
Acid Sphingomyelinase, a Lysosomal and Secretory Phospholipase C, Is Key for Cellular Phospholipid Catabolism


Abstract
:1. Introduction
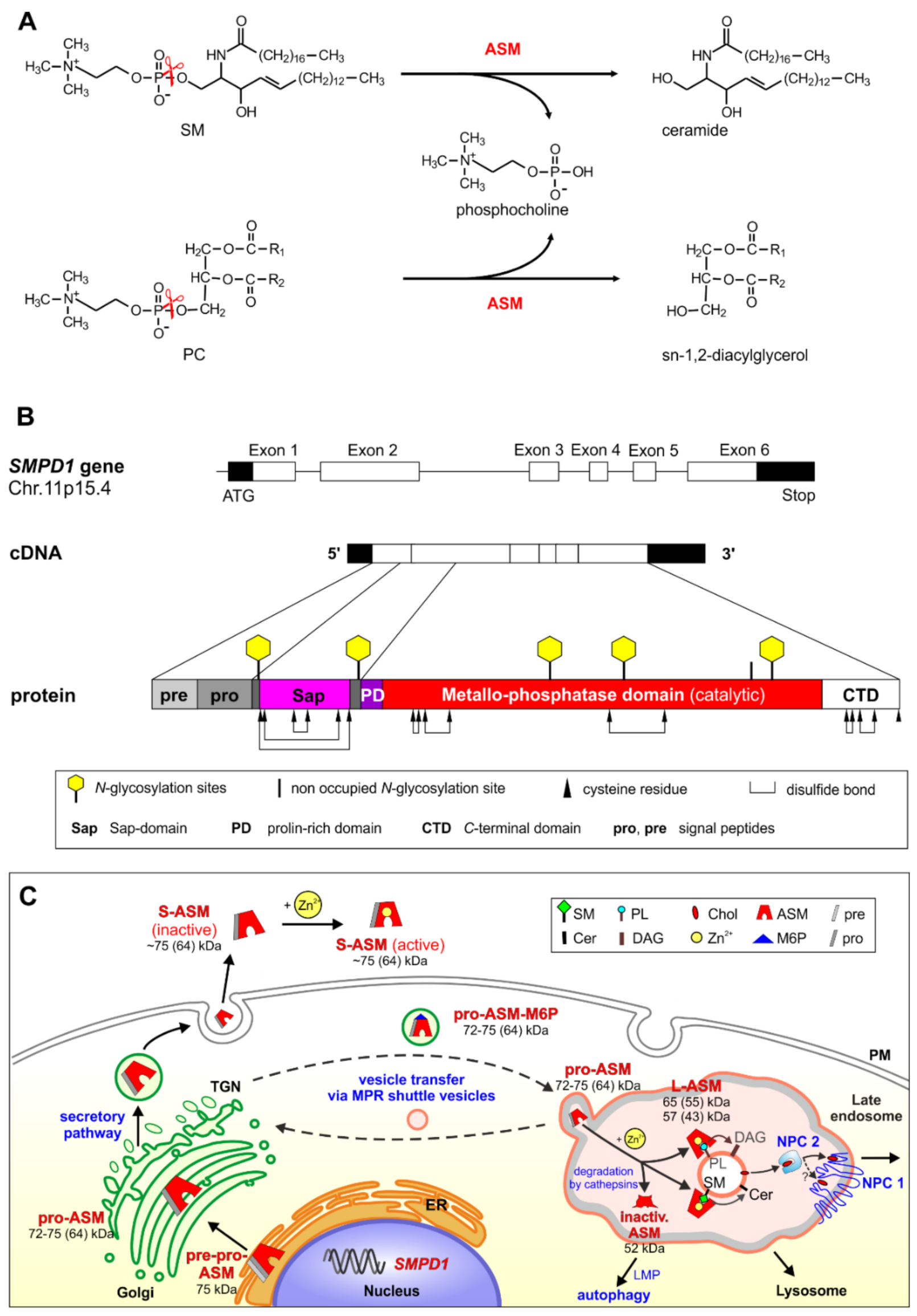
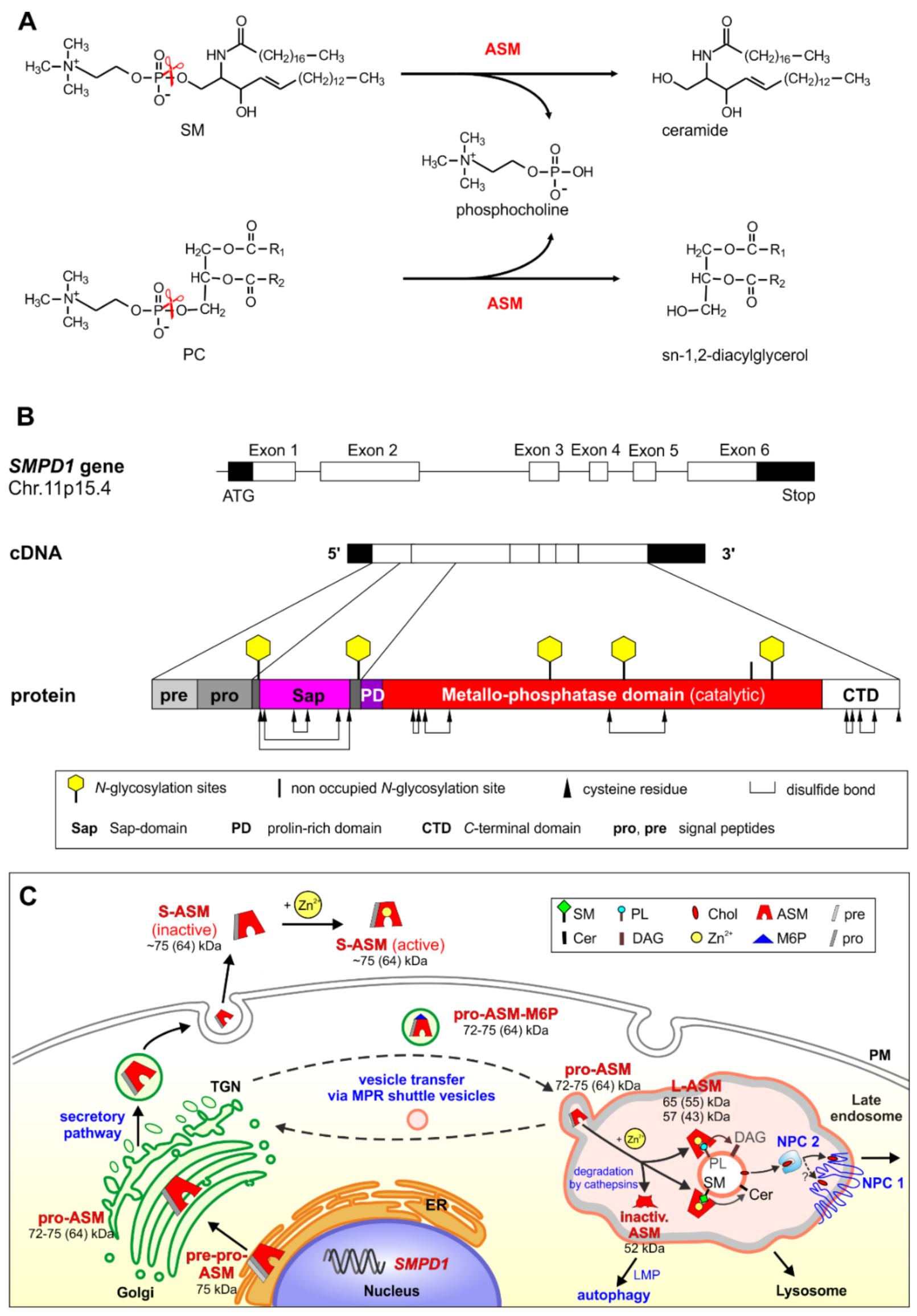
2. Biosynthesis, Cellular Processing, Trafficking and Structure of Human ASM
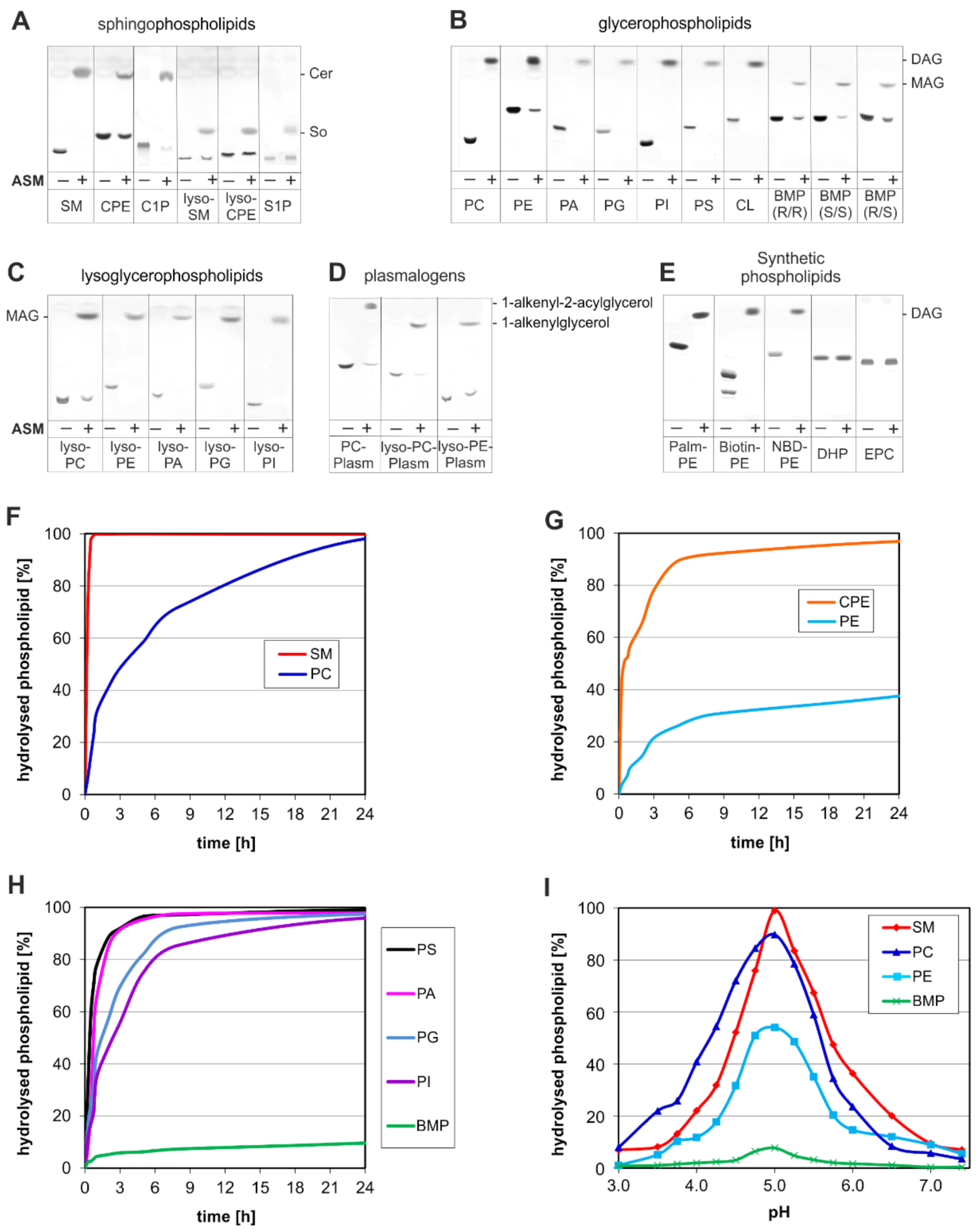
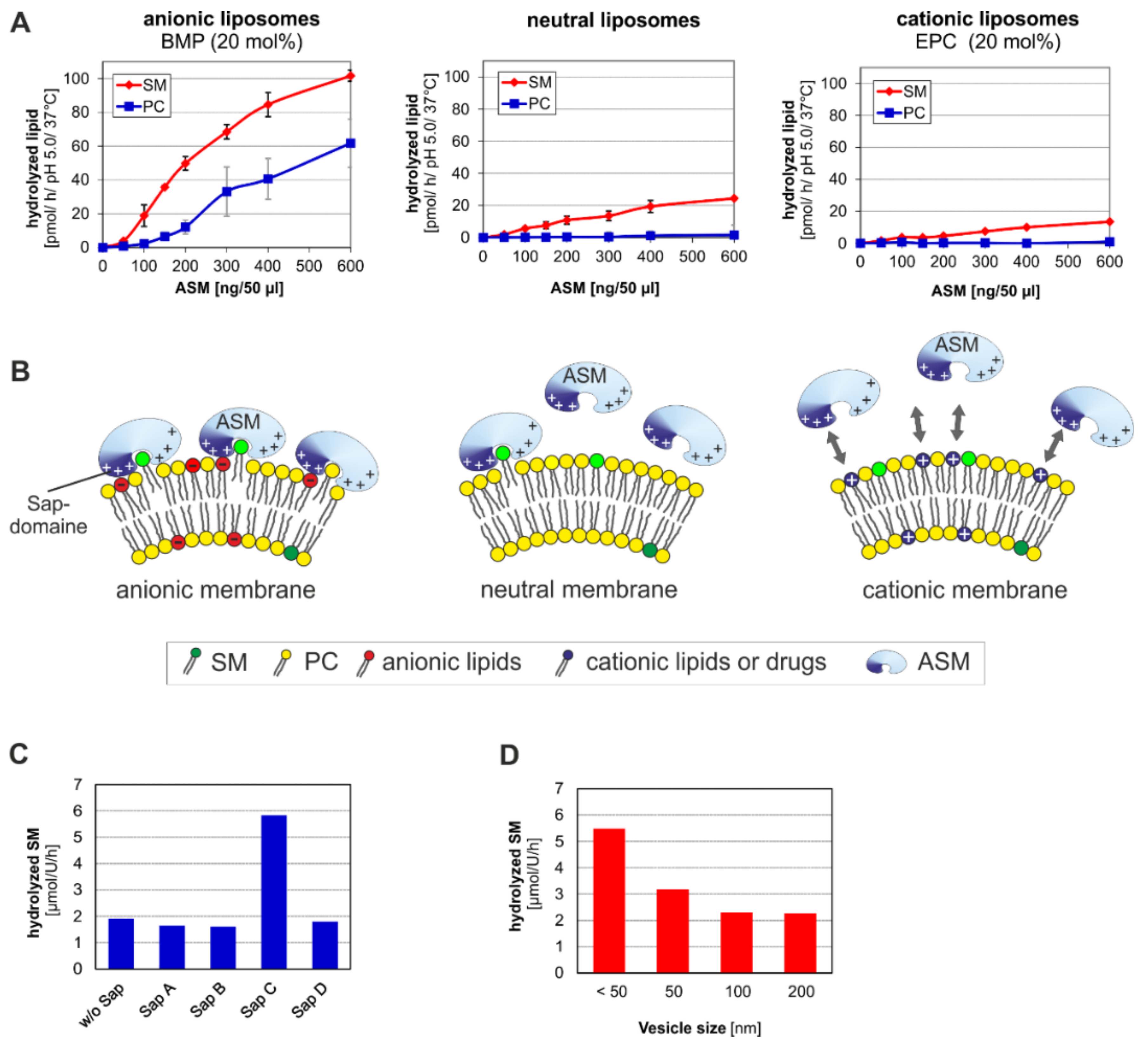
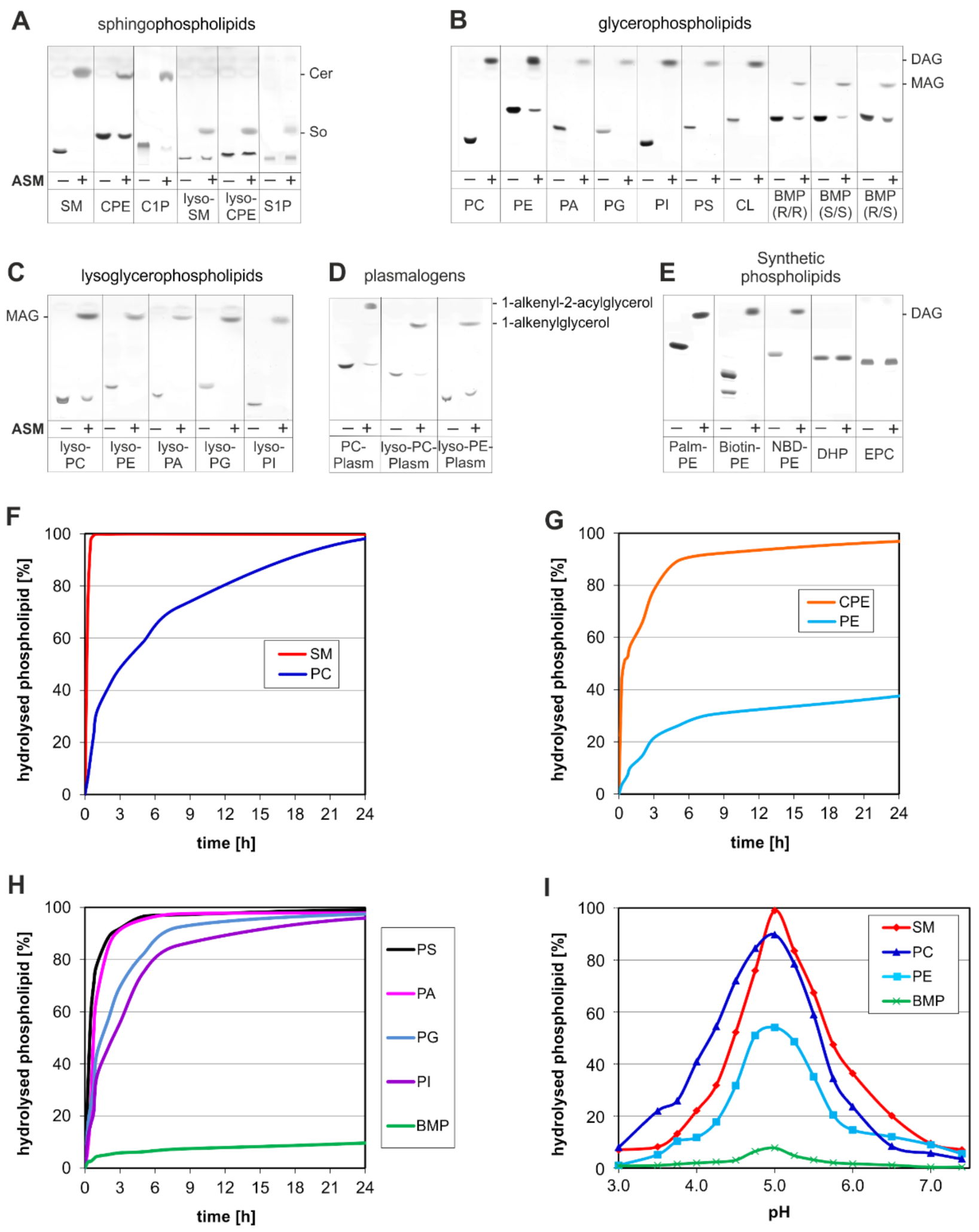
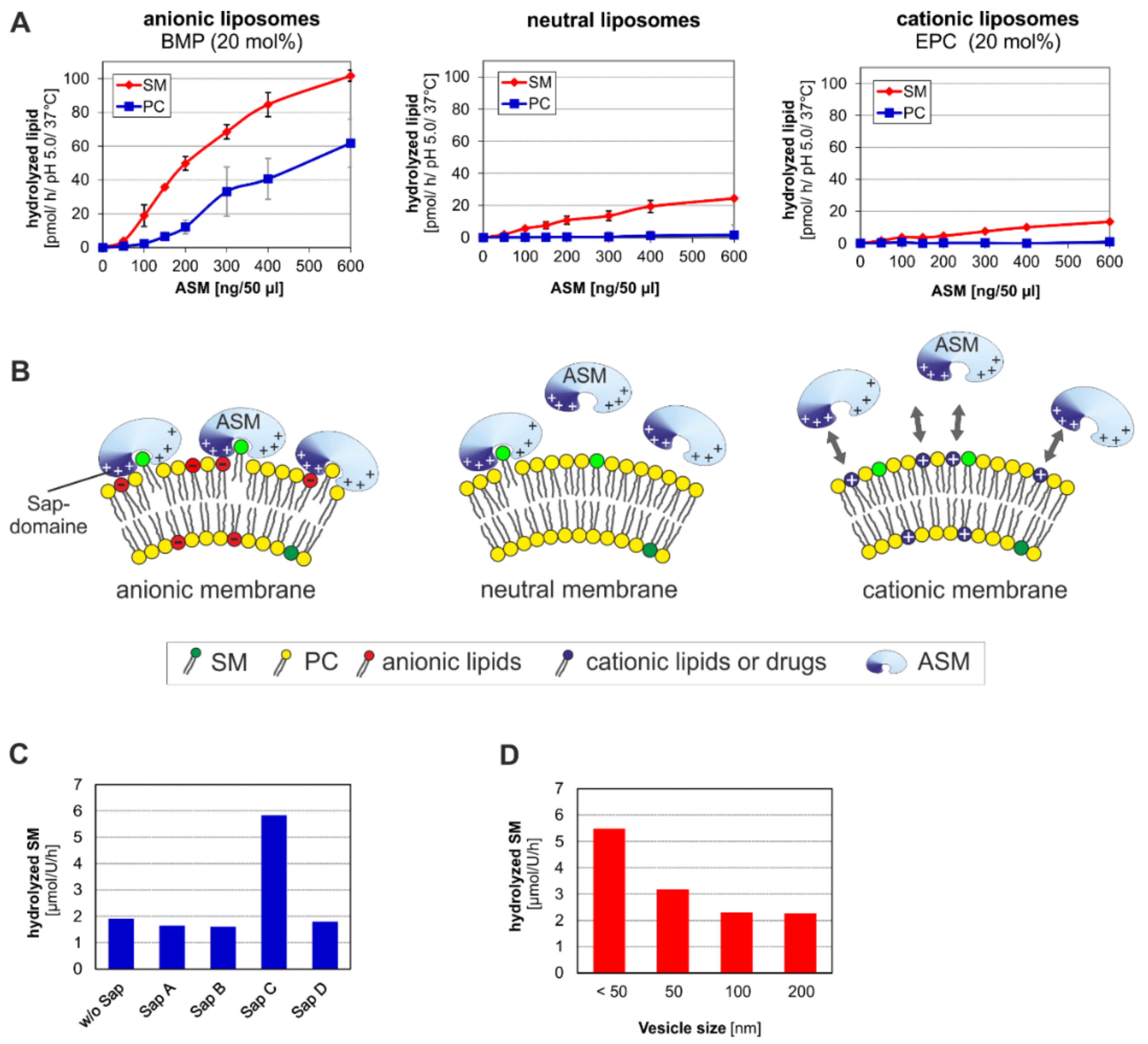
3. ASM, an Endolysosomal and Secretory Phospholipase C, Cleaving Membrane Lipids
- (a)
- the surface potential (measured by zeta potential) of the liposomal vesicles [48], modelling the ILVs of the lysosomal compartment,
- (b)
- the curvature of the membrane vesicles (e.g., the ILVs) [44],
- (c)
- the hydrolysis stimulating lipid binding proteins (the sphingolipid activator proteins) [49],
- (d)
- (e)
4. Emerging Functions of ASM, a Promiscuous Phospholipase C, Cleaving Membrane Lipids, Including Cer1P and BMP

{kind=link}
{kind=link}
{kind=link}
| Regulating Lipid | Sphingomyelin | References | Phosphatidylcholine | References |
|---|---|---|---|---|
| BMP | + | [42,48] | + | [48] |
| Anionic PLs | + | [42,48,74] | + | [48] |
| Cationic lipids/drugs | − | [48] | − | [48] |
| Cholesterol | ◯ | [48] | + (BMP) − (other anionic lipids) | [48] |
| Ceramide | (+) | [48] | − | [48] |
| Diacylglycerol | + | [12,48] | − | [48] |
| Fatty acids | + | [12,48] | − | [48] |
| Monoacylglycerol | + | [12,48] | − | [48] |
| Phosphatidylinositol-3,5-bisphosphate | − | [75] | ||
| Phosphatidylinositol-4,5-bisphosphate | − | [12] | ||
| Phosphatidylinositol-3,4,5-triphosphate | − | [76] | ||
| Sphingosin-1-phosphate | − 1 | [77] |
5. Topology and Regulation of PL-Cleaving Activity of ASM
5.1. Lipid Sorting and Maturation of ILVs
| Sphingomyelin 1 | Cholesterol 2 | Ceramide 3 | Diacylglycerol 4 | References | |
|---|---|---|---|---|---|
| Lipid transfer | |||||
| Cholesterol transfer by NPC2 | − | + | [48,96] | ||
| GM1 transfer by GM2AP 5 | − | − | − | [46,50] | |
| Membrane solubilization by | |||||
| Sap A | − | [92] | |||
| Sap B | − | [91] | |||
| GM2AP 5 | − | − | + | + | [46,50] |
| Lysosomal lipid degradation | |||||
| GM2 by Hex A/GM2AP 5 | − | − | + | + | [46,50] |
| GlcCer by GBA1 6 | − | + | + | + | [43] |
| SM by ASM | ◯ | + | + | [48] | |
| Further cellular processes | |||||
| Membrane fusion | − | + | + | [97] |
