
Generalized Pustular Psoriasis: Divergence of Innate and Adaptive Immunity



Abstract
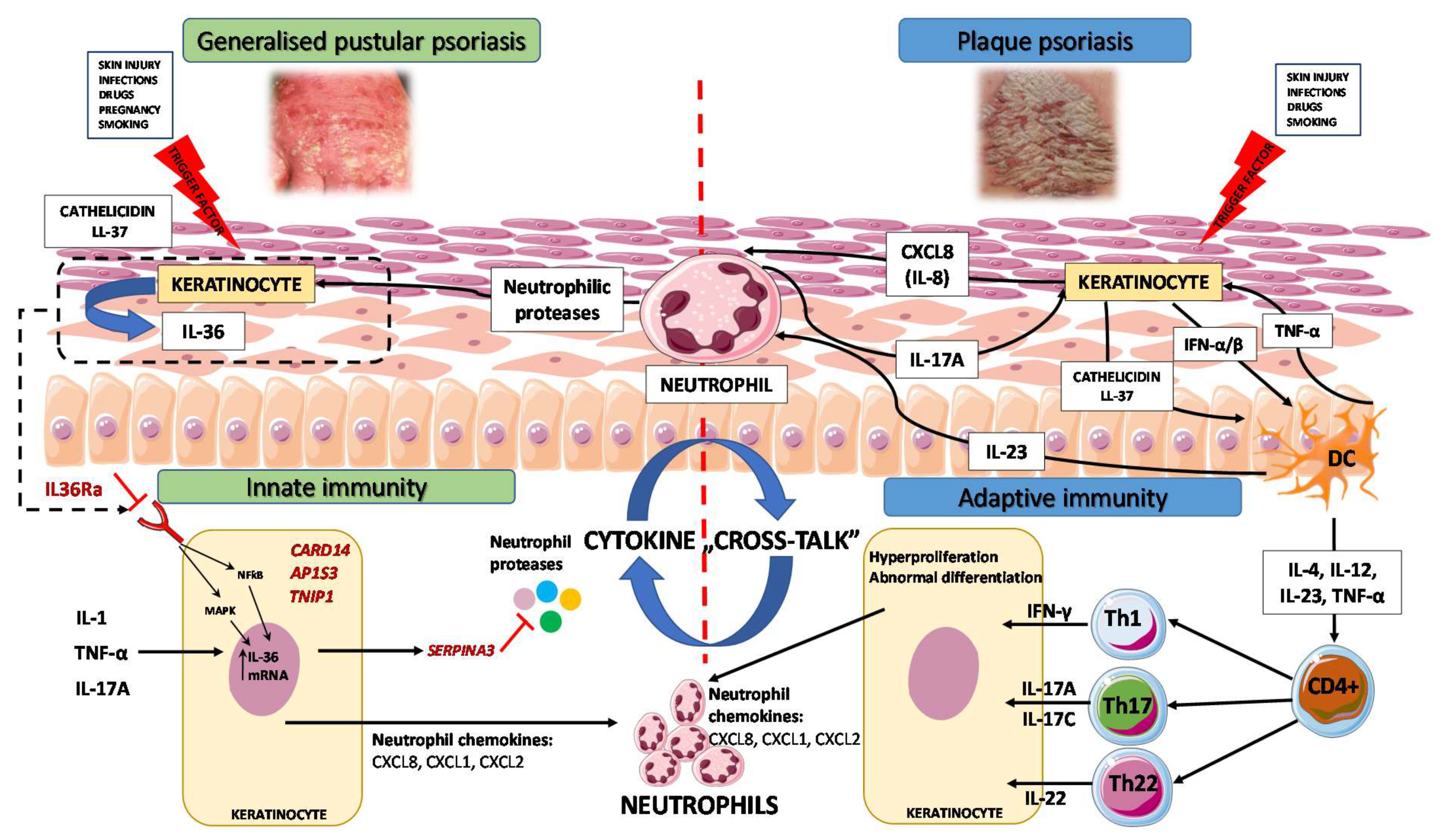
:1. Introduction
2. Gene Mutations in GPP
2.1. Mutations of IL-36 Receptor Antagonist
2.2. CARD14 Mutations/Variants
2.3. AP1S3 Mutations
2.4. TNIP1 Mutations
2.5. SERPINA3 Mutations
2.6. MPO Mutation
3. Immunopathogenesis
3.1. Autoinflammation and Autoimmunity in GPP
3.2. GPP as an Autoinflammatory Keratinization Disorder
3.3. IL-1/IL-36 Inflammatory Axis
3.4. IL-17/IL-36 Axis as a Bridge between Innate and Adaptive Immunity
4. Biologic Therapeutics for GPP in the Light of Novel Genetic and Immunological Findings
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baker, H.; Ryan, T.J. Generalized pustular psoriasis. A clinical and epidemiological study of 104 cases. Br. J. Dermatol. 1968, 80, 771–793. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.J.; Baker, H. The prognosis of generalized pustular psoriasis. Br. J. Dermatol. 1971, 85, 407–411. [Google Scholar] [CrossRef]
- Zelickson, B.D.; Muller, S.A. Generalized pustular psoriasis. A review of 63 cases. Arch. Dermatol. 1991, 127, 1339–1345. [Google Scholar] [CrossRef] [Green Version]
- Ohkawara, A.; Yasuda, H.; Kobayashi, H.; Inaba, Y.; Ogawa, H.; Hashimoto, I.; Imamura, S. Generalized pustular psoriasis in Japan: Two distinct groups formed by differences in symptoms and genetic background. Acta Derm. Venereol. 1996, 76, 68–71. [Google Scholar]
- Augey, F.; Renaudier, P.; Nicolas, J.F. Generalized pustular psoriasis (Zumbusch): A French epidemiological survey. Eur. J. Dermatol. 2006, 16, 669–673. [Google Scholar]
- Ito, T.; Takahashi, H.; Kawada, A.; Iizuka, H.; Nakagawa, H.; Japanese Society for Psoriasis Research. Epidemiological survey from 2009 to 2012 of psoriatic patients in Japanese Society for Psoriasis Research. J. Dermatol. 2018, 45, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Nakamura, K.; Kaneko, F.; Nakagawa, H.; Iizuka, H.; Japanese Society for Psoriasis Research. Analysis of psoriasis patients registered with the Japanese Society for Psoriasis Research from 2002–2008. J. Dermatol. 2011, 38, 1125–1129. [Google Scholar] [CrossRef]
- Twelves, S.; Mostafa, A.; Dand, N.; Burri, E.; Farkas, K.; Wilson, R.; Cooper, H.L.; Irvine, A.D.; Oon, H.H.; Kingo, K.; et al. Clinical and genetic differences between pustular psoriasis subtypes. J. Allergy Clin. Immunol. 2019, 143, 1021–1026. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Cho, H.H.; Kim, W.J.; Mun, J.H.; Song, M.; Kim, H.S.; Ko, H.C.; Kim, M.B.; Kim, H.; Kim, B.S. Clinical features and course of generalized pustular psoriasis in Korea. J. Dermatol. 2015, 42, 674–678. [Google Scholar] [CrossRef]
- Langley, R.G.; Krueger, G.G.; Griffiths, C.E. Psoriasis: Epidemiology, clinical features, and quality of life. Ann. Rheum. Dis. 2005, 64 (Suppl. 2), ii18–ii23, discussion ii24–ii25. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, C.; Barker, J. Psoriasis. In Rook’s Textbook of Dermatology, 8th ed.; Burns, T., Cox, N., Griffiths, C., Eds.; Wiley-Blackwell: Chichester, UK, 2010. [Google Scholar]
- Borges-Costa, J.; Silva, R.; Goncalves, L.; Filipe, P.; Soares de Almeida, L.; Marques Gomes, M. Clinical and laboratory features in acute generalized pustular psoriasis: A retrospective study of 34 patients. Am. J. Clin. Dermatol. 2011, 12, 271–276. [Google Scholar] [CrossRef]
- Viguier, M.; Allez, M.; Zagdanski, A.M.; Bertheau, P.; de Kerviler, E.; Rybojad, M.; Morel, P.; Dubertret, L.; Lémann, M.; Bachelez, H. High frequency of cholestasis in generalized pustular psoriasis: Evidence for neutrophilic involvement of the biliary tract. Hepatology 2004, 40, 452–458. [Google Scholar] [CrossRef]
- Bachelez, H. Pustular psoriasis and related pustular skin diseases. Br. J. Dermatol. 2018, 178, 614–618. [Google Scholar] [CrossRef]
- Choon, S.E.; Lai, N.M.; Mohammad, N.A.; Nanu, N.M.; Tey, K.E.; Chew, S.F. Clinical profile, morbidity, and outcome of adult-onset generalized pustular psoriasis: Analysis of 102 cases seen in a tertiary hospital in Johor, Malaysia. Int. J. Dermatol. 2014, 53, 676–684. [Google Scholar] [CrossRef]
- Navarini, A.A.; Burden, A.D.; Capon, F.; Mrowietz, U.; Puig, L.; Köks, S.; Kingo, K.; Smith, C.; Barker, J.N.; ERASPEN Network. European consensus statement on phenotypes of pustular psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1792–1799. [Google Scholar] [CrossRef] [Green Version]
- Umezawa, Y.; Ozawa, A.; Kawasima, T.; Shimizu, H.; Terui, T.; Tagami, H.; Ikeda, S.; Ogawa, H.; Kawada, A.; Tezuka, T.; et al. Therapeutic guidelines for the treatment of generalized pustular psoriasis (GPP) based on a proposed classification of disease severity. Arch. Dermatol. Res. 2003, 295 (Suppl. 1), S43–S54. [Google Scholar] [CrossRef]
