
The Importance of Digging into the Genetics of SMN Genes in the Therapeutic Scenario of Spinal Muscular Atrophy



Abstract
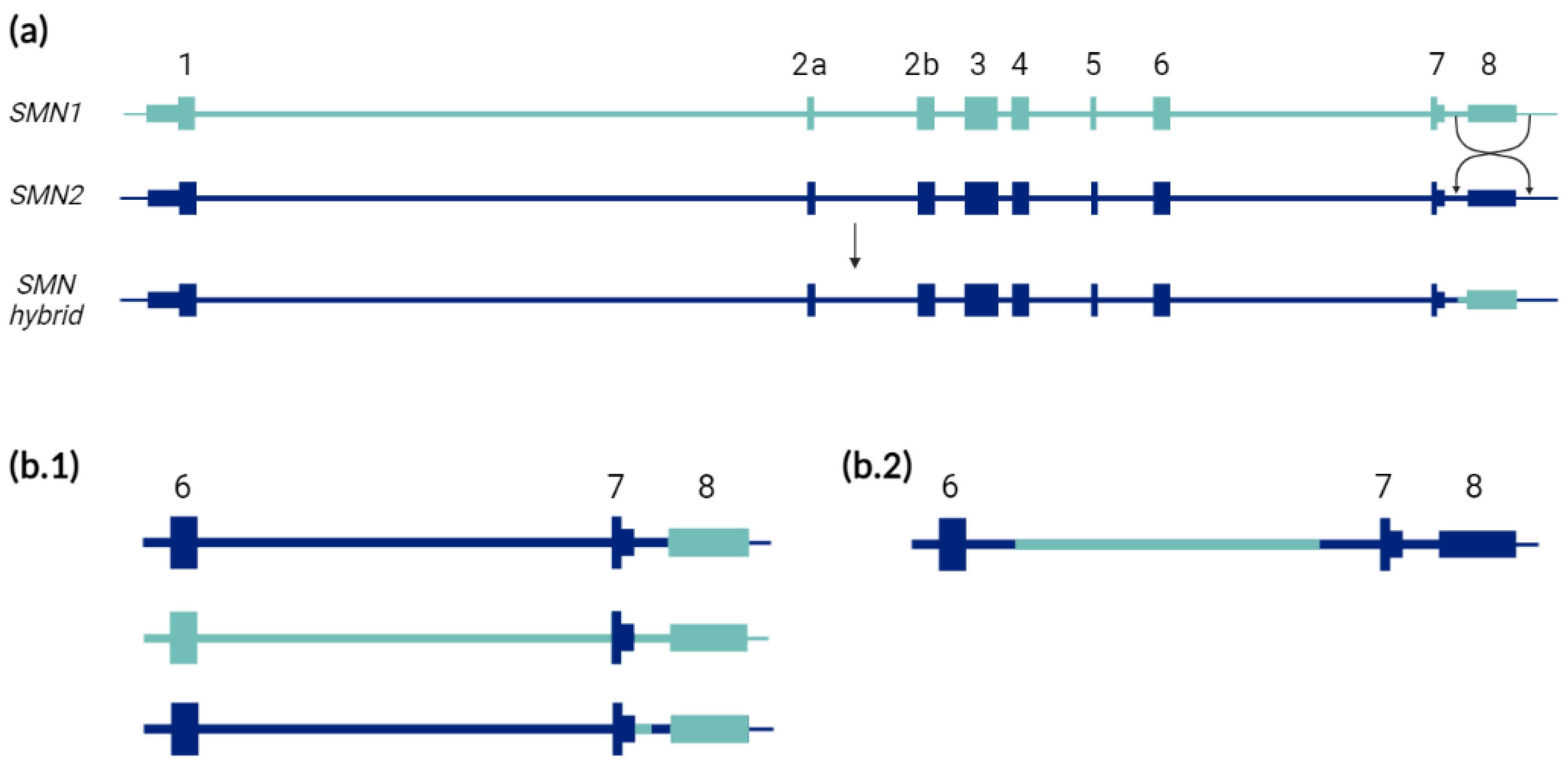
:1. SMA Is a Disease of Two Genes, a Determinant SMN1 and a Modifier SMN2
2. The Known Validated Genotypes
3. The Unknown or Yet Non-Validated Genotypes
4. Evolving Therapies and the Importance of SMN2
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef] [Green Version]
- Sugarman, E.; Nagan, N.; Zhu, H.; Akmaev, V.; Zhou, Z.; Rohlfs, E.; Flynn, K.; Hendrickson, B.; Scholl, T.; Sirko-Osadsa, D.; et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72,400 specimens. Eur. J. Hum. Genet. 2012, 20, 27–32. [Google Scholar] [CrossRef]
- Prior, T.W.; Leach, M.E.; Finanger, E. Spinal Muscular Atrophy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2000; updated 3 December 2020; Available online: Website (accessed on 28 July 2021).
