
Iterated Virtual Screening-Assisted Antiviral and Enzyme Inhibition Assays Reveal the Discovery of Novel Promising Anti-SARS-CoV-2 with Dual Activity


 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
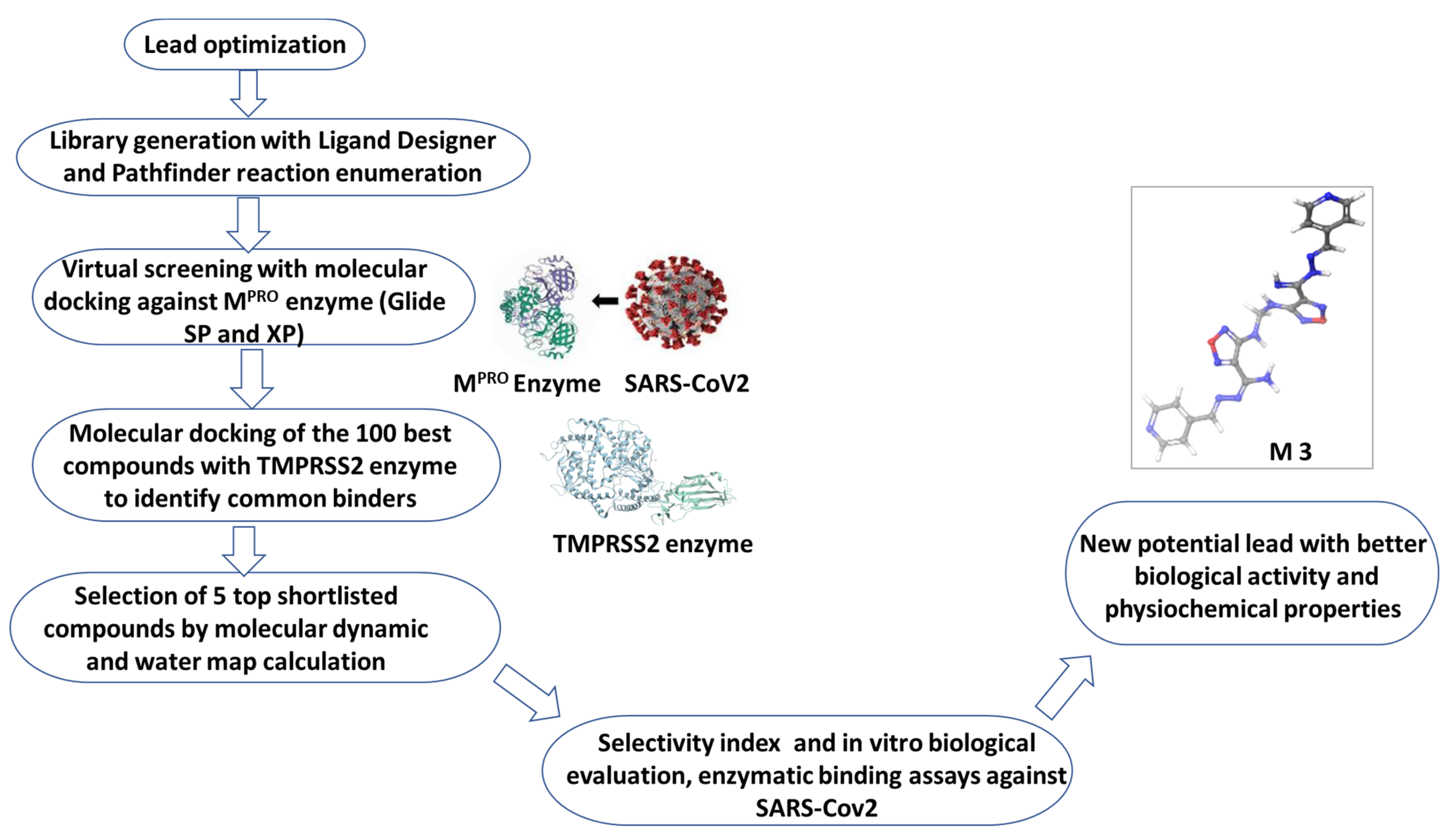
2.1. Rational Lead Discovery of Potential Anti-SARS-CoV-2
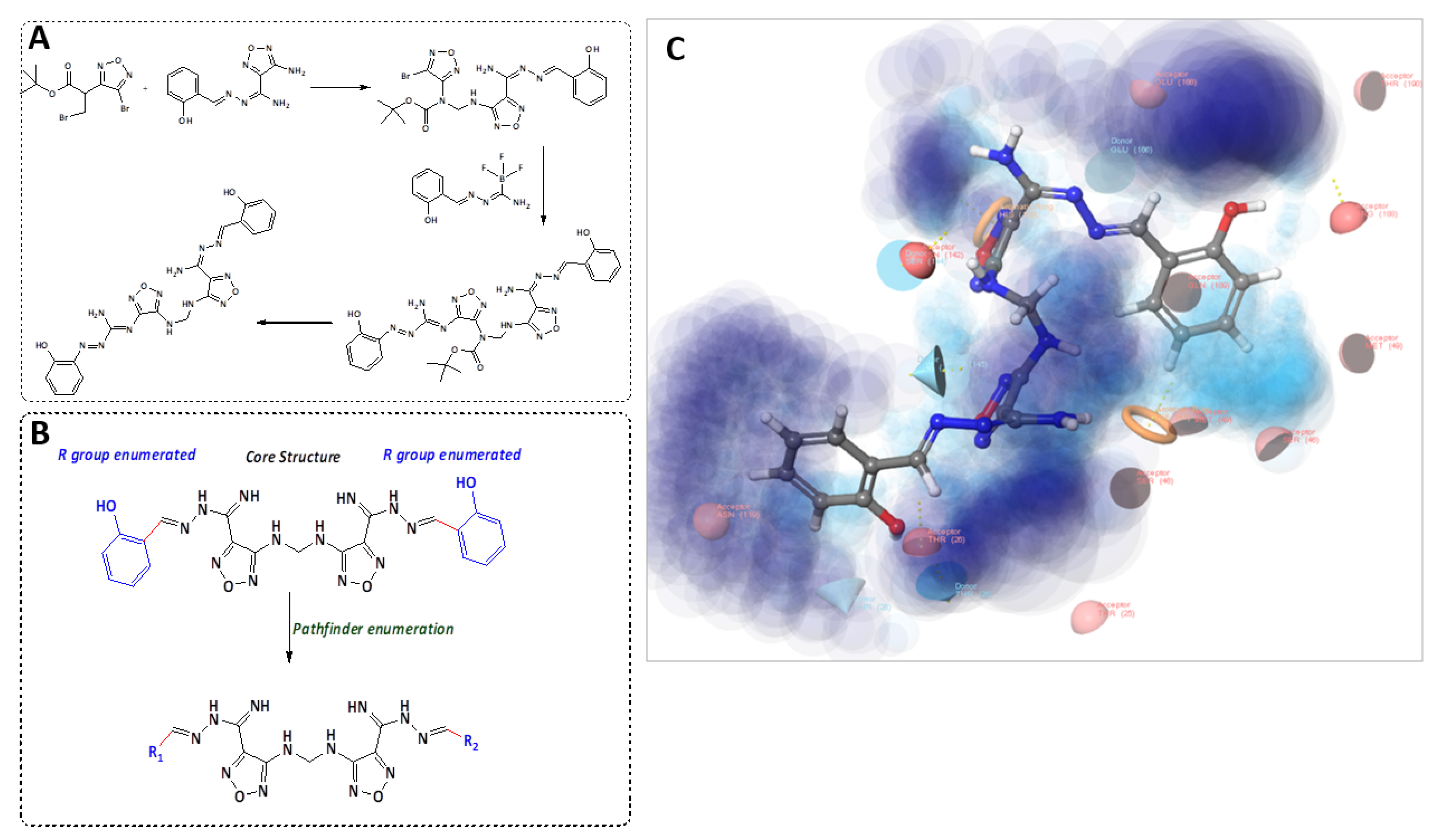
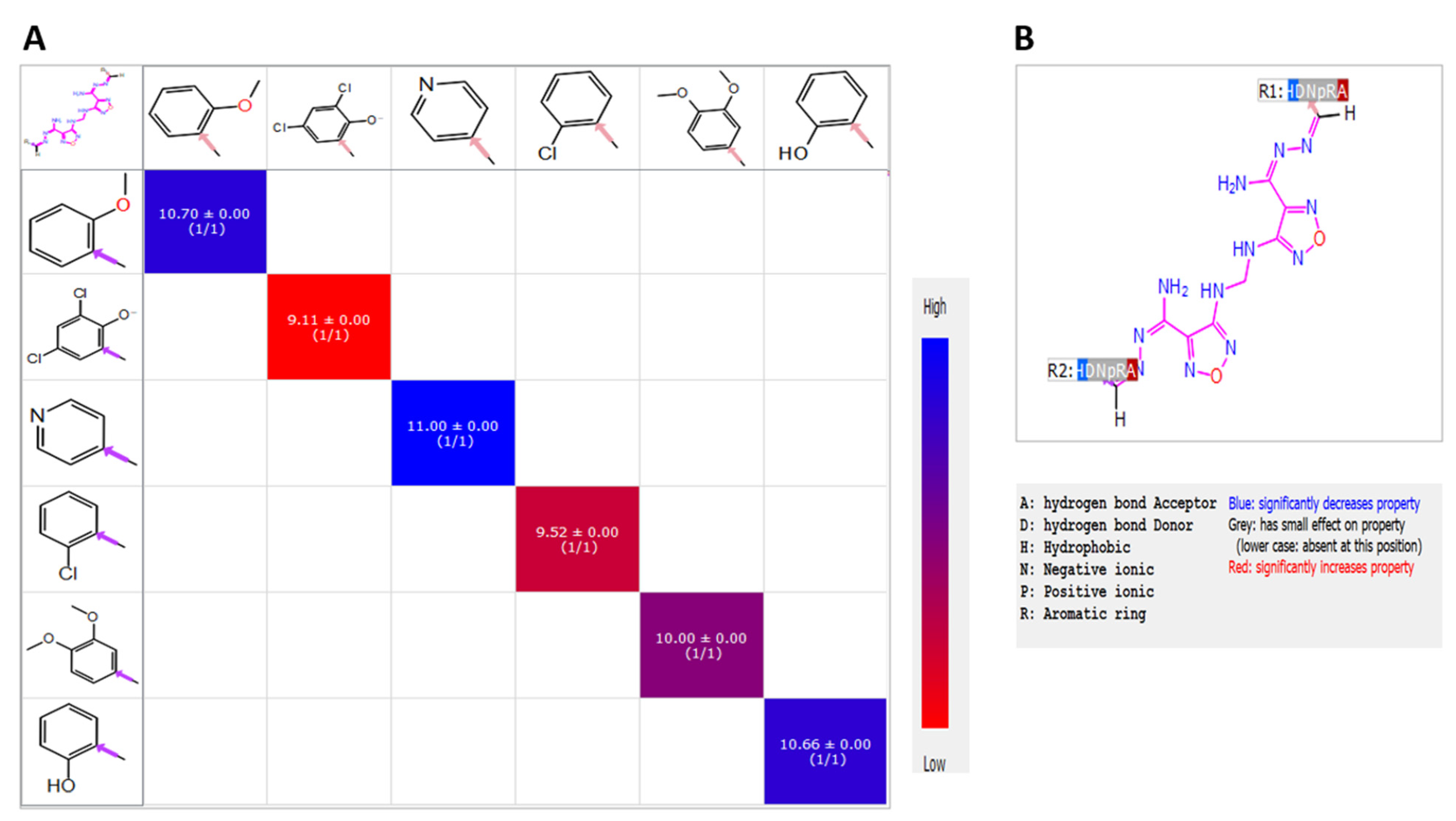
2.2. Candidate Election by Enumerated Structure Ligand Designer
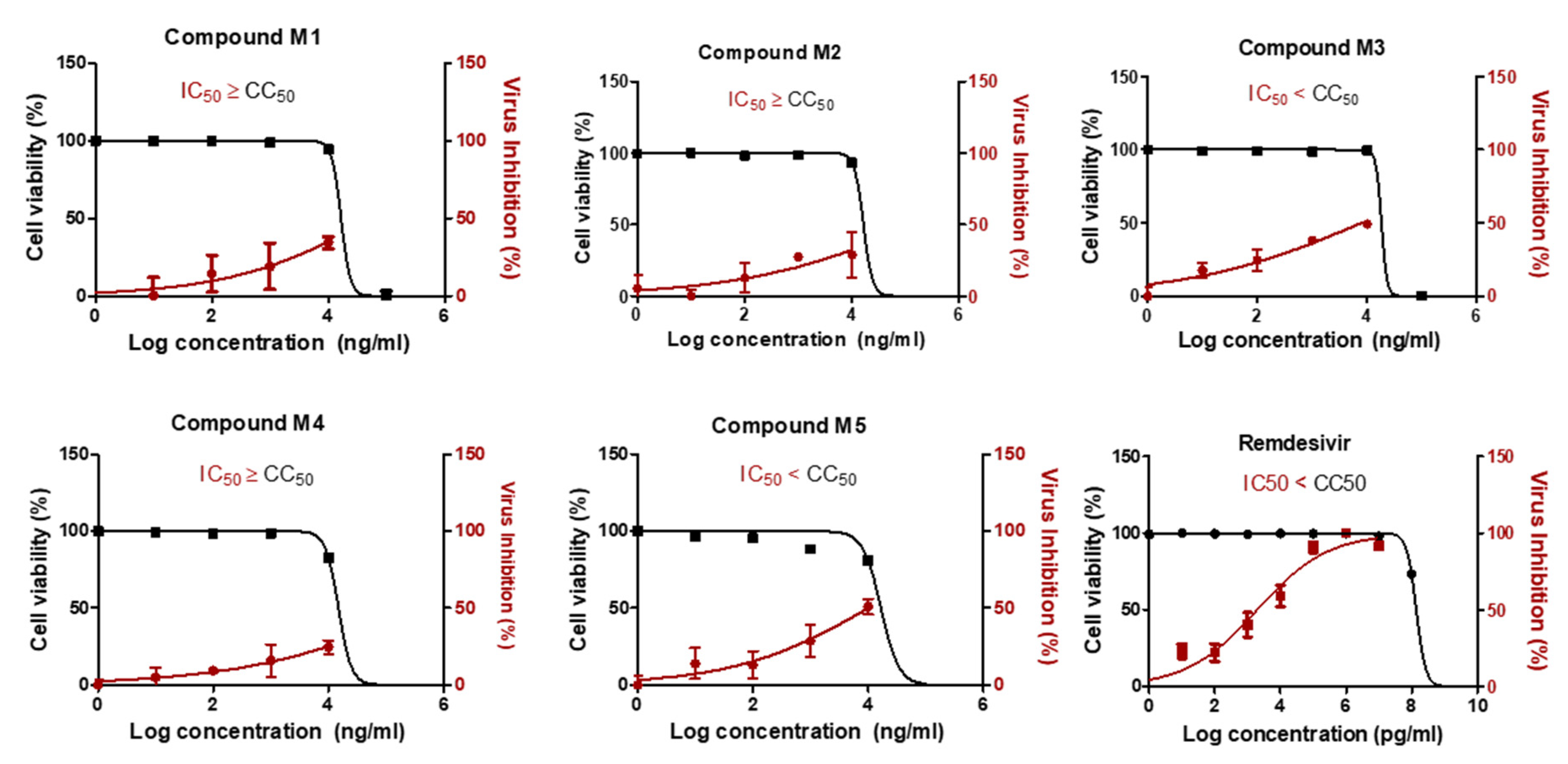
2.3. Two Candidate Compounds (M3 and M5) Showed Promising Anti-SARS-CoV-2 Activity In Vitro
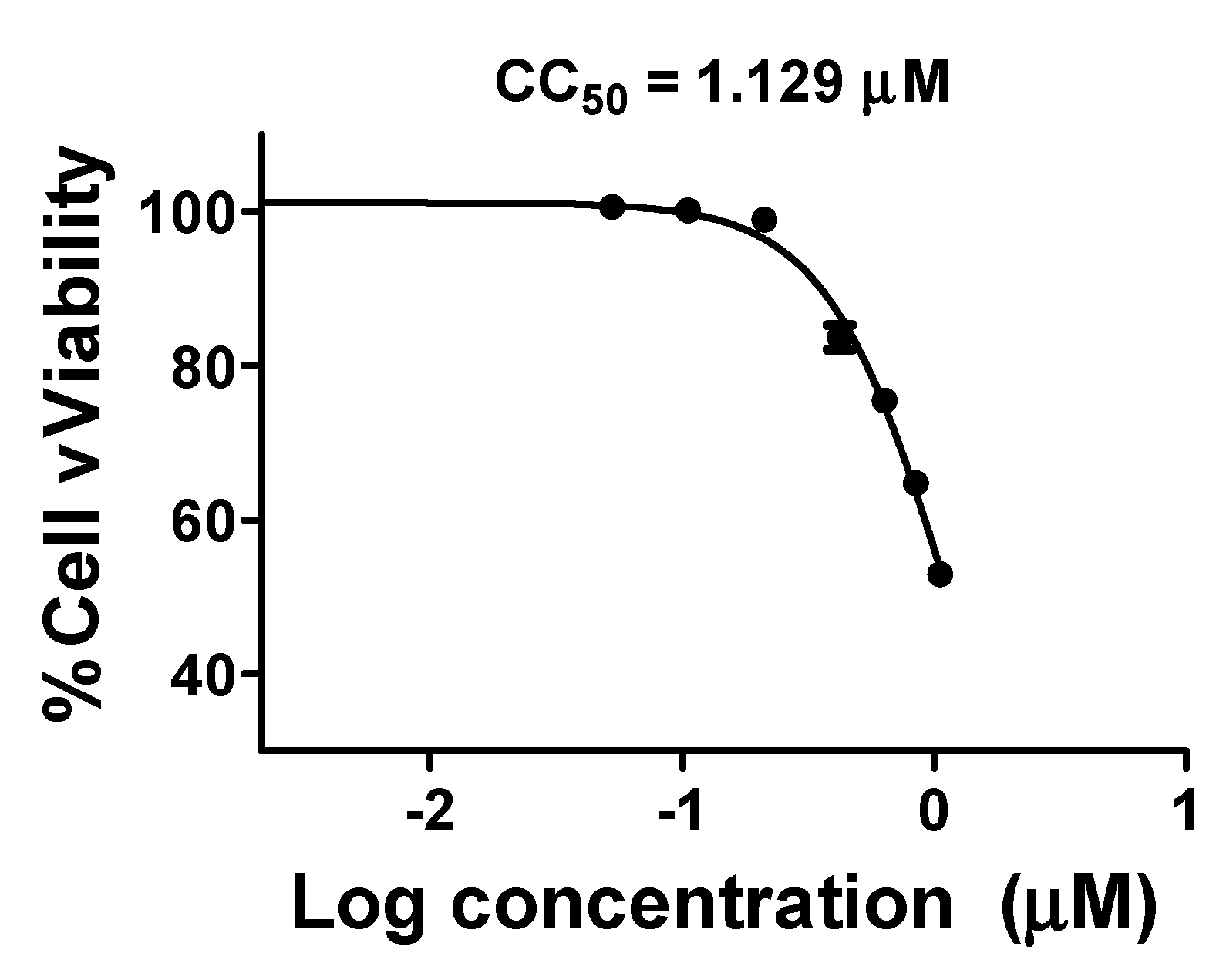
2.4. Compound M3 Possesses Dual Inhibition Activity against SARS-CoV-2 by Targeting Both the Viral and Human Proteases
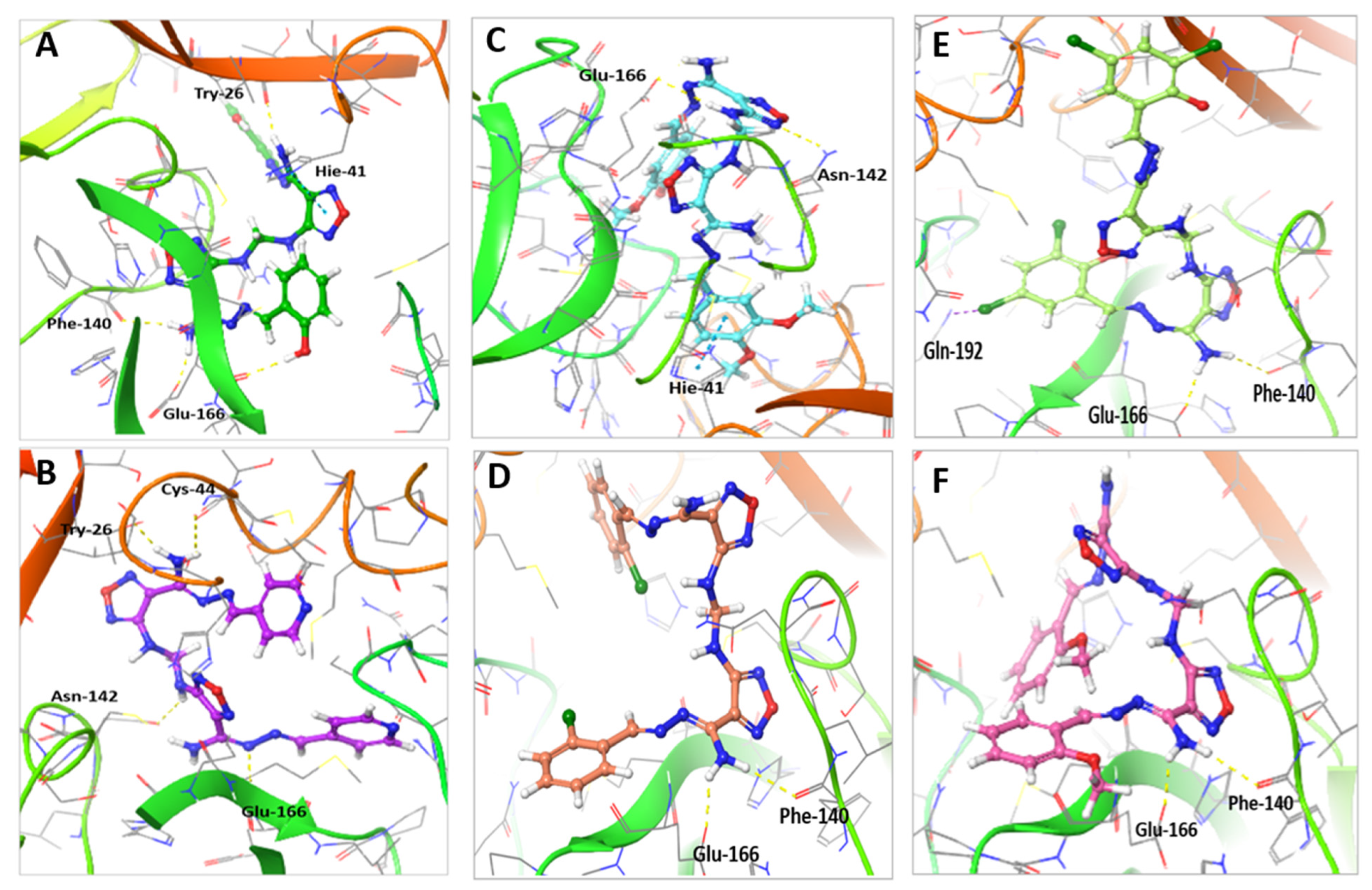
2.5. Molecular Docking Indicated the Binding Efficiency of M3 with SARS-CoV-2 Mpro and TMPRSS2 Protease
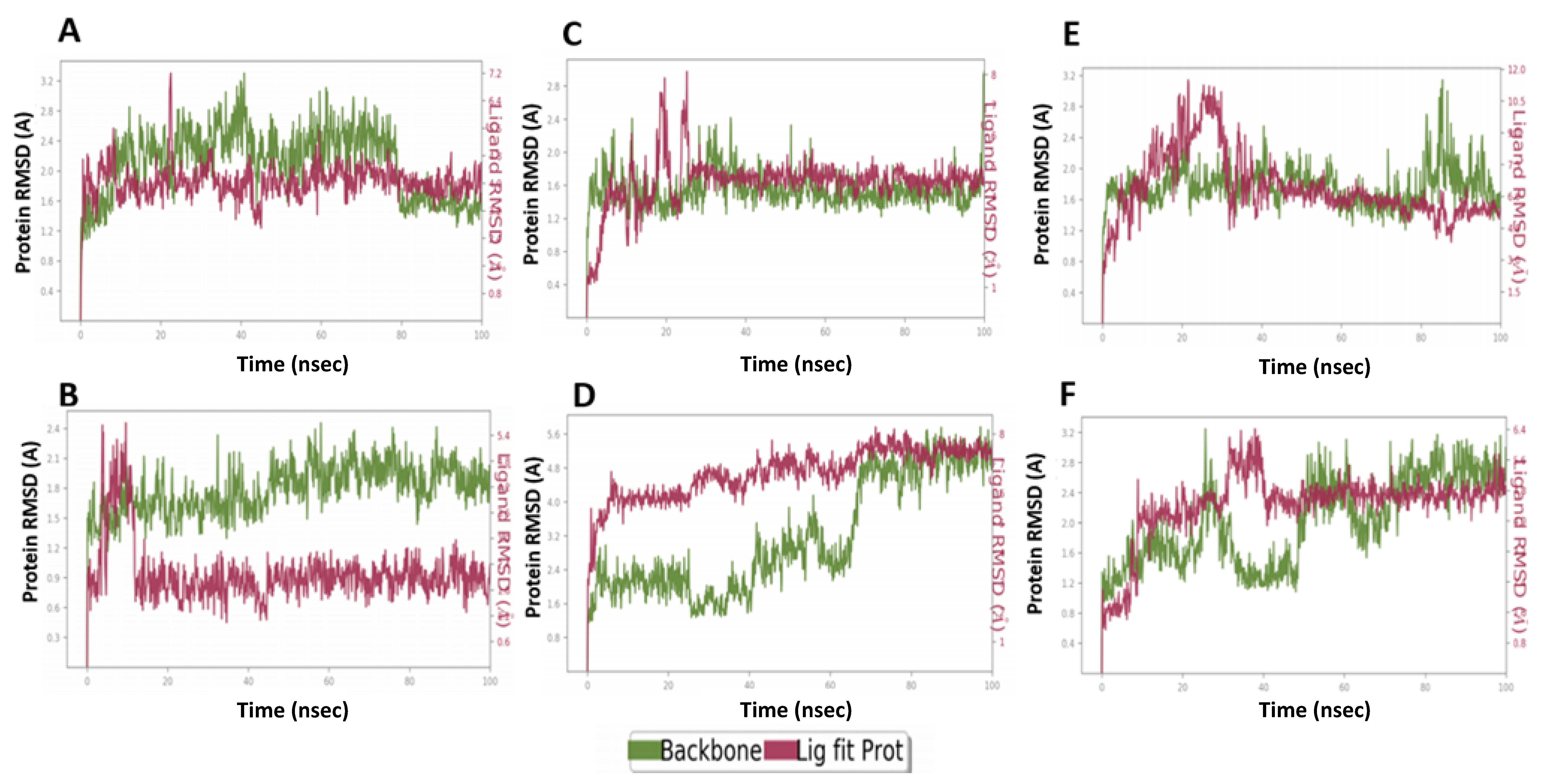
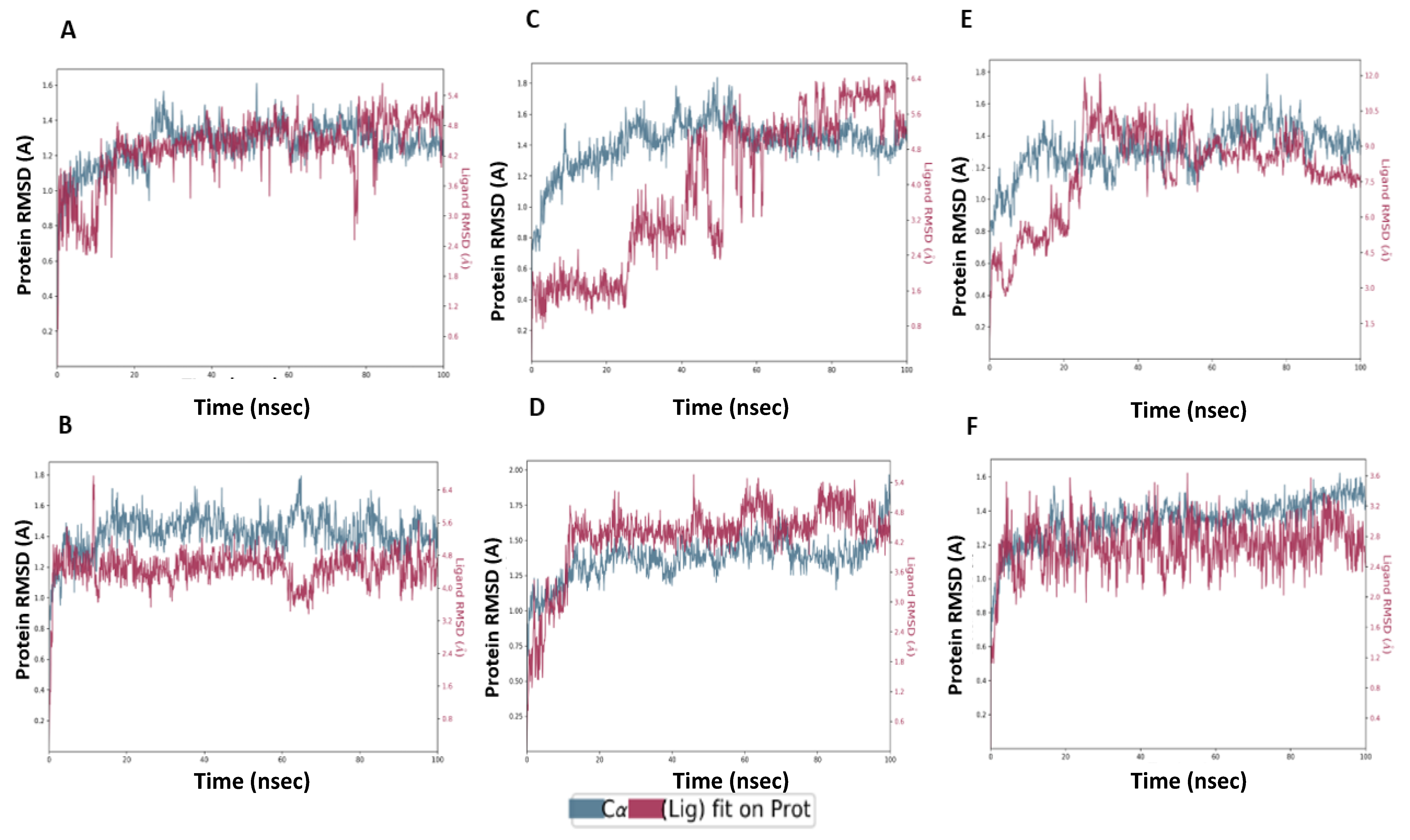
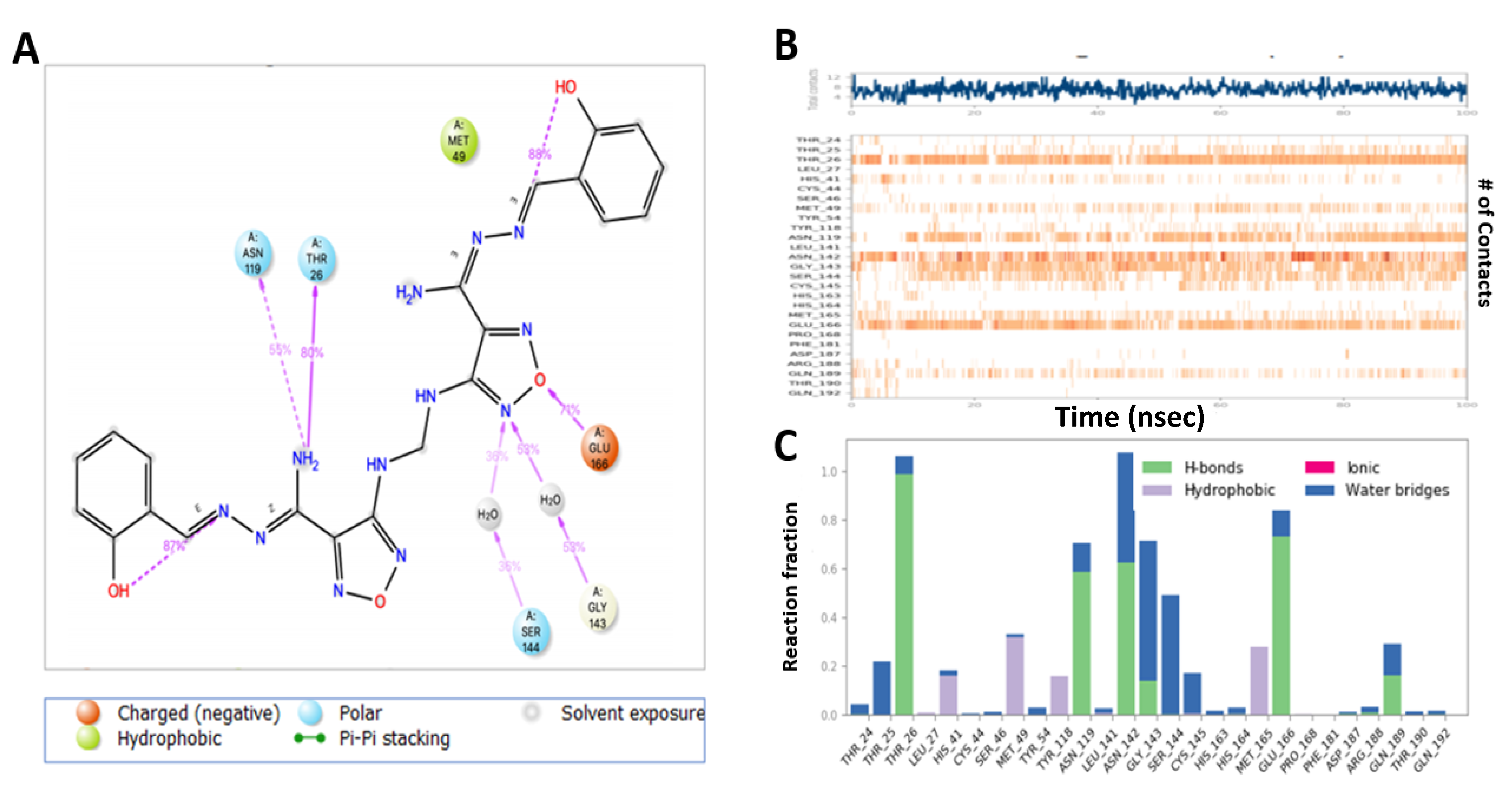