5.2. Activation and Inhibition of ASM
6. ASM Functions within Endosomes and Lysosomes
7. Can Secreted ASM Act at Cellular Membrane Surfaces Directly?
8. Medical Importance of Cellular ASM Activity
9. Assays for Studies of ASM Inhibitors
10. Major Depression and the Emerging Role of ASM and Acid Ceramidase in Lysosomal Lipid Turnover
11. Experimental Therapeutic Approaches for Niemann-Pick Disease Types A and B
12. The Role of ASM in Bacterial, Mycobacterial, Fungal, and Viral Infections
13. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASM | Acid sphingomyelinase |
| ASMD | Acid sphingomyelinase deficiency |
| BMP | Bis(monoacylglycero)phosphate |
| CAD | Cationic amphiphilic drugs |
| Cer1P | Ceramide-1-phosphate |
| GM2AP | GM2 activator protein |
| ILV | Intralysosomal luminal vesicles |
| NPC2 | Niemann-Pick disease protein C type 2 |
| PC | Phosphatidylcholine |
| PG | Phosphatidylglycerol |
| PL | Phospholipid |
| PLC | Phospholipase C |
| Sap | Saposin |
| SM | Sphingomyelin |
References
- Van Meer, G.; de Kroon, A.I. Lipid map of the mammalian cell. J. Cell Sci. 2011, 124, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Van Meer, G. Cellular lipidomics. EMBO J. 2005, 24, 3159–3165. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Maegawa, G.H.B.; Zhan, X.; Gao, X.; Wang, Y.; Xu, F.; Qiu, W.; Han, L.; Gu, X.; Zhang, H. Clinical, biochemical, and genotype-phenotype correlations of 118 patients with Niemann-Pick disease Types A/B. Hum. Mutat. 2021, 42, 614–625. [Google Scholar] [CrossRef]
- Niemann, A. Ein unbekanntes Krankheitsbild. Jahrb. Kinderheilkd. 1914, 79, 1–10. [Google Scholar]
- Pick, L. Über die lipoidzellige Splenohhehatomegalie typus Niemann-Pick als Stoffwechselerkrankung. Med. Klin. 1927, 23, 1483–1486. [Google Scholar]
- Klenk, E. Niemann-Pick’sche Krankheit und Amaurotische Idiotie. Hoppe-Seyler’s Z Physiol. Chem. 1939, 262, 128–143. [Google Scholar] [CrossRef]
- Thudichum, J.L.W. A Treatise on the Chemical Constitution of the Brain; Bailliere, Tindall and Cox: London, UK, 1884. [Google Scholar]
- Barnholz, Y.; Roitman, A.; Gatt, S. Enzymatic hydrolysis of sphingolipids. J. Biol. Chem. 1966, 241, 3731–3737. [Google Scholar] [CrossRef]
- Brady, R.O.; Kanfer, J.; Mock, M.; Fredrickson, D. The metabolism of sphingomyelin. Evidence of an enzymatic deficiency in Niemann-Pick disease. Proc. Natl. Acad. Sci. USA 1966, 55, 367–370. [Google Scholar] [CrossRef] [Green Version]