- Talbot, K.; Tizzano, E.F. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017, 24, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Bürglen, L.; Lefebvre, S.; Clermont, O.; Burlet, P.; Viollet, L.; Cruaud, C.; Munnich, A.; Melki, J. Structure and organization of the human survival motor neurone (SMN) gene. Genomics 1996, 32, 479–482. [Google Scholar] [CrossRef]
- Rochette, C.; Gilbert, N.; Simard, L. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet. 2001, 108, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Ruhno, C.; McGovern, V.L.; Avenarius, M.R.; Snyder, P.J.; Prior, T.W.; Nery, F.C.; Muhtaseb, A.; Roggenbuck, J.S.; Kissel, J.T.; Sansone, V.A.; et al. Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Hum. Genet. 2019, 138, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Blasco-Pérez, L.; Paramonov, I.; Leno, J.; Bernal, S.; Alias, L.; Fuentes-Prior, P.; Cuscó, I.; Tizzano, E.F. Beyond copy number: A new, rapid, and versatile method for sequencing the entire SMN2 gene in SMA patients. Hum. Mutat. 2021, 42, 787–795. [Google Scholar] [CrossRef]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler-Botija, C.; Cuscó, I.; Caselles, L.; López, E.; Baiget, M.; Tizzano, E.F. Implication of fetal SMN2 expression in type I SMA pathogenesis: Protection or pathological gain of function? J. Neuropathol. Exp. Neurol. 2005, 64, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Wirth, B.; Garbes, L.; Riessland, M. How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr. Opin. Genet. Dev. 2013, 23, 330–338. [Google Scholar] [CrossRef]
- Boza-Morán, M.G.; Martínez-Hernández, R.; Bernal, S.; Wanisch, K.; Also-Rallo, E.; Le Heron, A.; Alías, L.; Denis, C.; Girard, M.; Yee, J.; et al. Decay in survival motor neuron and plastin 3 levels during differentiation of iPSC-derived human motor neurons. Sci. Rep. 2015, 5, 11696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Arkblad, E.L.; Darin, N.; Berg, K.; Kimber, E.; Brandberg, G.; Lindberg, C.; Holmberg, E.; Tulinius, M.; Nordling, M. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul. Disord. 2006, 16, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Vijzelaar, R.; Snetselaar, R.; Clausen, M.; Mason, A.G.; Rinsma, M.; Zegers, M.; Molleman, N.; Boschloo, R.; Yilmaz, R.; Kuilboer, R.; et al. The frequency of SMN gene variants lacking exon 7 and 8 is highly population dependent. PLoS ONE 2019, 14, e0220211. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Sanchis-Juan, A.; French, C.E.; Connell, A.J.; Delon, I.; Kingsbury, Z.; Chawla, A.; Halpern, A.L.; Taft, R.J.; Bentley, D.R.; et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet. Med. 2020, 22, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Alías, L.; Bernal, S.; Barceló, M.J.; Also-Rallo, E.; Martínez-Hernández, R.; Rodríguez-Alvarez, F.J.; Hernández-Chico, C.; Baiget, M.; Tizzano, E.F. Accuracy of marker analysis, quantitative real-time polymerase chain reaction, and multiple ligation-dependent probe amplification to determine SMN2 copy number in patients with spinal muscular atrophy. Genet. Test. Mol. Biomark. 2011, 15, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Cuscó, I.; Barceló, M.J.; del Rio, E.; Martín, Y.; Hernández-Chico, C.; Bussaglia, E.; Baiget, M.; Tizzano, E.F. Characterisation of SMN hybrid genes in Spanish SMA patients: De novo, homozygous and compound heterozygous cases. Hum. Genet. 2001, 108, 222–229. [Google Scholar] [CrossRef]