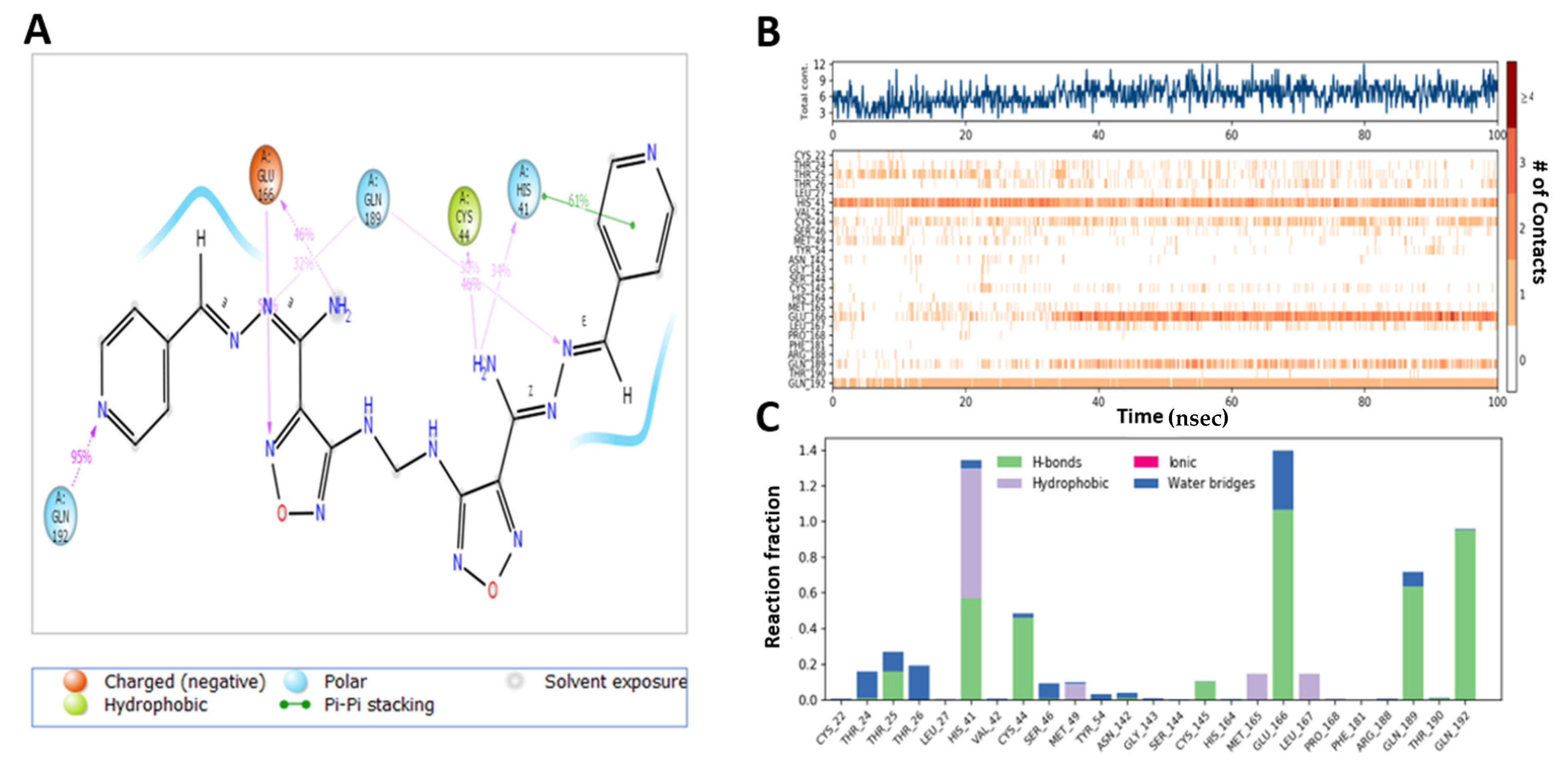
2.6. MD Simulation Confirmed the Stability of M3 Complex with the Target Enzymes, SARS-CoV-2 Mpro and Human TMPRSS2
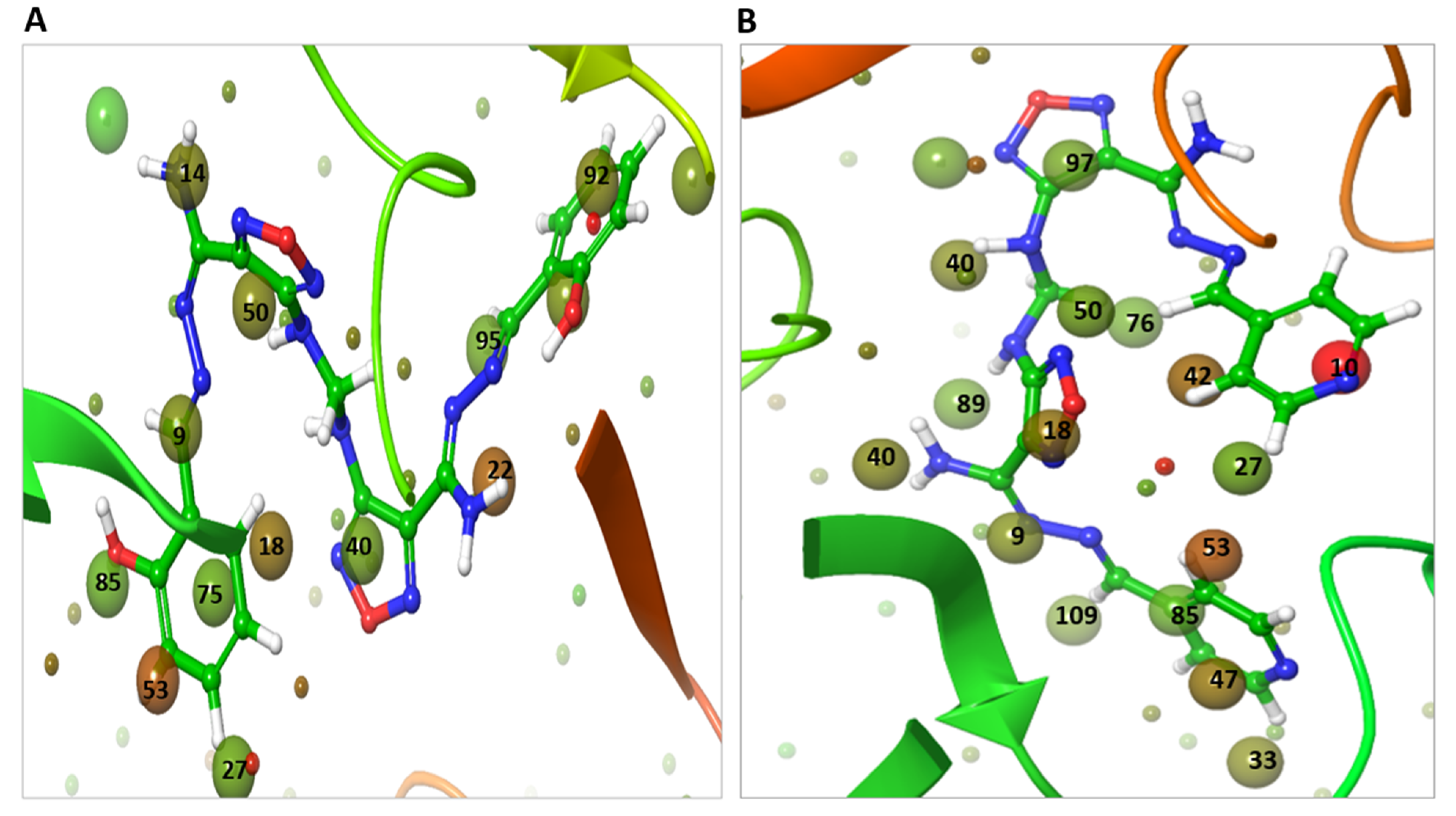
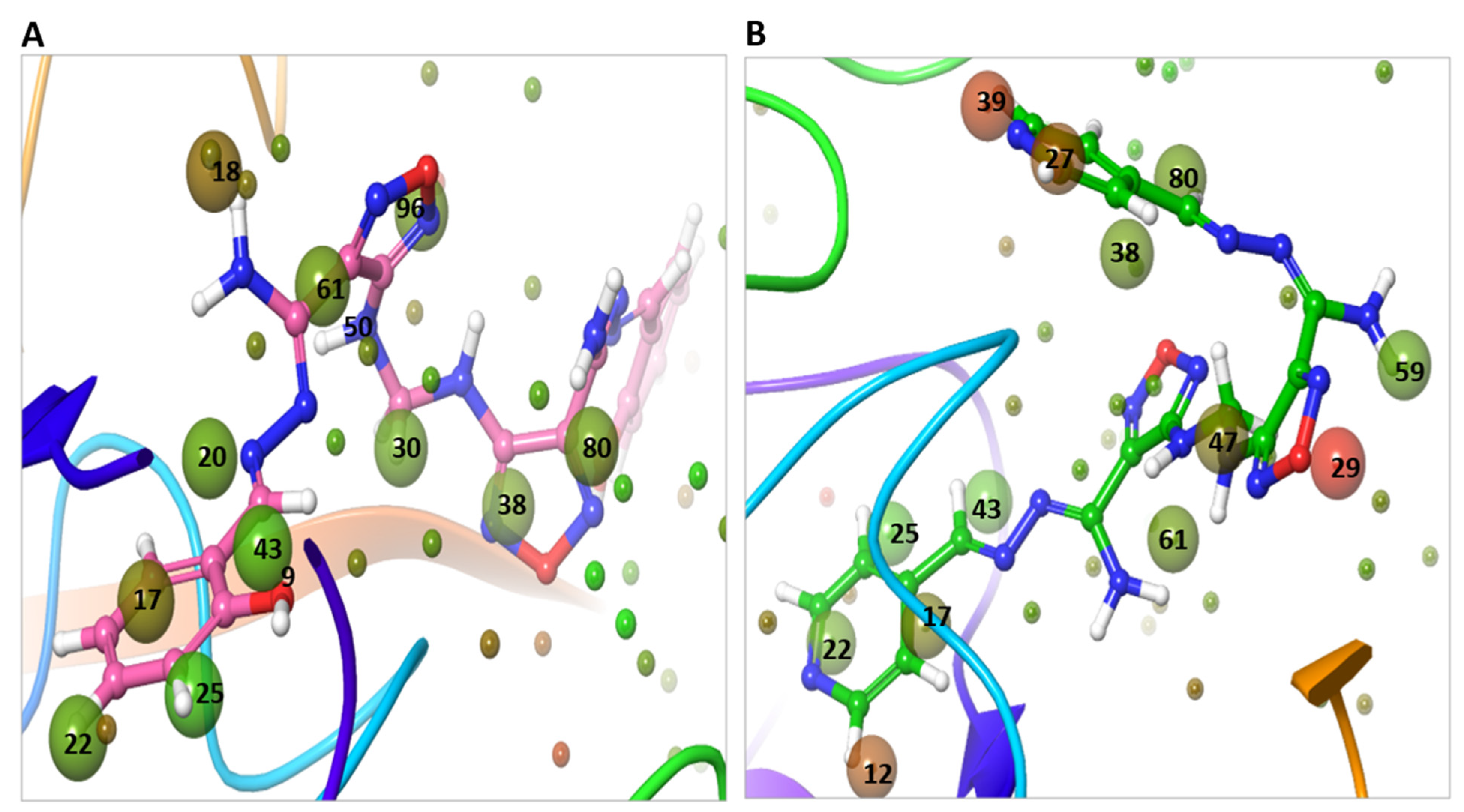
2.7. WaterMap Analysis Showed the Pronounced Efficacy of M3
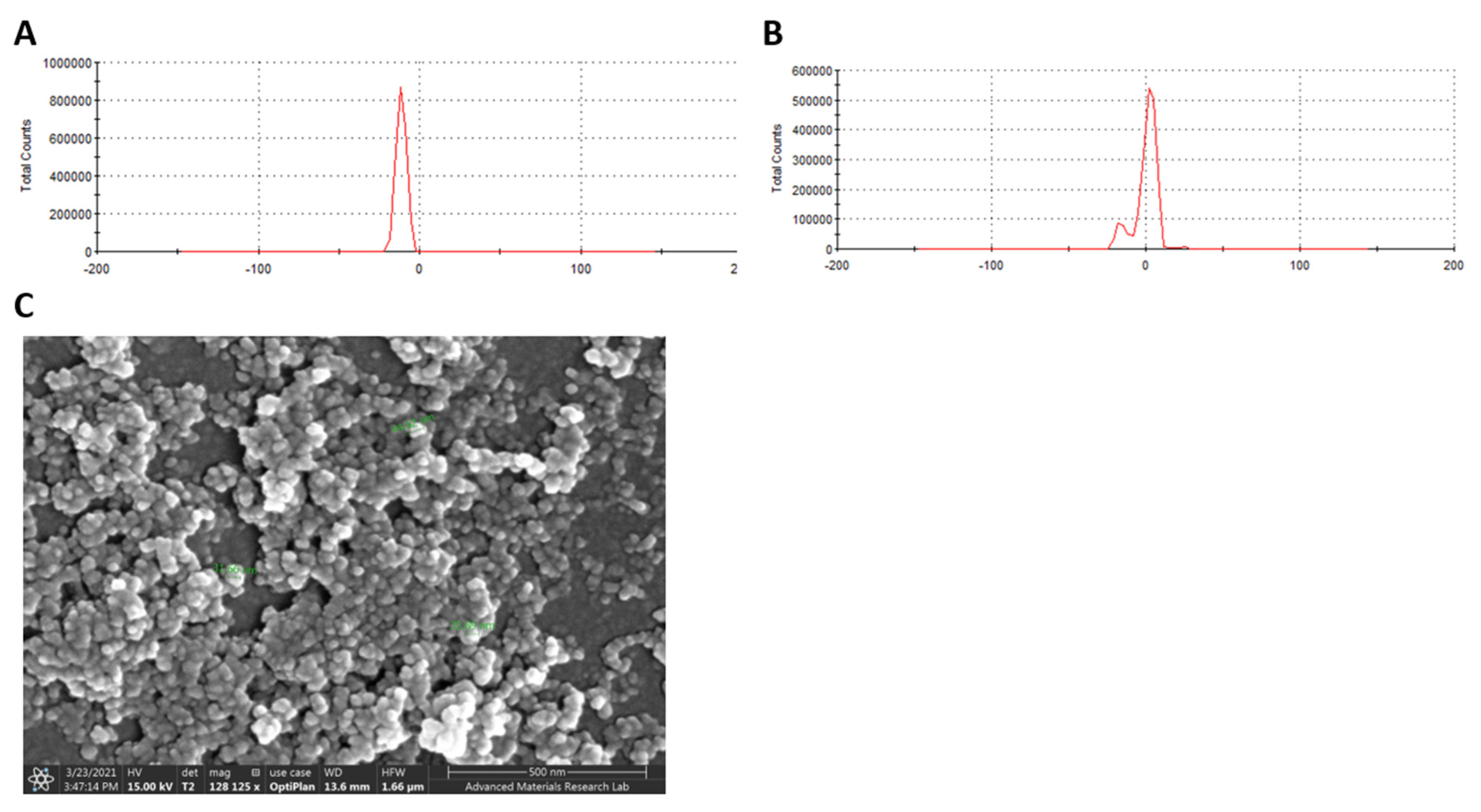
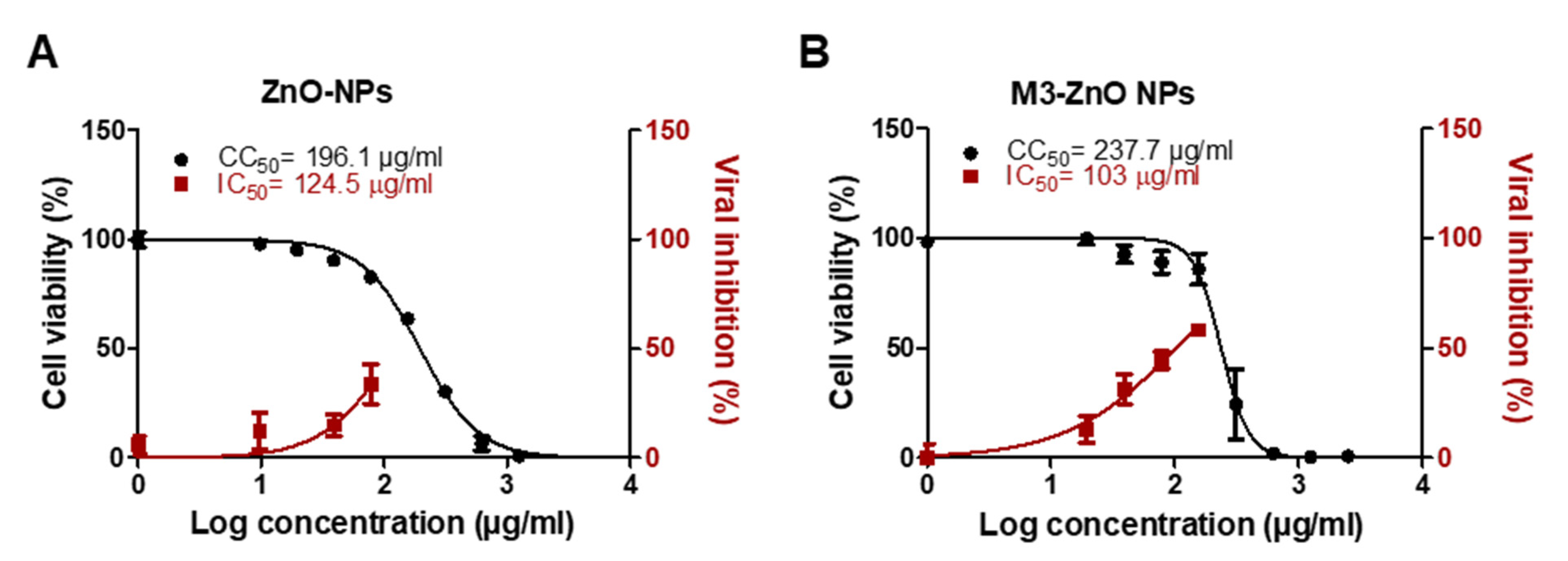
2.8. Adsorption of M3 Compound on ZnO NPs Enhanced the Efficiency against SARS-CoV-2
3. Discussion
4. Conclusions
5. Experimental Section
5.1. Material
5.2. Computational Studies
5.2.1. Protein Preparation
5.2.2. Ligand Preparation
5.2.3. Grid Generation
5.2.4. Molecular Docking
5.2.5. Induced Docking Fit (IDF)
5.2.6. Molecular Dynamics Simulation
5.2.7. WaterMap Analysis
5.2.8. PathFinder R-Group Enumeration
5.2.9. Ligand Designer
5.2.10. R-Group Analysis
5.3. Chemistry
5.4. In Vitro Evaluation of Anti-SARS-CoV-2 Activity
5.5. Cell Viability Assay
5.6. Main Protease (Mpro) Assay
5.7. Furin Protease Assay
5.8. TMPRSS2 Fluorogenic Assay