- Almutairi, D.; Sheasgreen, C.; Weizman, A.; Alavi, A. Generalized Pustular Psoriasis Induced by Infliximab in a Patient with Inflammatory Bowel Disease. J. Cutan. Med. Surg. 2018, 22, 507–510. [Google Scholar] [CrossRef]
- Wenk, K.S.; Claros, J.M.; Ehrlich, A. Flare of pustular psoriasis after initiating ustekinumab therapy. J. Dermatolog. Treat. 2012, 23, 212–214. [Google Scholar] [CrossRef]
- Kardaun, S.H.; Kuiper, H.; Fidler, V.; Jonkman, M.F. The histopathological spectrum of acute generalized exanthematous pustulosis (AGEP) and its differentiation from generalized pustular psoriasis. J. Cutan. Pathol. 2010, 37, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Sidoroff, A.; Dunant, A.; Viboud, C.; Halevy, S.; Bavinck, J.N.; Naldi, L.; Mockenhaupt, M.; Fagot, J.P.; Roujeau, J.C. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR). Br. J. Dermatol. 2007, 157, 989–996. [Google Scholar] [CrossRef]
- Li, Z.; Yang, Q.; Wang, S. Genetic polymorphism of IL36RN in Han patients with generalized pustular psoriasis in Sichuan region of China: A case–control study. Medicine 2018, 97, e11741. [Google Scholar] [CrossRef]
- Johnston, A.; Xing, X.; Wolterink, L.; Barnes, D.H.; Yin, Z.; Reingold, L.; Kahlenberg, J.M.; Harms, P.W.; Gudjonsson, J.E. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J. Allergy Clin. Immunol. 2017, 140, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, K.; Takemoto, A.; Yamaguchi, M.; Takahashi, H.; Shoda, Y.; Mitsuma, T.; Tsuda, K.; Nishida, E.; Togawa, Y.; Nakajima, K.; et al. The majority of generalized pustular psoriasis without psoriasis vulgaris is caused by deficiency of interleukin-36 receptor antagonist. J. Invest. Dermatol. 2013, 133, 2514–2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, A.; Ohkido, M.; Haruki, Y.; Kobayashi, H.; Ohkawara, A.; Ohno, Y.; Inaba, Y.; Ogawa, H. Treatments of generalized pustular psoriasis: A multicenter study in Japan. J. Dermatol. 1999, 26, 141–149. [Google Scholar] [CrossRef]
- Boehner, A.; Navarini, A.A.; Eyerich, K. Generalized pustular psoriasis—A model disease for specific targeted immunotherapy, systematic review. Exp. Dermatol. 2018, 27, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Imafuku, S.; Honma, M.; Okubo, Y.; Komine, M.; Ohtsuki, M.; Morita, A.; Seko, N.; Kawashima, N.; Ito, S.; Shima, T.; et al. Efficacy and safety of secukinumab in patients with generalized pustular psoriasis: A 52-week analysis from phase III open-label multicenter Japanese study. J. Dermatol. 2016, 43, 1011–1017. [Google Scholar] [CrossRef]
- Fujita, H.; Terui, T.; Hayama, K.; Akiyama, M.; Ikeda, S.; Mabuchi, T.; Ozawa, A.; Kanekura, T.; Kurosawa, M.; Komine, M.; et al. Japanese Dermatological Association Guidelines Development Committee for the Guidelines for the Management and Treatment of Generalized Pustular Psoriasis. Japanese guidelines for the management and treatment of generalized pustular psoriasis: The new pathogenesis and treatment of GPP. J. Dermatol. 2018, 45, 1235–1270. [Google Scholar] [PubMed]
- Yamasaki, K.; Nakagawa, H.; Kubo, Y.; Ootaki, K.; Japanese Brodalumab Study Group. Efficacy and safety of brodalumab in patients with generalized pustular psoriasis and psoriatic erythroderma: Results from a 52-week, open-label study. Br. J. Dermatol. 2017, 176, 741–751. [Google Scholar] [CrossRef]
- Sano, S.; Kubo, H.; Morishima, H.; Goto, R.; Zheng, R.; Nakagawa, H. Guselkumab, a human interleukin-23 monoclonal antibody in Japanese patients with generalized pustular psoriasis and erythrodermic psoriasis: Efficacy and safety analyses of a 52-week, phase 3, multicenter, open-label study. J. Dermatol. 2018, 45, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.M.; Jin, H.Z. Biologics in the treatment of pustular psoriasis. Expert Opin. Drug Saf. 2020, 19, 969–980. [Google Scholar] [CrossRef]
- Zhou, J.; Luo, Q.; Cheng, Y.; Wen, X.; Liu, J. An update on genetic basis of generalized pustular psoriasis (Review). Int. J. Mol. Med. 2021, 47, 118. [Google Scholar] [CrossRef] [PubMed]
- Plachouri, K.M.; Chourdakis, V.; Georgiou, S. The role of IL-17 and IL-17 receptor inhibitors in the management of generalized pustular psoriasis. Drugs Today 2019, 55, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Gooderham, M.J.; Van Voorhees, A.S.; Lebwohl, M.G. An update on generalized pustular psoriasis. Expert Rev. Clin. Immunol. 2019, 15, 907–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbidian, E.; Chaimani, A.; Afach, S.; Doney, L.; Dressler, C.; Hua, C.; Mazaud, C.; Phan, C.; Hughes, C.; Riddle, D.; et al. Systemic pharmacological treatments for chronic plaque psoriasis: A network meta-analysis. Cochrane Database Syst. Rev. 2020, 9, 1, CD011535. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Van Voorhees, A.S.; Hsu, S.; Korman, N.J.; Lebwohl, M.G.; Bebo, B.F., Jr.; Kalb, R.E. Treatment of pustular psoriasis: From the Medical Board of the National Psoriasis Foundation. J. Am. Acad. Dermatol. 2012, 67, 279–288. [Google Scholar] [CrossRef]