- Alías, L.; Bernal, S.; Fuentes-Prior, P.; Barceló, M.J.; Also, E.; Martínez-Hernández, R.; Rodríguez-Alvarez, F.J.; Martín, Y.; Aller, E.; Grau, E.; et al. Mutation update of spinal muscular atrophy in Spain: Molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum. Genet. 2009, 125, 29–39. [Google Scholar] [CrossRef]
- Hahnen, E.; Schönling, J.; Rudnik-Schöneborn, S.; Zerres, K.; Wirth, B. Hybrid survival motor neuron genes in patients with autosomal recessive spinal muscular atrophy: New insights into molecular mechanisms responsible for the disease. Am. J. Hum. Genet. 1996, 59, 1057–1065. [Google Scholar] [PubMed]
- Niba, E.T.E.; Nishio, H.; Wijaya, Y.O.S.; Lai, P.S.; Tozawa, T.; Chiyonobu, T.; Yamadera, M.; Okamoto, K.; Awano, H.; Takeshima, Y.; et al. Clinical phenotypes of spinal muscular atrophy patients with hybrid SMN gene. Brain Dev. 2021, 43, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, B.; Donohoe, C.; Akmaev, V.; Sugarman, E.; Labrousse, P.; Boguslavskiy, L.; Flynn, K.; Rohlfs, E.; Walker, A.; Allitto, B.; et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J. Med. Genet. 2009, 46, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Liu, L.; Peter, I.; Zhu, J.; Scott, S.A.; Zhao, G.; Eversley, C.; Kornreich, R.; Desnick, R.J.; Edelmann, L. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet. Med. 2014, 16, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Alías, L.; Barceló, M.J.; Bernal, S.; Martínez-Hernández, R.; Also-Rallo, E.; Vázquez, C.; Santana, A.; Millán, J.M.; Baiget, M.; Tizzano, E.F. Improving detection and genetic counseling in carriers of spinal muscular atrophy with two copies of the SMN1 gene. Clin. Genet. 2014, 85, 470–475. [Google Scholar] [CrossRef]
- Alías, L.; Bernal, S.; Calucho, M.; Martínez, E.; March, F.; Gallano, P.; Fuentes-Prior, P.; Abuli, A.; Serra-Juhe, C.; Tizzano, E.F. Utility of two SMN1 variants to improve spinal muscular atrophy carrier diagnosis and genetic counselling. Eur. J. Hum. Genet. 2018, 26, 1554–1557. [Google Scholar] [CrossRef]
- Coratti, G.; Messina, S.; Lucibello, S.; Pera, M.C.; Montes, J.; Pasternak, A.; Bovis, F.; Escudero, J.E.; Mazzone, E.S.; Mayhew, A.; et al. Clinical Variability in Spinal Muscular Atrophy Type III. Ann. Neurol. 2020, 88, 1109–1117. [Google Scholar] [CrossRef]
- Osredkar, D.; Jílková, M.; Butenko, T.; Loboda, T.; Golli, T.; Fuchsová, P.; Rohlenová, M.; Haberlova, J. Children and young adults with spinal muscular atrophy treated with nusinersen. Eur. J. Paediatr. Neurol. 2021, 30, 1–8. [Google Scholar] [CrossRef]
- Ou, S.; Ho, C.; Lee, W.; Lin, K.; Jones, C.; Jong, Y. Natural history in spinal muscular atrophy Type I in Taiwanese population: A longitudinal study. Brain Dev. 2021, 43, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.; Pinto, W.; Ricarte, A.; Badia, B.; Seneor, D.; Teixeira, D.; Caetano, L.; Gonçalves, E.; Chieia, M.; Farias, I.; et al. Clinical and radiological profile of patients with spinal muscular atrophy type 4. Eur. J. Neurol. 2021, 28, 609–619. [Google Scholar] [CrossRef]
- Zhang, Y.; He, J.; Zhang, Y.; Li, L.; Tang, X.; Wang, L.; Guo, J.; Jin, C.; Tighe, S.; Zhang, Y.; et al. The analysis of the association between the copy numbers of survival motor neuron gene 2 and neuronal apoptosis inhibitory protein genes and the clinical phenotypes in 40 patients with spinal muscular atrophy: Observational study. Medicine (Baltim.) 2020, 99, e18809. [Google Scholar] [CrossRef] [PubMed]