5.9. Preparation of Compound-Loaded ZnO Nanoparticles
5.10. Characterization of Compound-ZnO Nanoparticles
5.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Forni, G.; Mantovani, A. COVID-19 vaccines: Where we stand and challenges ahead. Cell Death Differ. 2021, 28, 626–639. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidis, N.C.; López-Cortés, A.; González, E.V.; Grimaldos, A.B.; Prado, E.O. SARS-CoV-2 vaccines strategies: A comprehensive review of phase 3 candidates. NPJ Vaccines 2021, 6, 1–17. [Google Scholar] [CrossRef]
- Kunzelmann, K. Getting hands on a drug for Covid-19: Inhaled and intranasal niclosamide. Lancet Reg. Health Eur. 2021, 4, 100094. [Google Scholar] [CrossRef] [PubMed]
- Sacco, M.D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J.A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 2020, 6, eabe0751. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Good, S.S.; Westover, J.; Jung, K.H.; Zhou, X.J.; Moussa, A.; La Colla, P.; Collu, G.; Canard, B.; Sommadossi, J.P. AT-527, a double prodrug of a guanosine nucleotide analog, is a potent inhibitor of SARS-CoV-2 in vitro and a promising oral antiviral for treatment of COVID-19. Antimicrob. Agents Chemother. 2021, 65, e02479-20. [Google Scholar] [CrossRef]
- Driouich, J.S.; Cochin, M.; Lingas, G.; Moureau, G.; Touret, F.; Petit, P.R.; Piorkowski, G.; Barthélémy, K.; Laprie, C.; Coutard, B.; et al. Favipiravir antiviral efficacy against SARS-CoV-2 in a hamster model. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Wahl, A.; Gralinski, L.E.; Johnson, C.E.; Yao, W.; Kovarova, M.; Dinnon, K.H.; Liu, H.; Madden, V.J.; Krzystek, H.M.; De, C.; et al. SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature 2021, 591, 451–457. [Google Scholar] [CrossRef]
- Vandyck, K.; Deval, J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Curr. Opin. Virol. 2021, 49, 36–40. [Google Scholar] [CrossRef]
- Fontanet, A.; Autran, B.; Lina, B.; Kieny, M.P.; Karim, S.S.A.; Sridhar, D. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 2021, 397, 952–954. [Google Scholar] [CrossRef]
