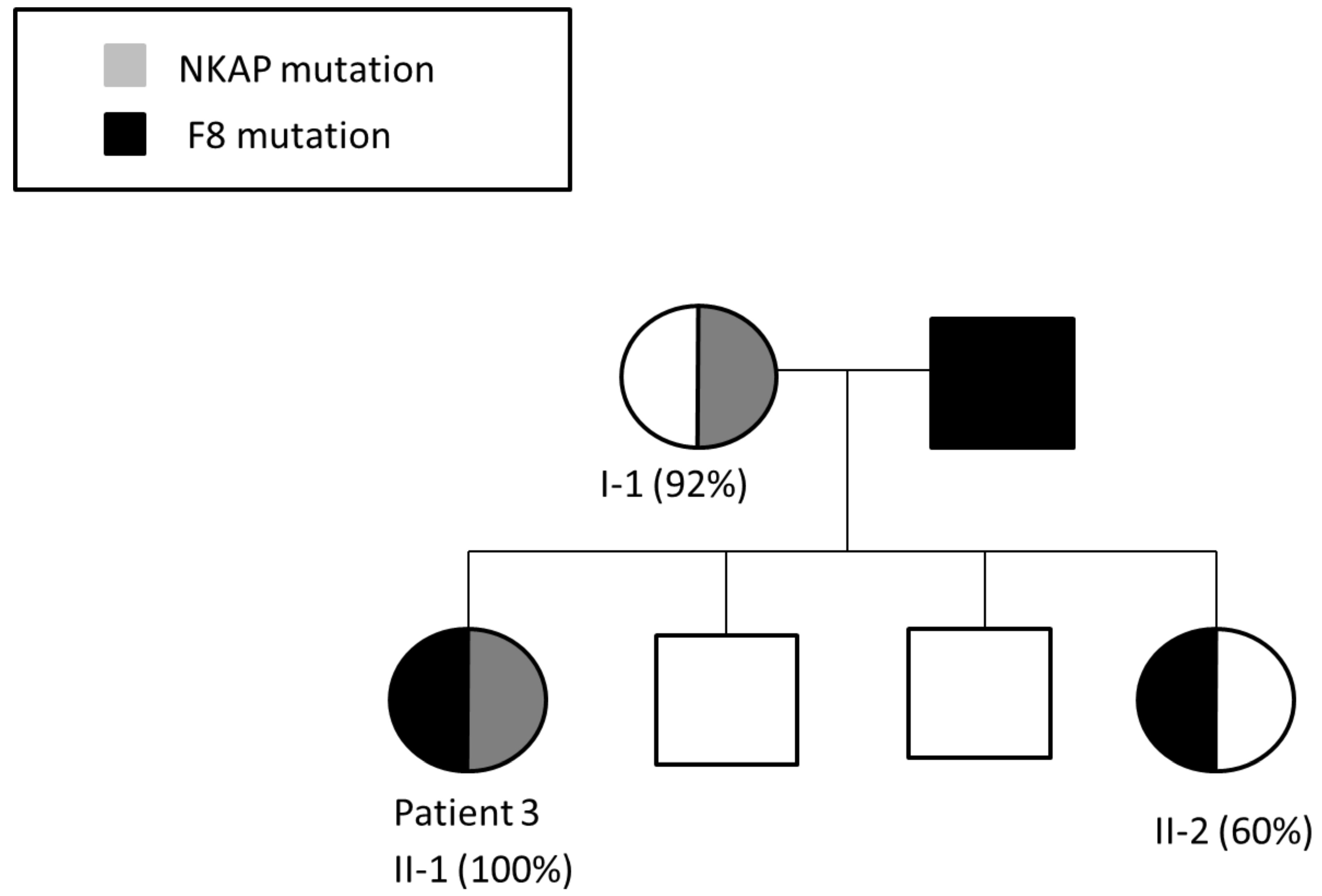
3.3. Patient 3
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heard, E.; Avner, P. Role play in X-inactivation. Hum. Mol. Genet. 1994, 3, 1481–1485. [Google Scholar] [CrossRef]
- Belmont, J.W. Genetic control of X inactivation and processes leading to X inactivation skewing. Am. J. Hum. Genet. 1996, 58, 1101–1108. [Google Scholar] [PubMed]
- Migeon, B.R. Why females are mosaics, X-chromosome inactivation, and sex differences in disease. Gend. Med. 2007, 4, 97–105. [Google Scholar] [CrossRef]
- Sharp, A.J.; Stathaki, E.; Migliavacca, E.; Brahmachary, M.; Montgomery, S.B.; Dupre, Y.; Antonarakis, S.E. DNA methylation profiles of human active and inactive X chromosomes. Genome. Res. 2011, 21, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Favier, R.; Lavergne, J.-M.; Costa, J.-M.; Caron, C.; Mazurier, C.; Viémont, M.; Delpech, M.; Valleix, S. Unbalanced X-chromosome inactivation with a novel FVIII gene mutation resulting in severe hemophilia A in a female. Blood 2000, 96, 4373–4375. [Google Scholar] [CrossRef]
- Pavlova, A.; Brondke, H.; Müsebeck, J.; Pollmann, H.; Srivastava, A.; Oldenburg, J. Molecular mechanisms underlying hemophilia A phenotype in seven females. J. Thromb. Haemost. 2009, 7, 976–982. [Google Scholar] [CrossRef]
- Morey, C.; Avner, P. Genetics and epigenetics of the X chromosome. Ann. N. Y. Acad. Sci. 2010, 1214, E18–E33. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.B.; Rizza, C.R.; Chediak, J.; Mannucci, P.M.; Briet, E.; Ljung, R.; Kasper, C.K.; Essien, E.M.; Green, P.P. Carrier detection in hemophilia A: A cooperative international study. I. The carrier phenotype. Blood 1986, 67, 1554–1559. [Google Scholar] [CrossRef]
- Miller, C.H.; Bean, C.J. Genetic causes of haemophilia in women and girls. Haemophilia 2021, 27, e164–e179. [Google Scholar] [CrossRef]
- Di Michele, D.M.; Gibb, C.; Lefkowitz, J.M.; Ni, Q.; Gerber, L.M.; Ganguly, A. Severe and moderate haemophilia A and B in US females. Haemophilia 2014, 20, e136–e143. [Google Scholar] [CrossRef]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.; Rosenblatt, H.M.; Belmont, J. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992, 51, 1229–1239. [Google Scholar]
- Hatakeyama, C.; Anderson, C.L.; Beever, C.L.; Penaherrera, M.S.; Brown, C.J.; Robinson, W.P. The dynamics of X-inactivation skewing as women age. Clin. Genet. 2004, 66, 327–332. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Li, M.; Li, J.; Li, M.J.; Pan, Z.; Hsu, J.S.; Liu, D.J.; Zhan, X.; Wang, J.; Song, Y.; Sham, P.C. Robust and rapid algorithms facilitate large-scale whole genome sequencing downstream analysis in an integrative framework. Nucleic. Acids. Res. 2017, 45, e75. [Google Scholar] [CrossRef] [PubMed]