- Lusakowska, A.; Jedrzejowska, M.; Kaminska, A.; Janiszewska, K.; Grochowski, P.; Zimowski, J.; Sierdzinski, J.; Kostera-Pruszczyk, A. Observation of the natural course of type 3 spinal muscular atrophy: Data from the polish registry of spinal muscular atrophy. Orphanet J. Rare Dis. 2021, 16. [Google Scholar] [CrossRef] [PubMed]
- Aragon-Gawinska, K.; Seferian, A.; Daron, A.; Gargaun, E.; Vuillerot, C.; Cances, C.; Ropars, J.; Chouchane, M.; Cuppen, I.; Hughes, I.; et al. Nusinersen in patients older than 7 months with spinal muscular atrophy type 1: A cohort study. Neurology 2018, 91, e1312–e1318. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Kong, X.; Zhao, Z.; Zhao, X. Mutation analysis of 419 family and prenatal diagnosis of 339 cases of spinal muscular atrophy in China. BMC Med. Genet. 2020, 21, 133. [Google Scholar] [CrossRef]
- Prior, T.W.; Krainer, A.R.; Hua, Y.; Swoboda, K.J.; Snyder, P.C.; Bridgeman, S.J.; Burghes, A.H.M.; Kissel, J.T. A Positive Modifier of Spinal Muscular Atrophy in the SMN2 Gene. Am. J. Hum. Genet. 2009, 85, 408–413. [Google Scholar] [CrossRef] [Green Version]
- Vezain, M.; Saugier-Veber, P.; Goina, E.; Touraine, R.; Manel, V.; Toutain, A.; Fehrenbach, S.; Frébourg, T.; Pagani, F.; Tosi, M.; et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum. Mutat. 2010, 31, E1110–E1125. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S.H.; Sun, J.; Krainer, A.R.; Hua, Y.; Prior, T.W. A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 2768–2780. [Google Scholar] [CrossRef] [Green Version]
- Bernal, S.; Alías, L.; Barceló, M.J.; Also-Rallo, E.; Martínez-Hernández, R.; Gámez, J.; Guillén-Navarro, E.; Rosell, J.; Hernando, I.; Rodríguez-Alvarez, F.J.; et al. The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J. Med. Genet. 2010, 47, 640–642. [Google Scholar] [CrossRef] [Green Version]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.M.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Bowen, B.M.; Truty, R.; Aradhya, S.; Bristow, S.L.; Johnson, B.A.; Morales, A.; Tan, C.A.; Westbrook, M.J.; Winder, T.L.; Chavez, J.C. SMA Identified: Clinical and Molecular Findings From a Sponsored Testing Program for Spinal Muscular Atrophy in More Than 2,000 Individuals. Front. Neurol. 2021, 12, 676. [Google Scholar] [CrossRef]
- Cuscó, I.; Bernal, S.; Blasco-Pérez, L.; Calucho, M.; Alias, L.; Fuentes-Prior, P.; Tizzano, E.F. Practical guidelines to manage discordant situations of SMN2 copy number in patients with spinal muscular atrophy. Neurol. Genet. 2020, 6, e530. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Bai, J.; Cao, Y.; Wang, H.; Jin, Y.; Du, J.; Ge, X.; Zhang, W.; Li, Y.; He, S.; et al. Mutation Spectrum of the Survival of Motor Neuron 1 and Functional Analysis of Variants in Chinese Spinal Muscular Atrophy. J. Mol. Diagn. 2016, 18, 741–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijaya, Y.; Ar Rohmah, M.; Niba, E.; Morisada, N.; Noguchi, Y.; Saito, K.; Nishio, H.; Shinohara, M. Phenotypes of SMA patients retaining SMN1 with intragenic mutation. Brain Dev. 2021, 43, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Cuscó, I.; Barceló, M.; del Río, E.; Baiget, M.; Tizzano, E. Detection of novel mutations in the SMN Tudor domain in type I SMA patients. Neurology 2004, 63, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Kotani, T.; Sutomo, R.; Sasongko, T.; Sadewa, A.; Gunadi; Minato, T.; Fujii, E.; Endo, S.; Lee, M.; Ayaki, E.; et al. A novel mutation at the N-terminal of SMN Tudor domain inhibits its interaction with target proteins. J. Neurol. 2007, 254, 624–630. [Google Scholar] [CrossRef]