- Schuchman, E.H.; Desnick, R.J. Niemann-Pick disease types A and B: Sphingomyelinase deficiencies. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3589–3610. [Google Scholar]
- Vanier, M.T. Complex lipid trafficking in Niemann-Pick disease type C. J. Inherit. Metab. Dis. 2015, 38, 187–199. [Google Scholar] [CrossRef]
- Quintern, L.E.; Weitz, G.; Nehrkorn, H.; Tager, J.M.; Schram, A.W.; Sandhoff, K. Acid sphingomyelinase from human urine: Purification and characterization. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1987, 922, 323–336. [Google Scholar] [CrossRef]
- Quintern, L.E.; Zenk, T.S.; Sandhoff, K. The urine from patients with peritonitis as a rich source for purifying human acid sphingomyelinase and other lysosomal enzymes. Biochim. Biophys. Acta 1989, 1003, 121–124. [Google Scholar] [CrossRef]
- Quintern, L.E.; Schuchman, E.H.; Levran, O.; Suchi, M.; Ferlinz, K.; Reinke, H.; Sandhoff, K.; Desnick, R.J. Isolation of cDNA clones encoding human acid sphingomyelinase: Occurrence of alternatively processed transcripts. EMBO J. 1989, 8, 2469–2473. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Suchi, M.; Takahashi, T.; Sandhoff, K.; Desnick, R.J. Human acid sphingomyelinase. Isolation, nucleotide sequence and expression of the full-length and alternatively spliced cDNAs. J. Biol. Chem. 1991, 266, 8531–8539. [Google Scholar] [CrossRef]
- Ferlinz, K.; Hurwitz, R.; Sandhoff, K. Molecular basis of acid sphingomyelinase deficiency in a patient with Niemann-Pick disease type A. Biochem. Biophys. Res. Commun. 1991, 179, 1187–1191. [Google Scholar] [CrossRef]
- Levran, O.; Desnick, R.; Schuchman, E. Niemann-Pick disease: A frequent missense mutation in the acid sphingomyelinase gene of Ashkenazi Jewish type A and B patients. Proc. Natl. Acad. Sci. USA 1991, 88, 3748–3752. [Google Scholar] [CrossRef] [Green Version]
- Schuchman, E.H.; Levran, O.; Pereira, L.V.; Desnick, R.J. Structural organization and complete nucleotide sequence of the gene encoding human acid sphingomyelinase (SMPD1). Genomics 1992, 12, 197–205. [Google Scholar] [CrossRef]
- Lansmann, S.; Schuette, C.G.; Bartelsen, O.; Hoernschemeyer, J.; Linke, T.; Weisgerber, J.; Sandhoff, K. Human acid sphingomyelinase. Eur. J. Biochem. 2003, 270, 1076–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlinz, K.; Hurwitz, R.; Moczall, H.; Lansmann, S.; Schuchman, E.H.; Sandhoff, K. Functional characterization of the N-glycosylation sites of human acid sphingomyelinase by site-directed mutagenesis. Eur. J. Biochem. 1997, 243, 511–517. [Google Scholar] [CrossRef]