- Collamer, A.N.; Battafarano, D.F. Psoriatic skin lesions induced by tumor necrosis factor antagonist therapy: Clinical features and possible immunopathogenesis. Semin. Arthritis Rheum. 2010, 40, 233–240. [Google Scholar] [CrossRef]
- Kucharekova, M.; Winnepenninckx, V.; Frank, J.; Poblete-Gutiérrez, P. Generalized pustulosis induced by adalimumab in a patient with rheumatoid arthritis—A therapeutic challenge. Int. J. Dermatol. 2008, 47 (Suppl. 1), 25–28. [Google Scholar] [CrossRef]
- Liang, Y.; Sarkar, M.K.; Tsoi, L.C.; Gudjonsson, J.E. Psoriasis: A mixed autoimmune and autoinflammatory disease. Curr. Opin. Immunol. 2017, 49, 1–8. [Google Scholar] [CrossRef]
- Liang, Y.; Xing, X.; Beamer, M.A.; Swindell, W.R.; Sarkar, M.K.; Roberts, L.W.; Voorhees, J.J.; Kahlenberg, J.M.; Harms, P.W.; Johnston, A.; et al. Six-transmembrane epithelial antigens of the prostate comprise a novel inflammatory nexus in patients with pustular skin disorders. J. Allergy Clin. Immunol. 2017, 139, 1217–1227. [Google Scholar] [CrossRef]
- Aksentijevich, I.; Masters, S.L.; Ferguson, P.J.; Dancey, P.; Frenkel, J.; van Royen-Kerkhoff, A.; Laxer, R.; Tedgård, U.; Cowen, E.W.; Pham, T.H.; et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N. Engl. J. Med. 2009, 360, 2426–2437. [Google Scholar] [CrossRef] [Green Version]
- Jesus, A.A.; Osman, M.; Silva, C.A.; Kim, P.W.; Pham, T.H.; Gadina, M.; Yang, B.; Bertola, D.R.; Carneiro-Sampaio, M.; Ferguson, P.J.; et al. A novel mutation of IL1RN in the deficiency of interleukin-1 receptor antagonist syndrome: Description of two unrelated cases from Brazil. Arthritis Rheum. 2011, 63, 4007–4017. [Google Scholar] [CrossRef] [Green Version]
- Minkis, K.; Aksentijevich, I.; Goldbach-Mansky, R.; Magro, C.; Scott, R.; Davis, J.G.; Sardana, N.; Herzog, R. Interleukin 1 receptor antagonist deficiency presenting as infantile pustulosis mimicking infantile pustular psoriasis. Arch. Dermatol. 2012, 148, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.; Jia, S.; Geoffrey, R.; Lorier, R.; Suchi, M.; Broeckel, U.; Hessner, M.J.; Verbsky, J. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N. Engl. J. Med. 2009, 360, 2438–2444. [Google Scholar] [CrossRef] [Green Version]
- Schnellbacher, C.; Ciocca, G.; Menendez, R.; Aksentijevich, I.; Goldbach-Mansky, R.; Duarte, A.M.; Rivas-Chacon, R. Deficiency of interleukin-1 receptor antagonist responsive to anakinra. Pediatr. Dermatol. 2013, 30, 758–760. [Google Scholar] [CrossRef] [Green Version]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Simpson, M.A.; Pink, A.E.; Di Meglio, P.; Smith, C.H.; Pullabhatla, V.; Knight, J.; Spain, S.L.; Nestle, F.O.; Burden, A.D.; et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 2011, 89, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Blumberg, H.; Dinh, H.; Trueblood, E.S.; Pretorius, J.; Kugler, D.; Weng, N.; Kanaly, S.T.; Towne, J.E.; Willis, C.R.; Kuechle, M.K.; et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J. Exp. Med. 2007, 204, 2603–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrowietz, U.; Burden, A.D.; Pinter, A.; Reich, K.; Schäkel, K.; Baum, P.; Datsenko, Y.; Deng, H.; Padula, S.J.; Thoma, C.; et al. Spesolimab, an Anti-Interleukin-36 Receptor Antibody, in Patients with Palmoplantar Pustulosis: Results of a Phase IIa, Multicenter, Double-Blind, Randomized, Placebo-Controlled Pilot Study. Dermatol. Ther. 2021, 11, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Setta-Kaffetzi, N.; Simpson, M.A.; Navarini, A.A.; Patel, V.M.; Lu, H.C.; Allen, M.H.; Duckworth, M.; Bachelez, H.; Burden, A.D.; Choon, S.E.; et al. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am. J. Hum. Genet. 2014, 94, 790–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahil, S.K.; Twelves, S.; Farkas, K.; Setta-Kaffetzi, N.; Burden, A.D.; Gach, J.E.; Irvine, A.D.; Képíró, L.; Mockenhaupt, M.; Oon, H.H.; et al. AP1S3 Mutations Cause Skin Autoinflammation by Disrupting Keratinocyte Autophagy and Up-Regulating IL-36 Production. J. Investig. Dermatol. 2016, 136, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Berki, D.M.; Liu, L.; Choon, S.E.; David Burden, A.; Griffiths, C.E.M.; Navarini, A.A.; Tan, E.S.; Irvine, A.D.; Ranki, A.; Ogo, T.; et al. Activating CARD14 Mutations Are Associated with Generalized Pustular Psoriasis but Rarely Account for Familial Recurrence in Psoriasis Vulgaris. J. Investig. Dermatol. 2015, 135, 2964–2970. [Google Scholar] [CrossRef] [Green Version]
- Mössner, R.; Wilsmann-Theis, D.; Oji, V.; Gkogkolou, P.; Löhr, S.; Schulz, P.; Körber, A.; Prinz, J.C.; Renner, R.; Schäkel, K.; et al. The genetic basis for most patients with pustular skin disease remains elusive. Br. J. Dermatol. 2018, 178, 740–748. [Google Scholar] [CrossRef] [Green Version]