- Yamamoto, T.; Sato, H.; Lai, P.S.; Nurputra, D.K.; Harahap, N.I.F.; Morikawa, S.; Nishimura, N.; Kurashige, T.; Ohshita, T.; Nakajima, H.; et al. Intragenic mutations in SMN1 may contribute more significantly to clinical severity than SMN2 copy numbers in some spinal muscular atrophy (SMA) patients. Brain Dev. 2014, 36, 914–920. [Google Scholar] [CrossRef]
- Takarada, T.; Rochmah, M.A.; Harahap, N.; Shinohara, M.; Saito, T.; Saito, K.; Lai, P.; Bouike, Y.; Takeshima, Y.; Awano, H.; et al. SMA mutations in SMN Tudor and C-terminal domains destabilize the protein. Brain Dev. 2017, 39, 606–612. [Google Scholar] [CrossRef]
- Parsons, D.; McAndrew, P.; Monani, U.; Mendell, J.; Burghes, A.; Prior, T. An 11 base pair duplication in exon 6 of the SMN gene produces a type I spinal muscular atrophy (SMA) phenotype: Further evidence for SMN as the primary SMA-determining gene. Hum. Mol. Genet. 1996, 5, 1727–1732. [Google Scholar] [CrossRef]
- Mendonça, R.; Rocha, A.; Lozano-Arango, A.; Diaz, A.; Castiglioni, C.; Silva, A.; Reed, U.; Kulikowski, L.; Paramonov, I.; Cuscó, I.; et al. Severe brain involvement in 5q spinal muscular atrophy type 0. Ann. Neurol. 2019, 86, 458–462. [Google Scholar] [CrossRef]
- Kubo, Y.; Nishio, H.; Saito, K. A new method for SMN1 and hybrid SMN gene analysis in spinal muscular atrophy using long-range PCR followed by sequencing. J. Hum. Genet. 2015, 60, 233–239. [Google Scholar] [CrossRef] [Green Version]
- Mendonça, R.; Matsui, C.; Polido, G.; Silva, A.; Kulikowski, L.; Dias, A.T.; Zanardo, E.; Solla, D.; Kok, F.; Reed, U.; et al. Intragenic variants in the SMN1 gene determine the clinical phenotype in 5q spinal muscular atrophy. Neurol. Genet. 2020, 6, e505. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Pastore, M.T.; Gavrilina, T.O.; Jablonka, S.; Le, T.T.; Andreassi, C.; DiCocco, J.M.; Lorson, C.; Androphy, E.J.; Sendtner, M.; et al. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J. Cell Biol. 2003, 160, 41–52. [Google Scholar] [CrossRef]
- Wadman, R.I.; Jansen, M.D.; Stam, M.; Wijngaarde, C.A.; Curial, C.A.D.; Medic, J.; Sodaar, P.; Schouten, J.; Vijzelaar, R.; Lemmink, H.H.; et al. Intragenic and structural variation in the SMN locus and clinical variability in spinal muscular atrophy. Brain Commun. 2020, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schorling, D.; Becker, J.; Pechmann, A.; Langer, T.; Wirth, B.; Kirschner, J. Discrepancy in redetermination of SMN2 copy numbers in children with SMA. Neurology 2019, 93, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Jedličková, I.; Přistoupilová, A.; Nosková, L.; Majer, F.; Stránecký, V.; Hartmannová, H.; Hodaňová, K.; Trešlová, H.; Hýblová, M.; Solár, P.; et al. Spinal muscular atrophy caused by a novel Alu-mediated deletion of exons 2a-5 in SMN1 undetectable with routine genetic testing. Mol. Genet. Genom. Med. 2020, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauke, J.; Riessland, M.; Lunke, S.; Eyüpoglu, I.; Blümcke, I.; El-Osta, A.; Wirth, B.; Hahnen, E. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum. Mol. Genet. 2009, 18, 304–317. [Google Scholar] [CrossRef]