- Hurwitz, R.; Ferlinz, K.; Vielhaber, G.; Moczall, H.; Sandhoff, K. Processing of human acid sphingomyelinase in normal and I-cell fibroblasts. J. Biol. Chem. 1994, 269, 5440–5445. [Google Scholar] [CrossRef]
- Ferlinz, K.; Hurwitz, R.; Vielhaber, G.; Suzuki, K.; Sandhoff, K. Occurrence of two molecular forms of human acid sphingomyelinase. Biochem. J. 1994, 301, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Lefrancois, S.; Zeng, J.; Hassan, A.J.; Canuel, M.; Morales, C.R. The lysosomal trafficking of sphingolipid activator proteins (SAPs) is mediated by sortilin. EMBO J. 2003, 22, 6430–6437. [Google Scholar] [CrossRef]
- Wähe, A.; Kasmapour, B.; Schmaderer, C.; Liebl, D.; Sandhoff, K.; Nykjaer, A.; Griffiths, G.; Gutierrez, M.G. Golgi-to-phagosome transport of acid sphingomyelinase and prosaposin is mediated by sortilin. J. Cell Sci. 2010, 123, 2502–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, X.; Morales, C.R. The lysosomal trafficking of acid sphingomyelinase is mediated by sortilin and mannose 6-phosphate receptor. Traffic 2006, 7, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Romiti, E.; Vasta, V.; Meacci, E.; Farnararo, M.; Linke, T.; Ferlinz, K.; Sandhoff, K.; Bruni, P. Characterization of sphingomyelinase activity released by thrombin-stimulated platelets. Mol. Cell Biochem. 2000, 205, 75–81. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Idkowiak-Baldys, J.; Simbari, F.; Canals, D.; Roddy, P.; Riner, C.D.; Clarke, C.J.; Hannun, Y.A. A novel mechanism of lysosomal acid sphingomyelinase maturation: Requirement for carboxyl-terminal proteolytic processing. J. Biol. Chem. 2011, 286, 3777–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, R.; Ferlinz, K.; Sandhoff, K. The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol. Chem. Hoppe-Seyler 1994, 375, 447–450. [Google Scholar] [CrossRef]
- Moles, A.; Tarrats, N.; Fernández-Checa, J.C.; Marí, M. Cathepsin B overexpression due to acid sphingomyelinase ablation promotes liver fibrosis in Niemann-Pick disease. J. Biol. Chem. 2012, 287, 1178–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saftig, P.; Sandhoff, K. Cancer: Killing from the inside. Nature 2013, 502, 312–313. [Google Scholar] [CrossRef]