- Towne, J.E.; Renshaw, B.R.; Douangpanya, J.; Lipsky, B.P.; Shen, M.; Gabel, C.A.; Sims, J.E. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J. Biol. Chem. 2011, 286, 42594–42602. [Google Scholar] [CrossRef] [Green Version]
- Towne, J.E.; Garka, K.E.; Renshaw, B.R.; Virca, G.D.; Sims, J.E. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 2004, 279, 13677–13688. [Google Scholar] [CrossRef] [Green Version]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef]
- Bassoy, E.Y.; Towne, J.E.; Gabay, C. Regulation and function of interleukin-36 cytokines. Immunol. Rev. 2018, 281, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Capon, F. IL36RN mutations in generalized pustular psoriasis: Just the tip of the iceberg? J. Investig. Dermatol. 2013, 133, 2503–2504. [Google Scholar] [CrossRef] [Green Version]
- Farooq, M.; Nakai, H.; Fujimoto, A.; Fujikawa, H.; Matsuyama, A.; Kariya, N.; Aizawa, A.; Fujiwara, H.; Ito, M.; Shimomura, Y. Mutation analysis of the IL36RN gene in 14 Japanese patients with generalized pustular psoriasis. Hum. Mutat. 2013, 34, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Takeichi, T.; McGrath, J.A.; Sugiura, K. Autoinflammatory keratinization diseases. J. Allergy Clin. Immunol. 2017, 140, 1545–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, M.; Takeichi, T.; McGrath, J.A.; Sugiura, K. Autoinflammatory keratinization diseases: An emerging concept encompassing various inflammatory keratinization disorders of the skin. J. Dermatol. Sci. 2018, 90, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, M. Autoinflammatory Keratinization Diseases (AiKDs): Expansion of Disorders to Be Included. Front. Immunol. 2020, 11, 280. [Google Scholar] [CrossRef] [Green Version]
- Uppala, R.; Tsoi, L.C.; Harms, P.W.; Wang, B.; Bill, A.C.; Maverakis, E.; Kahlenberg, M.J.; Ward, N.L.; Gudjonsson, J.E. “Autoinflammatory psoriasis”—genetics and biology of pustular psoriasis. Cell. Mol. Immunol. 2021, 18, 307–317. [Google Scholar] [CrossRef]
- Hussain, S.; Berki, D.M.; Choon, S.E.; Burden, A.D.; Allen, M.H.; Arostegui, J.I.; Chaves, A.; Duckworth, M.; Irvine, A.D.; Mockenhaupt, M.; et al. IL36RN mutations define a severe autoinflammatory phenotype of generalized pustular psoriasis. J. Allergy Clin. Immunol. 2015, 135, 1067–1070.e9. [Google Scholar] [CrossRef]
- Wang, T.S.; Chiu, H.Y.; Hong, J.B.; Chan, C.C.; Lin, S.J.; Tsai, T.F. Correlation of IL36RN mutation with different clinical features of pustular psoriasis in Chinese patients. Arch. Dermatol. Res. 2016, 308, 55–63. [Google Scholar] [CrossRef]
- Bachelez, H. Pustular Psoriasis: The Dawn of a New Era. Acta Derm. Venereol. 2020, 100, adv00034. [Google Scholar] [CrossRef] [Green Version]
- Fuchs-Telem, D.; Sarig, O.; van Steensel, M.A.; Isakov, O.; Israeli, S.; Nousbeck, J.; Richard, K.; Winnepenninckx, V.; Vernooij, M.; Shomron, N.; et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am. J. Hum. Genet. 2012, 91, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Blonska, M.; Lin, X. CARMA1-mediated NF-kappaB and JNK activation in lymphocytes. Immunol. Rev. 2009, 228, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Cao, L.; Roberson, E.D.; Pierson, K.C.; Yang, C.F.; Joyce, C.E.; Ryan, C.; Duan, S.; Helms, C.A.; Liu, Y.; et al. PSORS2 is due to mutations in CARD14. Am. J. Hum. Genet. 2012, 90, 784–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeichi, T.; Akiyama, M. Generalized Pustular Psoriasis: Clinical Management and Update on Autoinflammatory Aspects. Am. J. Clin. Dermatol. 2020, 21, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Fang, H.; Zhang, J.; Jiang, M.; Xue, K.; Ma, J.; Zhang, J.; Lei, J.; Zhang, Y.; Li, B.; et al. Neutrophil exosomes enhance the skin autoinflammation in generalized pustular psoriasis via activating keratinocytes. FASEB J. 2019, 33, 6813–6828. [Google Scholar] [CrossRef]
- Sugiura, K.; Muto, M.; Akiyama, M. CARD14 c.526G > C (pAsp176His) is a significant risk factor for generalized pustular psoriasis with psoriasis vulgaris in the Japanese cohort. J. Investig. Dermatol. 2014, 134, 1755–1757. [Google Scholar] [CrossRef] [Green Version]
- Takeichi, T.; Sugiura, K.; Nomura, T.; Sakamoto, T.; Ogawa, Y.; Oiso, N.; Futei, Y.; Fujisaki, A.; Koizumi, A.; Aoyama, Y.; et al. Pityriasis Rubra Pilaris Type V as an Autoinflammatory Disease by CARD14 Mutations. JAMA Dermatol. 2017, 153, 66–70. [Google Scholar] [CrossRef]
- Heyninck, K.; Kreike, M.M.; Beyaert, R. Structure-function analysis of the A20-binding inhibitor of NF-kappa B activation, ABIN-1. FEBS Lett. 2003, 536, 135–140. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, Y.; Zhang, Z.; Lin, J.; Chen, G.; Han, L.; Fang, X.U.; Huang, Q.; Xu, J. Identification of Two Loci Associated with Generalized Pustular Psoriasis. J. Investig. Dermatol. 2015, 135, 2132–2134. [Google Scholar] [CrossRef] [Green Version]