- Woo, C.J.; Maier, V.K.; Davey, R.; Brennan, J.; Li, G.; Brothers, J.; Schwartz, B.; Gordo, S.; Kasper, A.; Okamoto, T.R.; et al. Gene activation of SMN by selective disruption of lncRNA-mediated recruitment of PRC2 for the treatment of spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2017, 114, E1509–E1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nizzardo, M.; Simone, C.; Dametti, S.; Salani, S.; Ulzi, G.; Pagliarani, S.; Rizzo, F.; Frattini, E.; Pagani, F.; Bresolin, N.; et al. Spinal muscular atrophy phenotype is ameliorated in human motor neurons by SMN increase via different novel RNA therapeutic approaches. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cuscó, I.; Barceló, M.; Rojas-García, R.; Illa, I.; Gámez, J.; Cervera, C.; Pou, A.; Izquierdo, G.; Baiget, M.; Tizzano, E. SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J. Neurol. 2006, 253, 21–25. [Google Scholar] [CrossRef]
- Medrano, S.; Monges, S.; Gravina, L.P.; Alías, L.; Mozzoni, J.; Aráoz, H.V.; Bernal, S.; Moresco, A.; Chertkoff, L.; Tizzano, E. Genotype–phenotype correlation of SMN locus genes in spinal muscular atrophy children from Argentina. Eur. J. Paediatr. Neurol. 2016, 20, 910–917. [Google Scholar] [CrossRef]
- Brkušanin, M.; Kosać, A.; Jovanović, V.; Pešović, J.; Brajušković, G.; Dimitrijević, N.; Todorović, S.; Romac, S.; Rašić, V.M.; Savić-Pavićević, D. Joint effect of the SMN2 and SERF1A genes on childhood-onset types of spinal muscular atrophy in Serbian patients. J. Hum. Genet. 2015, 60, 723–728. [Google Scholar] [CrossRef]
- Wirth, B. Spinal Muscular Atrophy: In the Challenge Lies a Solution. Trends Neurosci. 2021, 44, 306–322. [Google Scholar] [CrossRef]
- Oprea, G.; Kröber, S.; McWhorter, M.; Rossoll, W.; Müller, S.; Krawczak, M.; Bassell, G.; Beattie, C.; Wirth, B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, B.; Mendoza-Ferreira, N.; Torres-Benito, L. Chapter 12—Spinal muscular atrophy disease modifiers. In Spinal Muscular Atrophy: Disease Mechanisms and Therapy; Academic Press: Amsterdam, The Netherlands, 2017; pp. 191–210. [Google Scholar]
- Pedrotti, S.; Bielli, P.; Paronetto, M.; Ciccosanti, F.; Fimia, G.; Stamm, S.; Manley, J.; Sette, C. The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J. 2010, 29, 1235–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powis, R.A.; Karyka, E.; Boyd, P.; Côme, J.; Jones, R.A.; Zheng, Y.; Szunyogova, E.; Groen, E.J.; Hunter, G.; Thomson, D.; et al. Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight 2016, 1, e87908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheleznyakova, G.; Nilsson, E.; Kiselev, A.; Maretina, M.; Tishchenko, L.; Fredriksson, R.; Baranov, V.; Schiöth, H. Methylation levels of SLC23A2 and NCOR2 genes correlate with spinal muscular atrophy severity. PLoS ONE 2015, 10, e0121964. [Google Scholar] [CrossRef]
- Branchu, J.; Biondi, O.; Chali, F.; Collin, T.; Leroy, F.; Mamchaoui, K.; Makoukji, J.; Pariset, C.; Lopes, P.; Massaad, C.; et al. Shift from extracellular signal-regulated kinase to AKT/cAMP response element-binding protein pathway increases survival-motor-neuron expression in spinal-muscular-atrophy-like mice and patient cells. J. Neurosci. 2013, 33, 4280–4294. [Google Scholar] [CrossRef] [Green Version]