- Plenge, R.M.; Hendrich, B.D.; Schwartz, C.; Arena, J.F.; Naumova, A.; Sapienza, C.; Winter, R.M.; Willard, H.F. A promoter mutation in the XIST gene in two unrelated families with skewed X-chromosome inactivation. Nat. Genet. 1997, 17, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Naumova, A.K.; Plenge, R.M.; Bird, L.M.; Leppert, M.; Morgan, K.; Willard, H.F.; Sapienza, C. Heritability of X chromosome--inactivation phenotype in a large family. Am. J. Hum. Genet. 1996, 58, 1111–1119. [Google Scholar] [PubMed]
- Lupski, J.R.; Garcia, C.A.; Zoghbi, H.Y.; Hoffman, E.P.; Fenwick, R.G. Discordance of muscular dystrophy in monozygotic female twins: Evidence supporting asymmetric splitting of the inner cell mass in a manifesting carrier of Duchenne dystrophy. Am. J. Med. Genet. 1991, 40, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Winchester, B.; Young, E.; Geddes, S.; Genet, S.; Hurst, J.; Middelton-Price, H.; Williams, N.; Webb, M.; Habel, A.; Malcolm, S. Female twin with Hunter disease due to nonrandom inactivation of the X-chromosome: A consequence of twinning. Am. J. Med. Genet. 1992, 44, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, E.; Whitaker, J.; Mowery-Rushton, P.; Surti, U.; Lanasa, M.; Hoffman, E.P. Familial skewed X inactivation: A molecular trait associated with high spontaneous-abortion rate maps to Xq28. Am. J. Hum. Genet. 1997, 61, 160–170. [Google Scholar] [CrossRef][Green Version]
- Nguyen, D.K.; Disteche, C.M. High expression of the mammalian X chromosome in brain. Brain Res. 2006, 1126, 46–49. [Google Scholar] [CrossRef]
- Fieremans, N.; van Esch, H.; Holvoet, M.; van Goethem, G.; Devriendt, K.; Rosello, M.; Mayo, S.; Martinez, F.; Jhangiani, S.; Muzny, D.M.; et al. Identification of Intellectual Disability Genes in Female Patients with a Skewed X-Inactivation Pattern. Hum. Mutat. 2016, 37, 804–811. [Google Scholar] [CrossRef]
- Plenge, R.M.; Stevenson, R.A.; Lubs, H.A.; Schwartz, C.E.; Willard, H.F. Skewed X chromosome inactivation is a common feature of X-linked mental retardation disorders. Am. J. Hum. Genet. 2002, 71, 168–173. [Google Scholar] [CrossRef]
- Viggiano, E.; Ergoli, M.; Picillo, E.; Politano, L. Determining the role of skewed X-chromosome inactivation in developing muscle symptoms in carriers of Duchenne muscular dystrophy. Hum. Genet. 2016, 135, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Boonyawat, B.; Dhanraj, S.; al Abbas, F.; Zlateska, B.; Grunenbaum, E.; Roifman, C.M.; Steele, L.; Meyn, S.; Blanchette, V.; Scherer, S.W.; et al. Combined de-novo mutation and non-random X-chromosome inactivation causing Wiskott-Aldrich syndrome in a female with thrombocytopenia. J. Clin. Immunol. 