- Han, J.W.; Wang, Y.; Alateng, C.; Li, H.B.; Bai, Y.H.; Lyu, X.X.; Wu, R. Tumor Necrosis Factor-alpha Induced Protein 3 Interacting Protein 1 Gene Polymorphisms and Pustular Psoriasis in Chinese Han Population. Chin. Med. J. 2016, 129, 1519–1524. [Google Scholar] [CrossRef]
- Cooperman, B.S.; Stavridi, E.; Nickbarg, E.; Rescorla, E.; Schechter, N.M.; Rubin, H. Antichymotrypsin interaction with chymotrypsin. Partitioning of the complex. J. Biol. Chem. 1993, 268, 23616–23625. [Google Scholar] [CrossRef]
- Frey, S.; Sticht, H.; Wilsmann-Theis, D.; Gerschütz, A.; Wolf, K.; Löhr, S.; Haskamp, S.; Frey, B.; Hahn, M.; Ekici, A.B.; et al. Rare Loss-of-Function Mutation in SERPINA3 in Generalized Pustular Psoriasis. J. Investig. Dermatol. 2020, 140, 1451–1455.e13. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tu, J.; Hu, Y.; Song, G.; Yin, Z. Cathepsin G cleaves and activates IL-36γ and promotes the inflammation of psoriasis. Drug Des. Devel. Ther. 2019, 13, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Austin, G.E.; Chan, W.C.; Zhao, W.; Racine, M. Myeloperoxidase gene expression in normal granulopoiesis and acute leukemias. Leuk. Lymphoma. 1994, 15, 209–226. [Google Scholar] [CrossRef]
- De Argila, D.; Dominguez, J.D.; Lopez-Estebaranz, J.L.; Iglesias, L. Pustular psoriasis in a patient with myeloperoxidase deficiency. Dermatology 1996, 193, 270. [Google Scholar] [CrossRef]
- Vergnano, M.; Mockenhaupt, M.; Benzian-Olsson, N.; Paulmann, M.; Grys, K.; Mahil, S.K.; Chaloner, C.; Barbosa, I.A.; August, S.; Burden, A.D.; et al. Loss-of-Function Myeloperoxidase Mutations Are Associated with Increased Neutrophil Counts and Pustular Skin Disease. Am. J. Hum. Genet. 2020, 107, 539–543. [Google Scholar] [CrossRef]
- Kizaki, M.; Miller, C.W.; Selsted, M.E.; Koeffler, H.P. Myeloperoxidase (MPO) gene mutation in hereditary MPO deficiency. Blood 1994, 83, 1935–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, C.; Patriarca, P.; Solero, G.P.; Baralle, F.E.; Romano, M. Genetic studies on myeloperoxidase deficiency in Italy. Jpn. J. Infect. Dis. 2004, 57, S10–S12. [Google Scholar]
- Haskamp, S.; Bruns, H.; Hahn, M.; Hoffmann, M.; Gregor, A.; Löhr, S.; Hahn, J.; Schauer, C.; Ringer, M.; Flamann, C.; et al. Myeloperoxidase Modulates Inflammation in Generalized Pustular Psoriasis and Additional Rare Pustular Skin Diseases. Am. J. Hum. Genet. 2020, 107, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Viguier, M.; Guigue, P.; Pagès, C.; Smahi, A.; Bachelez, H. Successful treatment of generalized pustular psoriasis with the interleukin-1-receptor antagonist Anakinra: Lack of correlation with IL1RN mutations. Ann. Intern. Med. 2010, 153, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Hüffmeier, U.; Wätzold, M.; Mohr, J.; Schön, M.P.; Mössner, R. Successful therapy with anakinra in a patient with generalized pustular psoriasis carrying IL36RN mutations. Br. J. Dermatol. 2014, 170, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Papagoras, C.; Lefaki, I.; Giatromanolaki, A.; Kotsianidis, I.; Speletas, M.; Bocly, V.; Theodorou, I.; Dalla, V.; Ritis, K. Successful response in a case of severe pustular psoriasis after interleukin-1β inhibition. Br. J. Dermatol. 2017, 176, 212–215. [Google Scholar] [CrossRef]
- Mansouri, B.; Richards, L.; Menter, A. Treatment of two patients with generalized pustular psoriasis with the interleukin-1β inhibitor gevokizumab. Br. J. Dermatol. 2015, 173, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Bachelez, H.; Choon, S.E.; Marrakchi, S.; Burden, A.D.; Tsai, T.F.; Morita, A.; Turki, H.; Hall, D.B.; Shear, M.; Baum, P.; et al. Inhibition of the Interleukin-36 Pathway for the Treatment of Generalized Pustular Psoriasis. N. Engl. J. Med. 2019, 380, 981–983. [Google Scholar] [CrossRef]
- Choon, S.E.; Lebwohl, M.G.; Marrakchi, S.; Burden, A.D.; Tsai, T.F.; Morita, A.; Navarini, A.A.; Zheng, M.; Xu, J.; Turki, H.; et al. Study protocol of the global Effisayil 1 Phase II, multicentre, randomised, double-blind, placebo-controlled trial of spesolimab in patients with generalized pustular psoriasis presenting with an acute flare. BMJ Open 2021, 11, e043666. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. A 5-year Study to Test BI 655130 in Patients with Generalized Pustular Psoriasis Who Took Part in Previous Studies with BI 655130. NCT03886246. Available online: https://clinicaltrials.gov/ct2/show/NCT03886246 (accessed on 15 May 2021).
- ClinicalTrials.gov. A Study to Evaluate the Efficacy and Safety of ANB019 in Subjects with Generalized Pustular Psoriasis (GPP). NCT03619902. Available online: https://clinicaltrials.gov/ct2/show/NCT03619902 (accessed on 15 May 2021).