- Gabandé-Rodríguez, E.; Boya, P.; Labrador, V.; Dotti, C.G.; Ledesma, M.D. High sphingomyelin levels induce lysosomal damage and autophagy dysfunction in Niemann Pick disease type A. Cell Death Differ. 2014, 21, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Schissel, S.L.; Keesler, G.A.; Schuchman, E.H.; Williams, K.J.; Tabas, I. The cellular trafficking and zinc dependence of secretory and lysosomal sphingomyelinase, two products of the acid sphingomyelinase gene. J. Biol. Chem. 1998, 273, 18250–18259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Z.J.; Huang, J.; Poda, G.; Pomès, R.; Privé, G.G. Structure of human acid sphingomyelinase reveals the role of the saposin domain in activating substrate hydrolysis. J. Mol. Biol. 2016, 428, 3026–3042. [Google Scholar] [CrossRef] [PubMed]
- Henseler, M.; Klein, A.; Glombitza, G.J.; Suziki, K.; Sandhoff, K. Expression of the three alternative forms of the sphingolipid activator protein precursor in baby hamster kidney cells and functional assays in a cell culture system. J. Biol. Chem. 1996, 271, 8416–8423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkhardt, J.K.; Hüttler, S.; Klein, A.; Möbius, W.; Habermann, A.; Griffiths, G.; Sandhoff, K. Accumulation of sphingolipids in SAP-precursor (prosaposin)-deficient fibroblasts occurs as intralysosomal membrane structures and can be completely reversed by treatment with human SAP-precursor. Eur. J. Cell Biol. 1997, 73, 10–18. [Google Scholar]
- Ahn, V.E.; Faull, K.F.; Whitelegge, J.P.; Fluharty, A.L.; Privé, G.G. Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc. Natl. Acad. Sci. USA 2003, 100, 38–43. [Google Scholar] [CrossRef] [Green Version]
- Popovic, K.; Holyoake, J.; Pomes, R.; Prive, G.G. Structure of saposin A lipoprotein discs. Proc. Natl. Acad. Sci. USA 2012, 109, 2908–2912. [Google Scholar] [CrossRef] [Green Version]
- Gorelik, A.; Illes, K.; Heinz, L.X.; Superti-Furga, G.; Nagar, B. Crystal structure of mammalian acid sphingomyelinase. Nat. Commun. 2016, 7, 12196. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-F.; Metcalf, M.C.; Garman, S.C.; Edmunds, T.; Qiu, H.; Wei, R.R. Human acid sphingomyelinase structures provide insight to molecular basis of Niemann–Pick disease. Nat. Commun. 2016, 7, 13082. [Google Scholar] [CrossRef] [Green Version]