2013, 33, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiao, Q.Z.; Chen, Y.M.; Yi, S.; Liu, D.; Liu, Y.H.; Zhang, C.-M.; Wei, X.-F.; Zhou, Y.-Q.; Zhong, X.-M.; et al. DNA hypermethylation and X chromosome inactivation are major determinants of phenotypic variation in women heterozygous for G6PD mutations. Blood Cells Mol. Dis. 2014, 53, 241–245. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Schaaf, C.P.; Fang, P.; Roeder, E.; Kimonis, V.E.; Church, J.A.; Patel, A.; Cheung, S.W. Clinical characterization of int22h1/int22h2-mediated Xq28 duplication/deletion: New cases and literature review. BMC Med. Genet. 2015, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Garagiola, I.; Mortarino, M.; Siboni, S.M.; Boscarino, M.; Mancuso, M.E.; Biganzoli, M.; Santagostino, E.; Peyvandi, F. X Chromosome inactivation: A modifier of factor VIII and IX plasma levels and bleeding phenotype in haemophilia carriers. Eur. J. Hum. Genet. 2021, 29, 241–249. [Google Scholar] [CrossRef]
- Orstavik, K.H.; Scheibel, E.; Ingerslev, J.; Schwartz, M. Absence of correlation between X chromosome inactivation pattern and plasma concentration of factor VIII and factor IX in carriers of haemophilia A and B. Thromb Haemost. 2000, 83, 433–437. [Google Scholar]
- Beutler, E. PGK deficiency. Br. J. Haematol. 2007, 136, 3–11. [Google Scholar] [CrossRef]
- Keitt, A.S. Pyruvate kinase deficiency and related disorders of red cell glycolysis. Am. J. Med. 1966, 41, 762–785. [Google Scholar] [CrossRef]
- McCarrey, J.R.; Thomas, K. Human testis-specific PGK gene lacks introns and possesses characteristics of a processed gene. Nature 1987, 326, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Matsumaru, S.; Oguni, H.; Ogura, H.; Shimojima, K.; Nagata, S.; Kanno, H.; Yamamoto, T. A novel PGK1 mutation associated with neurological dysfunction and the absence of episodes of hemolytic anemia or myoglobinuria. Intractable Rare Dis. Res. 2017, 6, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Rafi, S.K.; Fernández-Jaén, A.; Álvarez, S.; Nadeau, O.W.; Butler, M.G. High functioning autism with missense mutations in Synaptotagmin-Like Protein 4 (SYTL4) and Transmembrane Protein 187 (TMEM187) genes: SYTL4- protein modeling, protein-protein interaction, expression profiling and microRNA studies. Int. J. Mol. Sci. 2019, 20, 3358. [Google Scholar] [CrossRef]
- Wang, H.; Ishizaki, R.; Xu, J.; Kasai, K.; Kobayashi, E.; Gomi, H.; Izumi, T. The Rab27a effector exophilin7 promotes fusion of secretory granules that have not been docked to the plasma membrane. Mol. Biol. Cell. 2013, 24, 319–330. [Google Scholar] [CrossRef]