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999, 97, 133–144. [Google Scholar] [CrossRef]
- Brydges, S.; Kastner, D.L. The systemic autoinflammatory diseases: Inborn errors of the innate immune system. Curr. Top. Microbiol. Immunol. 2006, 305, 127–160. [Google Scholar] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Carrier, Y.; Ma, H.L.; Ramon, H.E.; Napierata, L.; Small, C.; O’Toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M.; et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: Implications in psoriasis pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabay, C.; Towne, J.E. Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J. Leukoc. Biol. 2015, 97, 645–652. [Google Scholar] [CrossRef]
- Mudigonda, P.; Mudigonda, T.; Feneran, A.N.; Alamdari, H.S.; Sandoval, L.; Feldman, S.R. Interleukin-23 and interleukin-17: Importance in pathogenesis and therapy of psoriasis. Dermatol. Online J. 2012, 18, 1. [Google Scholar] [CrossRef]
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. 2015, 26, 25–33. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef]
- Vigne, S.; Palmer, G.; Lamacchia, C.; Martin, P.; Talabot-Ayer, D.; Rodriguez, E.; Ronchi, F.; Sallusto, F.; Dinh, H.; Sims, J.E.; et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood 2011, 118, 5813–5823. [Google Scholar] [CrossRef]
- Goldstein, J.D.; Bassoy, E.Y.; Caruso, A.; Palomo, J.; Rodriguez, E.; Lemeille, S.; Gabay, C. IL-36 signaling in keratinocytes controls early IL-23 production in psoriasis-like dermatitis. Life Sci. Alliance 2020, 3, e202000688. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, A.; Vollmer, S.; Besgen, P.; Galinski, A.; Summer, B.; Kawakami, Y.; Wollenberg, A.; Dornmair, K.; Spannagl, M.; Ruzicka, T.; et al. Unopposed IL-36 Activity Promotes Clonal CD4+ T-Cell Responses with IL-17A Production in Generalized Pustular Psoriasis. J. Investig. Dermatol. 2018, 138, 1338–1347. [Google Scholar] [CrossRef] [Green Version]
- Trent, J.T.; Kerdel, F.A. Successful treatment of Von Zumbusch pustular psoriasis with infliximab. J. Cutan. Med. Surg. 2004, 8, 224–228. [Google Scholar] [CrossRef]
- Martin, D.A.; Towne, J.E.; Kricorian, G.; Klekotka, P.; Gudjonsson, J.E.; Krueger, J.G.; Russell, C.B. The emerging role of IL-17 in the pathogenesis of psoriasis: Preclinical and clinical findings. J. Investig. Dermatol. 2013, 133, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Johansen, C.; Usher, P.A.; Kjellerup, R.B.; Lundsgaard, D.; Iversen, L.; Kragballe, K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br. J. Dermatol. 2009, 160, 319–324. [Google Scholar] [CrossRef]
- Ishigame, H.; Kakuta, S.; Nagai, T.; Kadoki, M.; Nambu, A.; Komiyama, Y.; Fujikado, N.; Tanahashi, Y.; Akitsu, A.; Kotaki, H.; et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 2009, 30, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Furue, K.; Yamamura, K.; Tsuji, G.; Mitoma, C.; Uchi, H.; Nakahara, T.; Kido-Nakahara, M.; Kadono, T.; Furue, M. Highlighting Interleukin-36 Signalling in Plaque Psoriasis and Pustular Psoriasis. Acta Derm. Venereol. 2018, 98, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Neuhauser, R.; Eyerich, K.; Boehner, A. Generalized pustular psoriasis-Dawn of a new era in targeted immunotherapy. Exp. Dermatol. 2020, 29, 1088–1096. [Google Scholar] [CrossRef]
- Croxford, A.L.; Karbach, S.; Kurschus, F.C.; Wörtge, S.; Nikolaev, A.; Yogev, N.; Klebow, S.; Schüler, R.; Reissig, S.; Piotrowski, C.; et al. IL-6 regulates neutrophil microabscess formation in IL-17A-driven psoriasiform lesions. J. Investig. Dermatol. 2014, 134, 728–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saggini, A.; Chimenti, S.; Chiricozzi, A. IL-6 as a druggable target in psoriasis: Focus on pustular variants. J. Immunol. Res. 2014, 2014, 964069. [Google Scholar] [CrossRef] [PubMed]
- Strober, B.; Kotowsky, N.; Medeiros, R.; Mackey, R.H.; Harrold, L.R.; Valdecantos, W.C.; Flack, M.; Golembesky, A.K.; Lebwohl, M. Unmet Medical Needs in the Treatment and Management of Generalized Pustular Psoriasis Flares: Evidence from a Survey of Corrona Registry Dermatologists. Dermatol. Ther. 2021, 11, 529–541. [Google Scholar] [CrossRef]
- Ettehadi, P.; Greaves, M.W.; Wallach, D.; Aderka, D.; Camp, R.D. Elevated tumour necrosis factor-alpha (TNF-alpha) biological activity in psoriatic skin lesions. Clin. Exp. Immunol. 1994, 96, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Qiu, L.; Xiao, T.; Chen, H.D. Juvenile generalized pustular psoriasis with IL36RN mutation treated with short-term infliximab. Dermatol. Ther. 2016, 29, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Peng, C.; Ding, Y.; Yi, X.; Gao, Y. Development of herpes zoster during infliximab treatment for pediatric generalized pustular psoriasis: A case report. Dermatol. Ther. 2019, 32, e12838. [Google Scholar] [CrossRef]
- Skrabl-Baumgartner, A.; Weger, W.; Salmhofer, W.; Jahnel, J. Childhood generalized pustular psoriasis: Longtime remission with combined infliximab and methotrexate treatment. Pediatr. Dermatol. 2015, 32, e13–e14. [Google Scholar]
- Tsang, V.; Dvorakova, V.; Enright, F.; Murphy, M.; Gleeson, C. Successful use of infliximab as first line treatment for severe childhood generalized pustular psoriasis. J. Eur. Acad. Dermatol. Venereol. 2016, 30, e117–e119. [Google Scholar] [CrossRef]
- Viguier, M.; Aubin, F.; Delaporte, E.; Pagès, C.; Paul, C.; Beylot-Barry, M.; Goujon, C.; Rybojad, M.; Bachelez, H.; Groupe de Recherche sur le Psoriasis de la Société Française de Dermatologie. Efficacy and safety of tumor necrosis factor inhibitors in acute generalized pustular psoriasis. Arch. Dermatol. 2012, 148, 1423–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulalhon, N.; Begon, E.; Lebbé, C.; Lioté, F.; Lahfa, M.; Bengoufa, D.; Morel, P.; Dubertret, L.; Bachelez, H. A follow-up study in 28 patients treated with infliximab for severe recalcitrant psoriasis: Evidence for efficacy and high incidence of biological autoimmunity. Br. J. Dermatol. 2007, 156, 329–336. [Google Scholar] [CrossRef]
- Matsumoto, A.; Komine, M.; Karakawa, M.; Kishimoto, M.; Ohtsuki, M. Adalimumab administration after infliximab therapy is a successful treatment strategy for generalized pustular psoriasis. J. Dermatol. 2017, 44, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Silfvast-Kaiser, A.; Paek, S.Y.; Menter, A. Anti-IL17 therapies for psoriasis. Expert Opin. Biol. Ther. 2019, 19, 45–54. [Google Scholar] [CrossRef]
- Daudén, E.; Santiago-et-Sánchez-Mateos, D.; Sotomayor-López, E.; García-Díez, A. Ustekinumab: Effective in a patient with severe recalcitrant generalized pustular psoriasis. Br. J. Dermatol. 2010, 163, 1346–1347. [Google Scholar] [CrossRef]
- Arakawa, A.; Ruzicka, T.; Prinz, J.C. Therapeutic Efficacy of Interleukin 12/Interleukin 23 Blockade in Generalized Pustular Psoriasis Regardless of IL36RN Mutation Status. JAMA Dermatol. 2016, 152, 825–828. [Google Scholar] [CrossRef]
- Storan, E.R.; O’Gorman, S.M.; Markham, T. Generalized pustular psoriasis treated with ustekinumab. Clin. Exp. Dermatol. 2016, 41, 689–690. [Google Scholar] [CrossRef]
- Markham, A. Guselkumab: First Global Approval. Drugs 2017, 77, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- McKeage, K.; Duggan, S. Risankizumab: First Global Approval. Drugs 2019, 79, 893–900. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. A Study to Assess Efficacy and Safety of Two Different Dose Regimens of Risankizumab Administered Subcutaneously in Japanese Subjects with Generalized Pustular Psoriasis or Erythrodermic Psoriasis. NCT03022045. Available online: https://clinicaltrials.gov/ct2/show/NCT03022045 (accessed on 12 May 2021).