- Ota, S.; Noguchi, A.; Kondo, D.; Nakajima, Y.; Ito, T.; Arai, H.; Takahashi, T. An early-onset neuronopathic form of acid sphingomyelinase deficiency: A SMPD1 p.C133Y mutation in the saposin domain of acid sphingomyelinase. Tohoku J. Exp. Med. 2020, 250, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlinz, K.; Linke, T.; Bartelsen, O.; Weiler, M.; Sandhoff, K. Stimulation of lysosomal sphingomyelin degradation by sphingolipid activator proteins. Chem. Phys. Lipids 1999, 102, 35–43. [Google Scholar] [CrossRef]
- Linke, T.; Wilkening, G.; Lansmann, S.; Moczall, H.; Bartelsen, O.; Weisgerber, J.; Sandhoff, K. Stimulation of acid sphingomyelinase activity by lysosomal lipids and sphingolipid activator proteins. Biol. Chem. 2001, 382, 283–290. [Google Scholar] [CrossRef]
- Abdul-Hammed, M.; Breiden, B.; Schwarzmann, G.; Sandhoff, K. Lipids regulate the hydrolysis of membrane bound glucosylceramide by lysosomal β-glucocerebrosidase. J. Lipid Res. 2017, 58, 563–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, T.; Wilkening, G.; Sadeghlar, F.; Mozcall, H.; Bernardo, K.; Schuchman, E.; Sandhoff, K. Interfacial regulation of acid ceramidase activity. Stimulation of ceramide degradation by lysosomal lipids and sphingolipid activator proteins. J. Biol. Chem. 2001, 276, 5760–5768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkening, G.; Linke, T.; Uhlhorn-Dierks, G.; Sandhoff, K. Degradation of membrane-bound ganglioside GM1. Stimulation by bis(monoacylglycero)phosphate and the activator proteins SAP-B and GM2-AP. J. Biol. Chem. 2000, 275, 35814–35819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anheuser, S.; Breiden, B.; Sandhoff, K. Membrane lipids and their degradation compounds control GM2 catabolism at intralysosomal luminal vesicles. J. Lipid Res. 2019, 60, 1099–1111. [Google Scholar] [CrossRef]
- Sandhoff, R.; Sandhoff, K. Emerging concepts of ganglioside metabolism. FEBS Lett. 2018, 592, 3835–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oninla, V.O.; Breiden, B.; Babalola, J.O.; Sandhoff, K. Acid sphingomyelinase activity is regulated by membrane lipids and facilitates cholesterol transfer by NPC2. J. Lipid Res. 2014, 55, 2606–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breiden, B.; Sandhoff, K. Lysosomal glycosphingolipid storage diseases. Annu. Rev. Biochem. 2019, 88, 461–485. [Google Scholar] [CrossRef]
- Anheuser, S.; Breiden, B.; Schwarzmann, G.; Sandhoff, K. Membrane lipids regulate ganglioside GM2 catabolism and GM2 activator protein activity. J. Lipid Res. 2015, 56, 1747–1761. [Google Scholar] [CrossRef] [Green Version]