- Elseginy, S.A.; Fayed, B.; Hamdy, R.; Mahrous, N.; Mostafa, A.; Almehdi, A.M.; Soliman, S.S. Promising anti-SARS-CoV-2 drugs by effective dual targeting against the viral and host proteases. Bioorganic Med. Chem. Lett. 2021, 43, 128099. [Google Scholar] [CrossRef]
- Jiang, J.; Pi, J.; Cai, J. The advancing of zinc oxide nanoparticles for biomedical applications. Bioinorg. Chem. Appl. 2018, 2018, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, M.; Patel, K.R.; Danish, H.H.; Lawson, D.H.; Khan, M.K. BRAF inhibitors and radiotherapy for melanoma brain metastases: Potential advantages and disadvantages of combination therapy. OncoTargets Ther. 2016, 9, 7149. [Google Scholar] [CrossRef] [Green Version]
- Gradman, A.H.; Basile, J.N.; Carter, B.L.; Bakris, G.L. Combination therapy in hypertension. J. Am. Soc. Hypertens. 2010, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, X.; Wu, X.; Zhu, C. A dual-inhibitor system for the effective antifibrillation of Aβ40 peptides by biodegradable EGCG–Fe(iii)/PVP nanoparticles. J. Mater. Chem. B 2019, 7, 1292–1299. [Google Scholar] [CrossRef]
- Chen, K.; Tan, Z.; He, M.; Li, J.; Tang, S.; Hewlett, I.; Yu, F.; Jin, Y.; Yang, M. Structure-activity relationships (SAR) research of thiourea derivatives as dual inhibitors targeting both HIV-1 capsid and human cyclophilin A. Chem. Biol. Drug Des. 2010, 76, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wu, K.-J.; Liu, J.-B.; Zhou, X.-M.; Leung, C.-H.; Ma, D.-L. A dual-functional molecular strategy for in situ suppressing and visualizing of neuraminidase in aqueous solution using iridium (iii) complexes. Chem. Commun. 2019, 55, 6353–6356. [Google Scholar] [CrossRef] [PubMed]
- Gadde, S. Multi-drug delivery nanocarriers for combination therapy. MedChemComm 2015, 6, 1916–1929. [Google Scholar] [CrossRef]
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. 2021. Available online: https://covid19.who.int/ (accessed on 16 January 2021).