- Rossi-Semerano, L.; Piram, M.; Chiaverini, C.; De Ricaud, D.; Smahi, A.; Koné-Paut, I. First clinical description of an infant with interleukin-36-receptor antagonist deficiency successfully treated with anakinra. Pediatrics 2013, 132, e1043–e1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiler, J.; McDermott, M.F. Gevokizumab, an anti-IL-1β mAb for the potential treatment of type 1 and 2 diabetes, rheumatoid arthritis and cardiovascular disease. Curr. Opin. Mol. Ther. 2010, 12, 755–769. [Google Scholar]
- Ratnarajah, K.; Jfri, A.; Litvinov, I.V.; Netchiporouk, E. Spesolimab: A Novel Treatment for Pustular Psoriasis. J. Cutan. Med. Surg. 2020, 24, 199–200. [Google Scholar] [CrossRef]
- AnaptysBio Reports Positive Topline Data from GALLOP Phase 2 Clinical Trial of Imsidolimab in Moderate-to-Severe Generalized Pustular Psoriasis (GPP). AnaptysBio. News Release. 13 October 2020. Available online: https://ir.anaptysbio.com/news-releases/news-release-details/anaptysbio-reports-positive-topline-data-gallop-phase-2-clinical (accessed on 8 April 2021).
- Morita, A.; Yamazaki, F.; Matsuyama, T.; Takahashi, K.; Arai, S.; Asahina, A.; Imafuku, S.; Nakagawa, H.; Hasegawa, Y.; Williams, D.; et al. Adalimumab treatment in Japanese patients with generalized pustular psoriasis: Results of an open-label phase 3 study. J. Dermatol. 2018, 45, 1371–1380. [Google Scholar] [CrossRef] [Green Version]
- Hansel, K.; Marietti, R.; Tramontana, M.; Bianchi, L.; Romita, P.; Giuffrida, R.; Stingeni, L. Childhood generalized pustular psoriasis: Successful long-term treatment with adalimumab. Dermatol. Ther. 2020, 33, e13294. [Google Scholar] [CrossRef]
- Ho, P.H.; Tsai, T.F. Successful treatment of refractory juvenile generalized pustular psoriasis with secukinumab monotherapy: A case report and review of published work. J. Dermatol. 2018, 45, 1353–1356. [Google Scholar] [CrossRef]
- Mizutani, Y.; Mizutani, Y.H.; Matsuyama, K.; Kawamura, M.; Fujii, A.; Shu, E.; Ohnishi, H.; Seishima, M. Generalized pustular psoriasis in pregnancy, successfully treated with certolizumab pegol. J. Dermatol. 2020, 47, e262–e263. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.L.; Georgakopoulos, J.R.; Ighani, A.; Yeung, J. Systemic Monotherapy Treatments for Generalized Pustular Psoriasis: A Systematic Review. J. Cutan. Med. Surg. 2018, 22, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Kromer, C.; Loewe, E.; Schaarschmidt, M.L.; Pinter, A.; Gerdes, S.; Herr, R.; Poortinga, S.; Moessner, R.; Wilsmann-Theis, D. Drug survival in the treatment of generalized pustular psoriasis: A retrospective multicenter study. Dermatol. Ther. 2021, 34, e14814. [Google Scholar] [CrossRef] [PubMed]
- Saeki, H.; Nakagawa, H.; Nakajo, K.; Ishii, T.; Morisaki, Y.; Aoki, T.; Cameron, G.S.; Osuntokun, O.O.; Japanese Ixekizumab Study Group. Efficacy and safety of ixekizumab treatment for Japanese patients with moderate to severe plaque psoriasis, erythrodermic psoriasis and generalized pustular psoriasis: Results from a 52-week, open-label, phase 3 study (UNCOVER-J). J. Dermatol. 2017, 44, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.M.; Spanou, Z.; Tang, H.; Schibler, F.; Pelivani, N.; Yawalkar, N. Rapid downregulation of innate immune cells, interleukin-12 and interleukin-23 in generalized pustular psoriasis with infliximab in combination with acitretin. Dermatology 2012, 225, 338–343. [Google Scholar] [CrossRef] [PubMed]
- De Rie, M.A.; Zonneveld, I.M.; Witkamp, L. Soluble interleukin-2 receptor (sIL-2R) is a marker of disease activity in psoriasis: A comparison of sIL-2R, sCD27, sCD4, sCD8 and sICAM-1. Acta Dermatol. Venereol. 1996, 76, 357–360. [Google Scholar]
- Salim, A.; Emerson, R.M.; Dalziel, K.L. Successful treatment of severe generalized pustular psoriasis with basiliximab (interleukin-2 receptor blocker). Br. J. Dermatol. 2000, 143, 1121–1122. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study to Test Whether BI 655130 (Spesolimab) Prevents Flare-Ups in Patients with Generalized Pustular Psoriasis. NCT04399837. Available online: https://clinicaltrials.gov/ct2/show/NCT04399837 (accessed on 16 May 2021).