- Fiordaliso, S.K.; Iwata-Otsubo, A.; Ritter, A.L.; Quesnel-Vallières, M.; Fujiki, K.; Nishi, E.; Hancarova, M.; Miyake, N.; Morton, J.E.V.; Lee, S.; et al. Missense mutations in NKAP cause a disorder of transcriptional regulation characterized by Marfanoid habitus and cognitive impairment. Am. J. Hum. Genet. 2019, 105, 987–995. [Google Scholar] [CrossRef]
- Pajerowski, A.G.; Shapiro, M.J.; Gwin, K.; Sundsbak, R.; Nelson-Holte, M.; Medina, K.; Shapiro, V.S. Adult hematopoietic stem cells require NKAP for maintenance and survival. Blood 2010, 116, 2684–2693. [Google Scholar] [CrossRef] [PubMed]
- Janczar, S.; Babol-Pokora, K.; Jatczak-Pawlik, I.; Taha, J.; Klukowska, A.; Laguna, P.; Windyga, J.; Odnoczko, E.; Zdziarska, J.; Iwaniec, T.; et al. Six molecular patterns leading to hemophilia A phenotype in 18 females from Poland. Thromb Res. 2020, 193, 9–14, 40. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, S.; Barg, A.A.; Dardik, R.; Luboshitz, J.; Bashari, D.; Avishai, E.; Kenet, G. Women with hemophilia: Case series of reproductive choices and review of literature. TH Open 2021, 5, e183–e187. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dardik, R.; Avishai, E.; Lalezari, S.; Barg, A.A.; Levy-Mendelovich, S.; Budnik, I.; Barel, O.; Khavkin, Y.; Kenet, G.; Livnat, T. Molecular Mechanisms of Skewed X-Chromosome Inactivation in Female Hemophilia Patients—Lessons from Wide Genome Analyses. Int. J. Mol. Sci. 2021, 22, 9074. https://doi.org/10.3390/ijms22169074
Dardik R, Avishai E, Lalezari S, Barg AA, Levy-Mendelovich S, Budnik I, Barel O, Khavkin Y, Kenet G, Livnat T. Molecular Mechanisms of Skewed X-Chromosome Inactivation in Female Hemophilia Patients—Lessons from Wide Genome Analyses. International Journal of Molecular Sciences. 2021; 22(16):9074. https://doi.org/10.3390/ijms22169074
Chicago/Turabian StyleDardik, Rima, Einat Avishai, Shadan Lalezari, Assaf A. Barg, Sarina Levy-Mendelovich, Ivan Budnik, Ortal Barel, Yulia Khavkin, Gili Kenet, and Tami Livnat. 2021. "Molecular Mechanisms of Skewed X-Chromosome Inactivation in Female Hemophilia Patients—Lessons from Wide Genome Analyses" International Journal of Molecular Sciences 22, no. 16: 9074. https://doi.org/10.3390/ijms22169074
APA StyleDardik, R., Avishai, E., Lalezari, S., Barg, A. A., Levy-Mendelovich, S., Budnik, I., Barel, O., Khavkin, Y., Kenet, G., & Livnat, T. (2021). Molecular Mechanisms of Skewed X-Chromosome Inactivation in Female Hemophilia Patients—Lessons from Wide Genome Analyses. International Journal of Molecular Sciences, 22(16), 9074. https://doi.org/10.3390/ijms22169074