- Matzner, U.; Breiden, B.; Schwarzmann, G.; Yaghootfam, A.; Fluharty, A.L.; Hasilik, A.; Sandhoff, K.; Gieselmann, V. Saposin B-dependent reconstitution of arylsulfatase A activity in vitro and in cell culture models of Metachromatic Leukodystrophy. J. Biol. Chem. 2009, 284, 9372–9381. [Google Scholar] [CrossRef] [Green Version]
- Breiden, B.; Sandhoff, K. Emerging mechanisms of drug-induced phospholipidosis. Biol. Chem. 2020, 401, 31–46. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Mechanism of secondary ganglioside and lipid accumulation in lysosomal disease. Int. J. Mol. Sci. 2020, 21, 2566. [Google Scholar] [CrossRef] [Green Version]
- Shayman, J.A.; Tesmer, J.J.G. Lysosomal phospholipase A2. Biochim. Biophys. Acta 2019, 1864, 932–940. [Google Scholar] [CrossRef]
- Freeman, S.J.; Shankaran, P.; Wolfe, L.S.; Callahan, J.W. Phosphatidylcholine and 4-methylumbelliferyl phosphorylcholine hydrolysis by purified placental sphingomyelinase. Can. J. Biochem. Cell Biol. 1985, 63, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Huterer, S.; Wherrett, J.R.; Poulos, A.; Callahan, J.W. Deficiency of phospholipase C acting on phosphatidylglycerol in Niemann-Pick disease. Neurology 1983, 33, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y.; Hostetler, K.Y. Properties of phospholipase C isolated from rat liver lysosomes. J. Biol. Chem. 1980, 255, 646–652. [Google Scholar] [CrossRef]
- Hostetler, K.Y.; Hall, L.B. Phospholipase C activity of rat tissues. Biochem. Biophys. Res. Commun. 1980, 96, 388–393. [Google Scholar] [CrossRef]
- Hostetler, K.Y.; Hall, L.B. Inhibition of kidney lysosomal phospholipases A and C by aminoglycoside antibiotics: Possible mechanism of aminoglycoside toxicity. Proc. Natl. Acad. Sci. USA. 1982, 79, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Eisen, D.; Bartolf, M.; Franson, R.C. Inhibition of lysosomal phospholipases C and A in rabbit alveolar macrophages, polymorphonuclear leukocytes and rat liver by sodium bisulfite. Biochim. Biophys. Acta 1984, 793, 10–17. [Google Scholar] [CrossRef]
- Beaudet, A.L.; Hampton, M.S.; Patel, K.; Sparrow, J.T. Acidic phospholipases in cultured human fibroblasts: Deficiency of phospholipase C in Niemann-Pick disease. Clin. Chim. Acta 1980, 108, 403–414. [Google Scholar] [CrossRef]