- Hung, H.C.; Ke, Y.Y.; Huang, S.Y.; Huang, P.N.; Kung, Y.A.; Chang, T.Y.; Yen, K.J.; Peng, T.T.; Chang, S.E.; Huang, C.T.; et al. Discovery of M protease inhibitors encoded by SARS-CoV-2. Antimicrob. Agents Chemother. 2020, 64, e00872-20. [Google Scholar] [CrossRef] [PubMed]
- Corum, J.; Grady, D.; Wee, S.-L.; Zimmer, C. Coronavirus vaccine tracker. The New York Times, 15 April 2020. [Google Scholar]
- Mbunge, E.; Akinnuwesi, B.; Fashoto, S.G.; Metfula, A.S.; Mashwama, P. A critical review of emerging technologies for tackling COVID-19 pandemic. Hum. Behav. Emerg. 2021, 3, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Belayneh, A. Off-label use of chloroquine and hydroxychloroquine for COVID-19 treatment in Africa against WHO recommendation. Res. Rep. Trop. Med. 2020, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef]
- Cai, Q.; Yang, M.; Liu, D.; Chen, J.; Shu, D.; Xia, J.; Liao, X.; Gu, Y.; Cai, Q.; Yang, Y. Experimental treatment with favipiravir for COVID-19: An open-label control study. Engineering 2020, 6, 1192–1198. [Google Scholar] [CrossRef]
- Horby, P.W.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Emberson, J.; Palfreeman, A.; Raw, J.; Elmahi, E.; Prudon, B. Lopinavir–ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. The Lancet 2020, 396, 1345–1352. [Google Scholar] [CrossRef]
- Ganesh, R.; Philpot, L.M.; Bierle, D.M.; Anderson, R.J.; Arndt, L.L.; Arndt, R.F.; Culbertson, T.L.; Destro Borgen, M.J.; Hanson, S.N.; Kennedy, B.D. Real-World Clinical Outcomes of Bamlanivimab and Casirivimab-Imdevimab among High-Risk Patients with Mild to Moderate Coronavirus Disease 2019. J. Infect. Dis. 2021, jiab377. [Google Scholar] [CrossRef]
- Orders, M. An EUA for Bamlanivimab and Etesevimab for COVID-19. Med. Lett. Drugs Ther. 2021, 63, 49. [Google Scholar]
- Orders, M. An EUA for Sotrovimab for Treatment of COVID-19. Med. Lett. Drugs Ther. 2021, 63, 97. [Google Scholar]
- Ramos-Guzmán, C.A.; Ruiz-Pernía, J.J.; Tuñón, I. Computational simulations on the binding and reactivity of a nitrile inhibitor of the SARS-CoV-2 main protease. ChemComm 2021. [Google Scholar] [CrossRef]
- Choudhary, S.; Malik, Y.S.; Tomar, S. Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front. Immunol. 2020, 11, 1664. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, G.; Kumar, V. In-silico design of a potential inhibitor of SARS-CoV-2 S protein. PLoS ONE 2020, 15, e0240004. [Google Scholar] [CrossRef]
- Teli, D.M.; Shah, M.B.; Chhabria, M.T. In silico screening of natural compounds as potential inhibitors of SARS-CoV-2 main protease and spike RBD: Targets for COVID-19. Front. Mol. Biosci. 2020, 7, 429. [Google Scholar]
- Wei, T.Z.; Wang, H.; Wu, X.Q.; Lu, Y.; Guan, S.H.; Dong, F.Q.; Zhu, G.L.; Bao, Y.Z.; Zhang, J.; Wang, G.Y.; et al. In silico screening of potential spike glycoprotein inhibitors of SARS-CoV-2 with drug repurposing strategy. Chin. J. Integr. Med. 2020, 26, 663–669. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020, 64, e00819-20. [Google Scholar] [CrossRef]
- Murgolo, N.; Therien, A.G.; Howell, B.; Klein, D.; Koeplinger, K.; Lieberman, L.A.; Adam, G.C.; Flynn, J.; McKenna, P.; Swaminathan, G.; et al. SARS-CoV-2 tropism, entry, replication, and propagation: Considerations for drug discovery and development. PLoS Pathog. 2021, 17, e1009225. [Google Scholar] [CrossRef] [PubMed]
- Abdool Karim, S.S.; de Oliveira, T. New SARS-CoV-2 variants—Clinical, public health, and vaccine implications. N. Eng. J. Med. 2021, 384, 1866–1868. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; Rohde, C.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef] [PubMed]
- Adedeji, A.O.; Sarafianos, S.G. Antiviral drugs specific for coronaviruses in preclinical development. Curr. Opin. Virol. 2014, 8, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Jena, N.R. Drug targets, mechanisms of drug action, and therapeutics against SARS-CoV-2. Chem. Phys. Impact 2021, 2, 100011. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Huang, Y.Y.; Wu, Y.; Liu, R.; Zhou, L.; Lin, Y.; Wu, D.; Zhang, L.; Liu, H.; et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc. Natl. Acad. Sci. USA 2020, 117, 27381–27387. [Google Scholar] [CrossRef]
- Wang, K.Y.; Liu, F.; Jiang, R.; Yang, X.; You, T.; Liu, X.; Xiao, C.Q.; Shi, Z.; Jiang, H.; Rao, Z.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 289–293. [Google Scholar]
- Kumar, V.; Dhanjal, J.K.; Bhargava, P.; Kaul, A.; Wang, J.; Zhang, H.; Kaul, S.C.; Wadhwa, R.; Sundar, D. Withanone and Withaferin-A are predicted to interact with transmembrane protease serine 2 (TMPRSS2) and block entry of SARS-CoV-2 into cells. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Nayak, T.R.; Hong, H.; Cai, W. Biomedical applications of zinc oxide nanomaterials. Curr. Mol. Med. 2013, 13, 1633–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhivya, R.; Ranjani, J.; Rajendhran, J.; Mayandi, J.; Annaraj, J. Enhancing the anti-gastric cancer activity of curcumin with biocompatible and pH sensitive PMMA-AA/ZnO nanoparticles. Mater. Sci. Eng. 2018, 82, 182–189. [Google Scholar] [CrossRef]
- El-Megharbel, S.M.; Alsawat, M.; Al-Salmi, F.A.; Hamza, R.Z. Utilizing of (zinc oxide nano-spray) for disinfection against “SARS-CoV-2” and testing its biological effectiveness on some biochemical parameters during (COVID-19 pandemic)—“ZnO nanoparticles have antiviral activity against (SARS-CoV-2)”. Mater. Sci. 2021, 11, 388. [Google Scholar]
- Tavakoli, A.; Ataei-Pirkooh, A.; Sadeghi, G.M.; Bokharaei-Salim, F.; Sahrapour, P.; Kiani, S.J.; Moghoofei, M.; Farahmand, M.; Javanmard, D.; Monavari, S.H. Polyethylene glycol-coated zinc oxide nanoparticle: An efficient nanoweapon to fight against herpes simplex virus type 1. Nanomedicine 2018, 13, 2675–2690. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.B.; Chess, J.J.; Wingett, D.G.; Punnoose, A. Serum proteins enhance dispersion stability and influence the cytotoxicity and dosimetry of ZnO nanoparticles in suspension and adherent cancer cell models. Nanoscale Res. Lett. 2015, 10, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taratula, O.; Galoppini, E.; Wang, D.; Chu, D.; Zhang, Z.; Chen, H.; Saraf, G.; Lu, Y. Binding studies of molecular linkers to ZnO and MgZnO nanotip films. J. Phys. Chem. B 2006, 110, 6506–6515. [Google Scholar] [CrossRef]
- Hariharan, R.; Senthilkumar, S.; Suganthi, A.; Rajarajan, M. Synthesis and characterization of doxorubicin modified ZnO/PEG nanomaterials and its photodynamic action. J. Photochem. Photobiol. B Biol. 2012, 116, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, H.; Tavakoli, A.; Moradi, A.; Tabarraei, A.; Bokharaei-Salim, F.; Zahmatkeshan, M.; Farahmand, M.; Javanmard, D.; Kiani, S.J.; Esghaei, M.; et al. Inhibition of H1N1 influenza virus infection by zinc oxide nanoparticles: Another emerging application of nanomedicine. J. Biomed. Sci. 2019, 26, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.; Connolly, C.; Schettino, G.; Butterworth, K.T.; Prise, K.M. Biological mechanisms of gold nanoparticle radiosensitization. Cancer Nanotechnol. 2017, 8, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqi, K.S.; Rahman, A.U.; Husen, A. Properties of zinc oxide nanoparticles and their activity against microbes. Nanoscale Res. Lett. 2018, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.R.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Giardina, S.F.; Werner, D.S.; Pingle, M.; Feinberg, P.B.; Foreman, K.W.; Bergstrom, D.E.; Arnold, L.D.; Barany, F. Novel, self-assembling dimeric inhibitors of human β tryptase. J. Med. Chem. 2020, 63, 3004–3027. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Ganai, S.A.; Abdullah, E.; Rashid, R.; Altaf, M. Combinatorial in silico strategy towards identifying potential hotspots during inhibition of structurally identical HDAC1 and HDAC2 enzymes for effective chemotherapy against neurological disorders. Front. Mol. Neurosci. 2017, 10, 357. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, E.; Rendleman, C.A.; Bowers, K.J.; Dror, R.O.; Hughes, D.H.; Gullingsrud, J.; Sacerdoti, F.D.; Shaw, D.E. Desmond Performance on a Cluster of Multicore Processors, D. E. Shaw Research Technical Report DESRES/TR--2008-01; D. E. Shaw: New York, NY, USA, 2008. [Google Scholar]