- Riessland, M.; Kaczmarek, A.; Schneider, S.; Swoboda, K.J.; Löhr, H.; Bradler, C.; Grysko, V.; Dimitriadi, M.; Hosseinibarkooie, S.; Torres-Benito, L.; et al. Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. Am. J. Hum. Genet. 2017, 100, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Little, D.; Valori, C.F.; Mutsaers, C.A.; Bennett, E.J.; Wyles, M.; Sharrack, B.; Shaw, P.J.; Gillingwater, T.H.; Azzouz, M.; Ning, K. PTEN Depletion Decreases Disease Severity and Modestly Prolongs Survival in a Mouse Model of Spinal Muscular Atrophy. Mol. Ther. 2015, 23, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Helmken, C.; Hofmann, Y.; Schoenen, F.; Oprea, G.; Raschke, H.; Rudnik-Schöneborn, S.; Zerres, Z.; Wirth, B. Evidence for a modifying pathway in SMA discordant families: Reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum. Genet. 2003, 114, 11–21. [Google Scholar] [CrossRef]
- Soler-Botija, C.; Ferrer, I.; Alvarez, J.; Baiget, M.; Tizzano, E. Downregulation of Bcl-2 proteins in type I spinal muscular atrophy motor neurons during fetal development. J. Neuropathol. Exp. Neurol. 2003, 62, 420–426. [Google Scholar] [CrossRef] [Green Version]
- Farooq, F.; Molina, F.A.; Hadwen, J.; MacKenzie, D.; Witherspoon, L.; Osmond, M.; Holcik, M.; MacKenzie, A. Prolactin increases SMN expression and survival in a mouse model of severe spinal muscular atrophy via the STAT5 pathway. J. Clin. Investig. 2011, 121, 3050. [Google Scholar] [CrossRef] [PubMed]
- Grondard, C.; Biondi, O.; Armand, A.-S.; Lécolle, S.; Gaspera, B.D.; Pariset, C.; Li, H.; Gallien, C.-L.; Vidal, P.-P.; Chanoine, C.; et al. Regular Exercise Prolongs Survival in a Type 2 Spinal Muscular Atrophy Model Mouse. J. Neurosci. 2005, 25, 7622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in Type 1 Spinal Muscular Atrophy. N. Engl. J. Med. 2021, 384, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Lee, B.H.; Collins, E.; Lewis, L.; Guntrum, D.; Eichinger, K.; Voter, K.; Abdel-Hamid, H.Z.; Ciafaloni, E. Combination therapy with nusinersen and AVXS-101 in SMA type 1. Neurology 2019, 93, 640–641. [Google Scholar] [CrossRef]
- Matesanz, S.E.; Curry, C.; Gross, B.; Rubin, A.I.; Linn, R.; Yum, S.W.; Kichula, E.A. Clinical Course in a Patient With Spinal Muscular Atrophy Type 0 Treated With Nusinersen and Onasemnogene Abeparvovec. J. Child Neurol. 2020, 35, 717–723. [Google Scholar] [CrossRef]
- Harada, Y.; Rao, V.K.; Arya, K.; Kuntz, N.L.; DiDonato, C.J.; Napchan-Pomerantz, G.; Agarwal, A.; Stefans, V.; Katsuno, M.; Veerapandiyan, A. Combination molecular therapies for type 1 spinal muscular atrophy. Muscle Nerve 2020, 62, 550–554. [Google Scholar] [CrossRef]
- Van Alstyne, M.; Tattoli, I.; Delestrée, N.; Recinos, Y.; Workman, E.; Shihabuddin, L.; Zhang, C.; Mentis, G.; Pellizzoni, L. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat. Neurosci. 2021, 24, 930–940. [Google Scholar] [CrossRef]