{kind=link}
| Genetic Variant | Encoded Molecule and Its Function in Relation to the Pathogenesis of GPP | Type of Mutation and Its Consequence | Associated Pustular Psoriasis Subtype | Clinical Features of Mutation Carriers vs. Noncarriers | References |
|---|---|---|---|---|---|
| IL36RN | IL-36 receptor antagonist (IL-36Ra); counteracts the pro-inflammatory effect of IL-36 cytokines | Loss-of-function; amplification of the downstream inflammatory responses | GPP, ACH, PPP | Low frequency of concurrent PsV, earlier age of onset, increased risk of systemic manifestations | [22,24,46,47,58,59,64,65] |
| CARD14 | Caspase recruitment domain family member 14 (CARD14); regulates epidermal NF-κB signal transduction | Gain-of-function; enhancement of NF-κB signaling | GPP, PPP | Concomitant PsV was found in most cases | [52,69,72,73] |
| AP1S3 | Adaptor protein complex 1 subunit sigma 3 (AP1S3); regulates the trafficking of autophagosomes | Loss-of-function; disruption of autophagy in keratinocytes resulting in overproduction of pro-inflammatory cytokines | GPP, ACH, PPP | [50,51] | |
| TNIP1 | TNF-alpha induced protein 3-interacting protein 1 (TNIP1); inhibits NF-κB activation | Loss-of-function; enhancement of NF-κB signaling | GPP, ACH, PPP | Concomitant PsV is less frequent | [74,75,76] |
| SERPINA3 | Serine protease inhibitor A3 (serpin A3); inhibits cathepsin G and thereby limits inflammation | Loss-of-function; uninhibited processing of IL-36 cytokines to their more active forms results in an increase of their pro-inflammatory activity and uncontrolled inflammation | GPP | Negative correlation of mutation frequency with age | [78] |
| MPO | Myeloperoxidase (MPO); modulates neutrophilic inflammatory response | Loss-of-function; increase in neutrophil accumulation and activity, as well as a reduction in the number of apoptotic neutrophils | GPP, ACH | More frequent concurrent PPP, tongue involvement, positive family history for inflammatory skin and joint diseases | [86] |
| Treatment Type | Drug | Therapeutic Target | Properties | Rationale for the Treatment of GPP | Efficacy Data |
|---|---|---|---|---|---|
| Anti-TNF-α | Etanercept | TNF-α | Recombinant DNA-derived TNF receptor IgG fusion protein | TNF-α is a pro-inflammatory cytokine that is significantly upregulated in GPP lesions [23]. | Case reports revealed that etanercept is effective in patients with GPP, with or without IL36RN gene mutation; 50 mg biweekly dosing of etanercept was more effective, with good efficacy and rapid effect. Etanercept was also successfully used in treating GPP in children [31]. A retrospective study found that some patients did not respond to etanercept therapy [120]. |
| Adalimumab | TNF-α | Fully human monoclonal antibody | Clinical efficacy demonstrated in a phase 3 open-label study (7 of 10 GPP patients who received adalimumab achieved clinical response after 16 weeks) [134]. Adalimumab might be used as a first-line drug for childhood GPP [135]. However, adalimumab-resistant GPP patients have also been reported [136]. | ||
| Infliximab | TNF-α | Chimeric (mouse/human) IgG1 monoclonal antibody | Infliximab was reported to have a rapid onset of action (pustule clearance in 1–3 days) and is the most widely used and recommended TNF-α inhibitor in GPP [36]. | ||
| Certolizumab pegol | TNF-α | Recombinant, humanized, PEGylated Fab′-only antibody | Efficacy data in GPP is limited, however it appears to be an option for the treatment of pregnant women [137]. | ||
| Anti-IL-17 | Secukinumab | IL-17A | Fully human IgG1κ monoclonal antibody | IL-17A is an important activator of innate immune mechanisms, including the recruitment and survival of neutrophils [33]. | Sustained clinical efficacy demonstrated in case reports and several small open-label phase 3 trials in GPP [27,138]. Secukinumab was found to have the longest drug survival of all biologic and non-biologic agents in the treatment of GPP [139]. |
| Ixekizumab | IL-17A | Humanized IgG4 monoclonal antibody | Clinical efficacy demonstrated in an open-label uncontrolled study and in an open-label, phase 3 study [27,140]. | ||
| Brodalumab | IL-17RA | Fully human IgG2 monoclonal antibody | Treatment success, as defined by “improved” or “remission” on the CGI-I 4-point scale, was achieved in 10 of 12 (83.3%) and 11 of 12 (91.7%) patients at week 12 and 52, respectively [29]. | ||
| Anti-IL-12/IL-23 | Ustekinumab | IL-12/23 p40 | Fully human IgG1κ monoclonal antibody | IL-12 stimulates Th1 cells and IFN-γ production, IL-23 leads to the activation of Th17 cells [141]. | Several case reports and one case series demonstrating clinical efficacy [124,125,126]. |
| Anti-IL-23 | Guselkumab | IL-23 p19 | Fully human IgG1λ monoclonal antibody | IL-23 is a key regulator of multiple effector cytokines that has been demonstrated to play a role in the pathogenesis of GPP. Th17 cells are a major source of pro-inflammatory cytokines, including IL-17A that can promote tissue inflammation via IL-23 stimulation [105]. | In a phase 3, single-arm, open-label a total of 7/9 (77.8%) GPP patients achieved treatment success at week 16 [30]. |
| Risankizumab | IL-23 p19 | Humanized IgG1 monoclonal antibody | Efficacy data from phase III clinical trial in Japanese GPP patients have not been published yet [129]. | ||
| Anti-IL-1/IL-36 | Anakinra | IL-1R | Recombinant human monoclonal antibody | IL-1/IL-36 inflammatory axis is a potent driver of disease pathology in GPP [23]. | Excellent efficacy in GPP patients has been reported in several case reports [87,88,130]. |
| Canakinumab | IL-1β | Recombinant human monoclonal antibody | Canakinumab therapy attenuated the lesions of a patient with severe GPP, who had failed to response to anakinra [89]. | ||
| Gevokizumab | IL-1β | Humanized IgG2 monoclonal antibody | In an open-label study performed in patients with severe, recalcitrant GPP, 79% and 65% reductions in GPP area and severity index scores were achieved, respectively, after 4 weeks [90]. | ||
| Anti-IL-2 | Basiliximab | IL-2Rα chain (CD25) | Chimeric (mouse/human) monoclonal antibody | Psoriatic skin lesions demonstrate a type 1 cytokine profile as demonstrated by the predominance of IL-2 and IFN-γ expression [142]. | Basiliximab was successfully used in the treatment of severe GPP [143]. |
| Anti-IL-36 | Spesolimab (BI 655130) | IL-36R | Humanized IgG1 monoclonal antibody | Overexpression of IL-36 inflammatory cytokines in skin lesions and loss-of-function mutations in the IL36RN gene, as well as mutations in other genes connected with the IL-36 pathway have been identified in genetic studies for patients with GPP [23,46]. | Phase I proof-of-concept study showed rapid improvements in skin and pustule clearance with a single dose of spesolimab in patients with an acute GPP flare [91].Further phase II (Effisayil 1) and phase IIb (Effisayil 2) multicenter, randomized, double-blind, placebo-controlled clinical trials to investigate the efficacy of spesolimab in GPP are ongoing [92,144]. |
| Imsidolimab (ANB019) | IL-36R | Humanized monoclonal antibody | 75% of patients (6/8) achieved the primary endpoint of improvement in the CGI scale after 28 days of imisdolimab monotherapy.Imsidolimab was generally well-tolerated, and most treatment-emergent adverse events were mild to moderate in severity [133]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samotij, D.; Szczęch, J.; Reich, A. Generalized Pustular Psoriasis: Divergence of Innate and Adaptive Immunity. Int. J. Mol. Sci. 2021, 22, 9048. https://doi.org/10.3390/ijms22169048
Samotij D, Szczęch J, Reich A. Generalized Pustular Psoriasis: Divergence of Innate and Adaptive Immunity. International Journal of Molecular Sciences. 2021; 22(16):9048. https://doi.org/10.3390/ijms22169048
Chicago/Turabian StyleSamotij, Dominik, Justyna Szczęch, and Adam Reich. 2021. "Generalized Pustular Psoriasis: Divergence of Innate and Adaptive Immunity" International Journal of Molecular Sciences 22, no. 16: 9048. https://doi.org/10.3390/ijms22169048
APA StyleSamotij, D., Szczęch, J., & Reich, A. (2021). Generalized Pustular Psoriasis: Divergence of Innate and Adaptive Immunity. International Journal of Molecular Sciences, 22(16), 9048. https://doi.org/10.3390/ijms22169048