- Zeiske, T.; Stafford, K.; Friesner, R.A.; Palmer, A.G. Starting-structure dependence of nanosecond timescale intersubstate transitions and reproducibility of MD-derived order parameters. Proteins Struct. Funct. Bioinform. 2013, 81, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearlstein, R.A.; Sherman, W.; Abel, R. Contributions of water transfer energy to protein-ligand association and dissociation barriers: Watermap analysis of a series of p38α MAP kinase inhibitors. Proteins Struct. Funct. Bioinform. 2013, 81, 1509–1526. [Google Scholar] [CrossRef]
- Cappel, D.; Sherman, W.; Beuming, T. Calculating water thermodynamics in the binding site of proteins—Applications of WaterMap to drug discovery. Curr. Top. Med. Chem. 2017, 17, 2586–2598. [Google Scholar] [CrossRef]
- Abel, R.; Young, T.; Farid, R.; Berne, B.J.; Friesner, R.A. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. [Google Scholar] [CrossRef] [Green Version]
- Young, T.; Abel, R.; Kim, B.; Berne, B.J.; Friesner, R.A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc. Natl. Acad. Sci. USA 2007, 104, 808–813. [Google Scholar] [CrossRef] [Green Version]
- Horbert, R.; Pinchuk, B.; Johannes, E.; Schlosser, J.; Schmidt, D.; Cappel, D.; Totzke, F.; Schächtele, C.; Peifer, C. Optimization of potent DFG-in inhibitors of platelet derived growth factor receptorβ (PDGF-Rβ) guided by water thermodynamics. J. Med. Chem. 2014, 58, 170–182. [Google Scholar] [CrossRef]
- Lazaridis, T. Inhomogeneous fluid approach to solvation thermodynamics. 1. Theory. J. Phys. Chem. B 1998, 102, 3531–3541. [Google Scholar] [CrossRef]
- Lazaridis, T. Inhomogeneous fluid approach to solvation thermodynamics. 2. Applications to simple fluids. J. Phys. Chem. B 1998, 102, 3542–3550. [Google Scholar] [CrossRef]
- Konze, K.D.; Bos, P.H.; Dahlgren, M.K.; Leswing, K.; Tubert-Brohman, I.; Bortolato, A.; Robbason, B.; Abel, R.; Bhat, S. Reaction-based enumeration, active learning, and free energy calculations to rapidly explore synthetically tractable chemical space and optimize potency of cyclin-dependent kinase 2 inhibitors. J. Chem. Inf. Model. 2019, 59, 3782–3793. [Google Scholar] [CrossRef]
- Mostafa, A.; Kandeil, A.; Elshaier, Y.A.M.M.; Kutkat, O.; Moatasim, Y.; Rashad, A.A.; Shehata, M.; Gomaa, M.R.; Mahrous, N.; Mahmoud, S.H.; et al. FDA-approved drugs with potent in vitro antiviral activity against severe acute respiratory syndrome coronavirus 2. Pharmaceuticals 2020, 13, 443. [Google Scholar] [CrossRef]
- Soliman, S.S.M.; Alhamidi, T.B.; Abdin, S.; Almehdi, A.M.; Semreen, M.H.; Alhumaidi, R.B.; Shakartalla, S.B.; Haider, M.; Husseiny, M.I.; Omar, H.A. Effective targeting of breast cancer cells (MCF7) via novel biogenic synthesis of gold nanoparticles using cancer-derived metabolites. PLoS ONE 2020, 15, e0240156. [Google Scholar] [CrossRef] [PubMed]
- Soliman, S.S.M.; Saeed, B.Q.; Elseginy, S.A.; Al-Marzooq, F.; Ahmady, I.M.; El-Keblawy, A.A.; Hamdy, R. Critical discovery and synthesis of novel antibacterial and resistance-modifying agents inspired by plant phytochemical defense mechanisms. Chem. Biol. Interact. 2020, 333, 109318. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Wang, J.; Lee, J.S.; Kim, D.; Zhu, L. Exploration of zinc oxide nanoparticles as a multitarget and multifunctional anticancer nanomedicine. ACS Appl. Mater. Interfaces 2017, 9, 39971–39984. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Docking Score with | |
|---|---|---|
| MPRO | TMPRSS2 | |
| M1 | −9.97 | −8.11 |
| M2 | −8.16 | −8.53 |
| M3 | −8.79 | −9.81 |
| M4 | −9.67 | −9.32 |
| M5 | −7.37 | −6.32 |
| Compound 13 | −7.01 | −9.86 |
| Compound | Moiety | Interaction | Amino Acid Residue |
|---|---|---|---|
| M1 | NH N of oxadiazole Phenyl ring | H-bond H-bond pi-pi stacking bond Hydrophobic bond | Glu-166 Asn-142 His-41 His-41, Met-49, Cys-145, Met-165 and Pro-168 |
| M2 | NH2 | 2H-bonds Hydrophobic bond | Phe-140 and Glu-166 His-41, Met-49, Cys-145, Met-165 and Gln-189 |
| M3 | NH2 NH N pyridyl moiety and CH | 2H-bonds H-bond H-bond Hydrophobic bond | Cys-44 and Thr-25 Asn-142 Glu-166 Met-49, Cys-44, Glu-166 and Met-165 |
| M4 | NH2 Cl | 2- H-bonds Halogen bond Hydrophobic bond | Glu-166 and Phe-140 Gln-192 Met-49, Glu-166, Cys-145, Met-165, Gln-189, Gln-192 and Pro-168 |
| M5 | NH2 | 2H-bonds Hydrophobic bond | Glu-166 and Phe-140 Cys-44, His-41, Met-49, Gln-189 and Pro-168 |
| Compound 13 | Phenolic OH NH2 NH2 N Phenolic OH Phenyl ring and CH | H-bond H-bond H-bond pi-pi stacking bond Hydrophobic bond | Glu-166 Phe-140 Thr-26 His-40 Met-44, Cys-44 and Met-165. |
| Compound | Moiety | Interaction | Amino Acid Residue |
|---|---|---|---|
| M1 | NH N | H-bond H-bond Hydrophobic bond | Gln-192 Ser-195 Arg-41, Cys-219 and Tyr-228 |
| M2 | NH2 NH | H-bond H-bond Hydrophobic bond | Gln-192 Ser-214 Tyr-36, Arg-41, Gln-192, Val-213 and Cys-219 |
| M3 | 2-NH N of 4-pyridyl moiety N pyridinyl moiety and CH | 2H-bonds H-bond H-bond Hydrophobic bond | Ser-214 Arg-41 Glu-218 His-57, Arg-41 and Cys-219 and Cys-42 |
| M4 | NH2 Cl N | 3-H-bonds 3-Halogen bond H-bond Hydrophobic bond | His-57, Ser-214, Gln-192 Ala-220, Ser-214, Ser-39 Arg-41 Arg-41, Val-213, Cys-219 and Tyr-228 |
| M5 | OCH3 NH | H-bond H-bond Hydrophobic bond | Arg-41 Gln-192 Arg-41, Tyr-149, Ala-190, Val-213 and Cys-219 |
| Compound 13 | Phenolic OH NH2 NH2 N Phenolic OH Phenyl ring and CH | H-bond H-bond H-bond H-bond H-bond Hydrophobic bond | His-40 Gln-192 Ser-214 Arg-41 Glu-218 Arg-41, Ala-190, Val-213, Cys-42 and Trp-36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamdy, R.; Fayed, B.; Mostafa, A.; Shama, N.M.A.; Mahmoud, S.H.; Mehta, C.H.; Nayak, Y.; M. Soliman, S.S. Iterated Virtual Screening-Assisted Antiviral and Enzyme Inhibition Assays Reveal the Discovery of Novel Promising Anti-SARS-CoV-2 with Dual Activity. Int. J. Mol. Sci. 2021, 22, 9057. https://doi.org/10.3390/ijms22169057
Hamdy R, Fayed B, Mostafa A, Shama NMA, Mahmoud SH, Mehta CH, Nayak Y, M. Soliman SS. Iterated Virtual Screening-Assisted Antiviral and Enzyme Inhibition Assays Reveal the Discovery of Novel Promising Anti-SARS-CoV-2 with Dual Activity. International Journal of Molecular Sciences. 2021; 22(16):9057. https://doi.org/10.3390/ijms22169057
Chicago/Turabian StyleHamdy, Rania, Bahgat Fayed, Ahmed Mostafa, Noura M. Abo Shama, Sara Hussein Mahmoud, Chetan Hasmukh Mehta, Yogendra Nayak, and Sameh S. M. Soliman. 2021. "Iterated Virtual Screening-Assisted Antiviral and Enzyme Inhibition Assays Reveal the Discovery of Novel Promising Anti-SARS-CoV-2 with Dual Activity" International Journal of Molecular Sciences 22, no. 16: 9057. https://doi.org/10.3390/ijms22169057
APA StyleHamdy, R., Fayed, B., Mostafa, A., Shama, N. M. A., Mahmoud, S. H., Mehta, C. H., Nayak, Y., & M. Soliman, S. S. (2021). Iterated Virtual Screening-Assisted Antiviral and Enzyme Inhibition Assays Reveal the Discovery of Novel Promising Anti-SARS-CoV-2 with Dual Activity. International Journal of Molecular Sciences, 22(16), 9057. https://doi.org/10.3390/ijms22169057