- Kirschner, J.; Butoianu, N.; Goemans, N.; Haberlova, J.; Kostera-Pruszczyk, A.; Mercuri, E.; van der Pol, W.L.; Quijano-Roy, S.; Sejersen, T.; Tizzano, E.F.; et al. European ad-hoc consensus statement on gene replacement therapy for spinal muscular atrophy. Eur. J. Paediatr. Neurol. 2020, 28, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Tizzano, E.F.; Finkel, R.S. Spinal muscular atrophy: A changing phenotype beyond the clinical trials. Neuromuscul. Disord. 2017, 27, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, V.; Guerra, M.; Di Rosa, V.; Compagnucci, C.; Sette, C. Combined treatment with the histone deacetylase inhibitor LBH589 and a splice-switch antisense oligonucleotide enhances SMN2 splicing and SMN expression in Spinal Muscular Atrophy cells. J. Neurochem. 2020, 153, 264–275. [Google Scholar] [CrossRef] [PubMed]
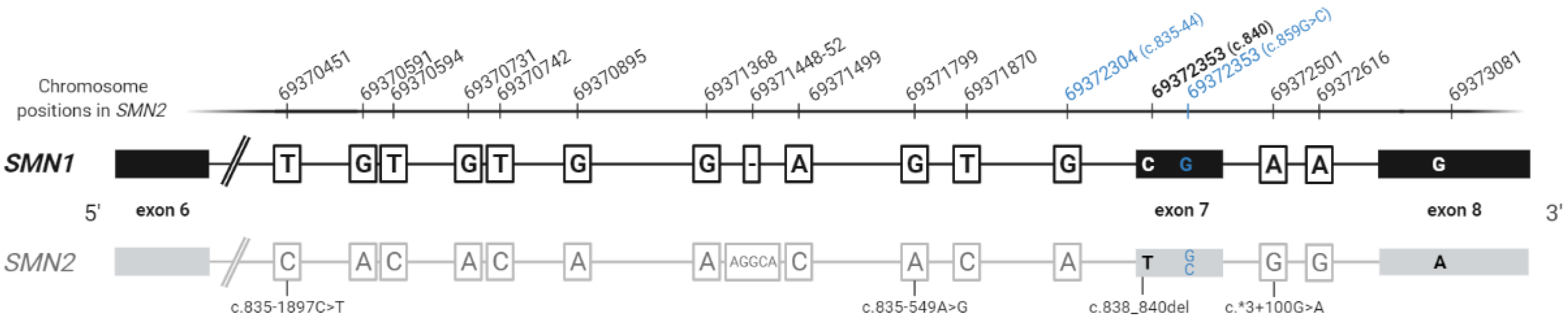
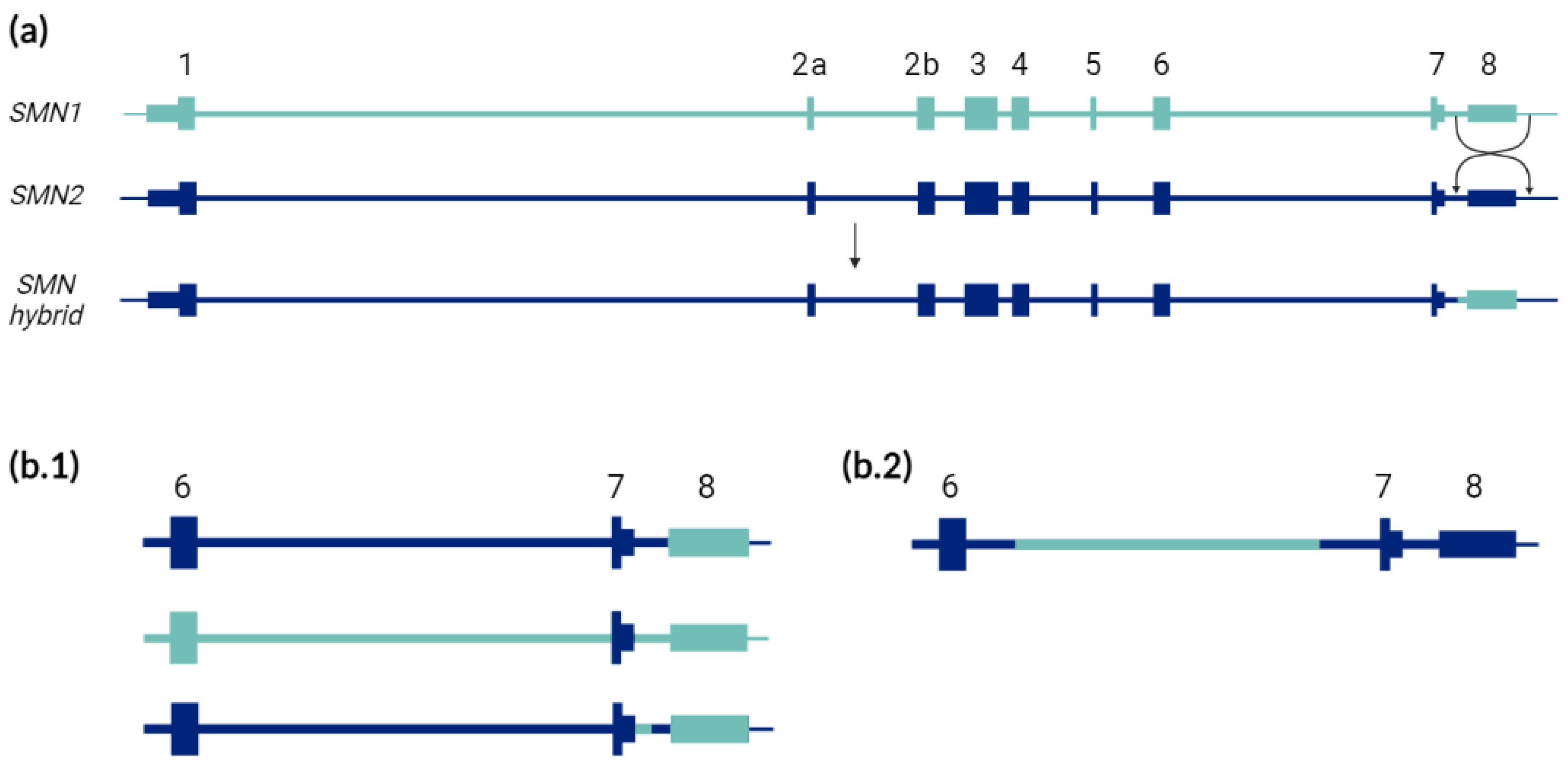
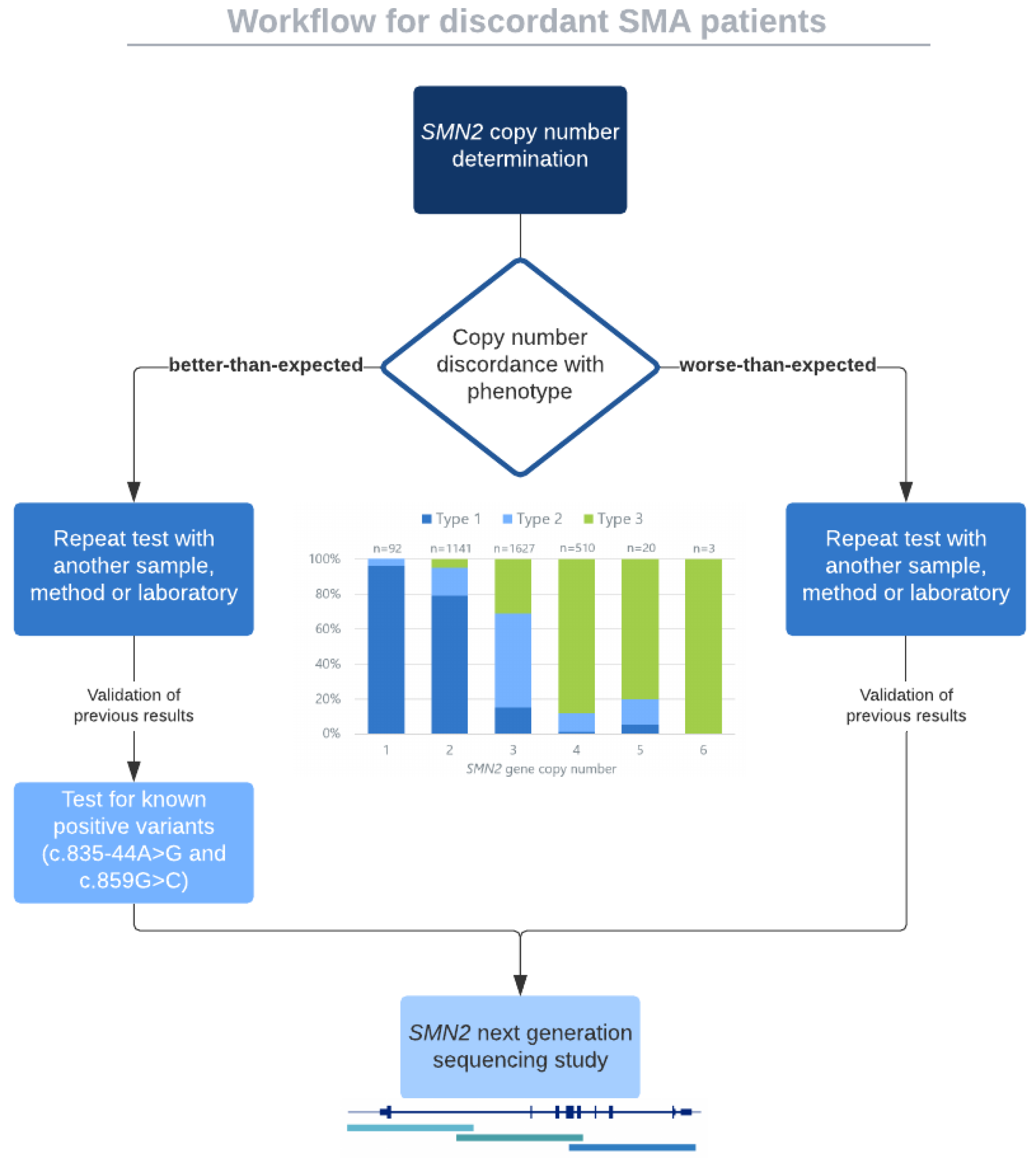

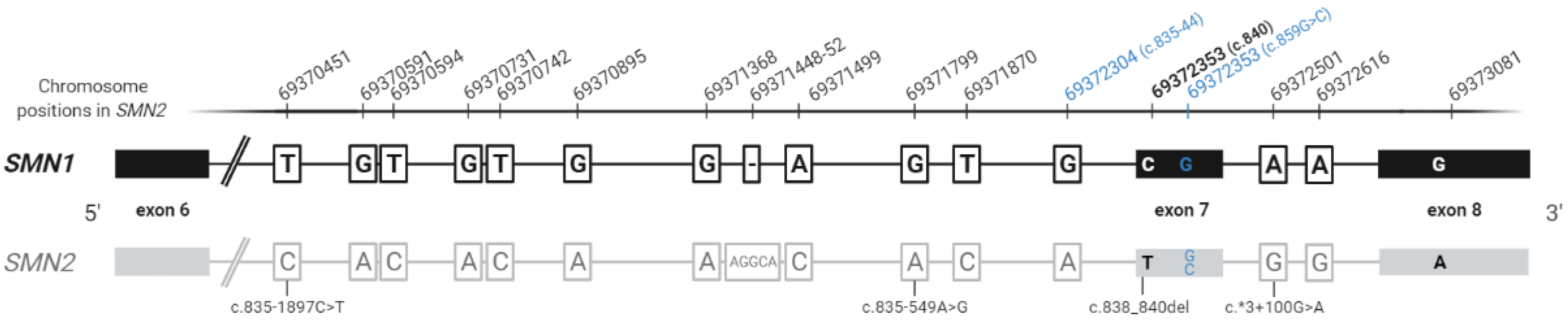{kind=link}
{kind=link}
{kind=link}
| Complex Biological Features of SMN Region | Technical Limitations |
|---|---|
| High homology between SMN1 and SMN2 | Difficulty in establishing if specific variants belong to SMN1 or SMN2 |
| Multiple copies of SMN genes | Inaccurate copy number determination |
| Partial deletions | Undetectable by routine analysis (exonic sequencing and MLPA) |
| SMN2/SMN1 hybrid structures | |
| Unknown variants in deep intronic regions |
| Modifier Type | Example | Effect | Reference |
|---|---|---|---|
| SMA locus | SMN2 copies and variants | SMA types | Calucho et al. 2018 [15] |
| Splicing regulators | hnRNP-A1/Sam68 | Exon 7 inclusion | Pedrotti et al. 2010 [66] |
| SMN degradation | UBA1 | ↑ Survival SMA mice | Powis et al. 2016 [67] |
| DNA methylation | SLC23A2/NCOR2 | SMA types differences | Zheleznyakova et al. 2015 [68] |
| Actin polymerization | PLS3 | Siblings differences | Oprea et al. 2008 [64] |
| Cytoskeleton dynamics | ERK | ↑ Survival SMA mice | Branchu et al. 2013 [69] |
| Endocytosis regulators | NCALD | Ameliorates SMA | Riessland et al. 2017 [70] |
| Neurogenesis regulators | PTEN | ↑ Survival SMA mice | Little et al. 2015 [71] |
| Axogenesis | ZPR1 | ↓ in SMA patients | Helmken et al. 2003 [72] |
| Apoptosis | Bcl2 | ↓ SMA motor neurons | Soler-Botija et al. 2003 [73] |
| Hormones/growth factors | Prolactin | ↑ Survival SMA mice | Farooq et al. 2011 [74] |
| Environmental factors | Exercise | ↑ Survival SMA mice | Grondard et al. 2005 [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa-Roger, M.; Blasco-Pérez, L.; Cuscó, I.; Tizzano, E.F. The Importance of Digging into the Genetics of SMN Genes in the Therapeutic Scenario of Spinal Muscular Atrophy. Int. J. Mol. Sci. 2021, 22, 9029. https://doi.org/10.3390/ijms22169029
Costa-Roger M, Blasco-Pérez L, Cuscó I, Tizzano EF. The Importance of Digging into the Genetics of SMN Genes in the Therapeutic Scenario of Spinal Muscular Atrophy. International Journal of Molecular Sciences. 2021; 22(16):9029. https://doi.org/10.3390/ijms22169029
Chicago/Turabian StyleCosta-Roger, Mar, Laura Blasco-Pérez, Ivon Cuscó, and Eduardo F. Tizzano. 2021. "The Importance of Digging into the Genetics of SMN Genes in the Therapeutic Scenario of Spinal Muscular Atrophy" International Journal of Molecular Sciences 22, no. 16: 9029. https://doi.org/10.3390/ijms22169029
APA StyleCosta-Roger, M., Blasco-Pérez, L., Cuscó, I., & Tizzano, E. F. (2021). The Importance of Digging into the Genetics of SMN Genes in the Therapeutic Scenario of Spinal Muscular Atrophy. International Journal of Molecular Sciences, 22(16), 9029. https://doi.org/10.3390/ijms22169029