- Lansmann, S.; Bartelsen, O.; Sandhoff, K. Purification and characterization of recombinant human acid sphingomyelinase expressed in insect Sf21 cells. Methods Enzymol. 2000, 311, 149–156. [Google Scholar] [CrossRef]
- Grabner, G.F.; Fawzy, N.; Schreiber, R.; Pusch, L.M.; Bulfon, D.; Koefeler, H.; Eichmann, T.O.; Lass, A.; Schweiger, M.; Marsche, G.; et al. Metabolic regulation of the lysosomal cofactor bis(monoacylglycero)phosphate in mice. J. Lipid Res. 2020, 61, 995–1003. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Hostetler, K.Y. Degradation of bis(monoacylglycero)phosphate by an acid phosphodiesterase in rat liver lysosomes. J. Biol. Chem. 1979, 254, 5997–6001. [Google Scholar] [CrossRef]
- Breilyn, M.S.; Zhang, W.; Yu, C.; Wasserstein, M.P. Plasma lyso-sphingomyelin levels are positively associated with clinical severity in acid sphingomyelinase deficiency. Mol. Genet. Metab. Rep. 2021, 28, 100780. [Google Scholar] [CrossRef]
- Thurberg, B.L. Autopsy pathology of infantile neurovisceral ASMD (Niemann-Pick Disease type A): Clinicopathologic correlations of a case report. Mol. Genet. Metab. Rep. 2020, 24, 100626. [Google Scholar] [CrossRef] [PubMed]
- Gulbins, E.; Kolesnick, R. Raft ceramide in molecular medicine. Oncogene 2003, 22, 7070–7077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann, N.; Mohamud Yusuf, A.; Martiny, C.; Zhang, X.; Kleinschnitz, C.; Gunzer, M.; Kolesnick, R.; Gulbins, E.; Hermann, D.M. Homozygous Smpd1 deficiency aggravates brain ischemia/reperfusion injury by mechanisms involving polymorphonuclear neutrophils, whereas heterozygous Smpd1 deficiency protects against mild focal cerebral ischemia. Basic Res. Cardiol. 2020, 115, 64. [Google Scholar] [CrossRef]
- Paris, F.; Fuks, Z.; Kang, A.; Capodieci, P.; Juan, G.; Ehleiter, D.; Haimovitz-Friedman, A.; Cordon-Cardo, C.; Kolesnick, R. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 2001, 293, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003, 300, 1155–1159. [Google Scholar] [CrossRef] [Green Version]
- Ordoñez, M.; Presa, N.; Dominguez-Herrera, A.; Trueba, M.; Gomez-Muñoz, A. Regulation of adipogenesis by ceramide 1-phosphate. Exp. Cell Res. 2018, 372, 150–157. [Google Scholar] [CrossRef]
- Mena, H.A.; Zubiry, P.R.; Dizier, B.; Mignon, V.; Parborell, F.; Schattner, M.; Boisson-Vidal, C.; Negrotto, S. Ceramide 1-phosphate protects endothelial colony-forming cells from apoptosis and increases vasculogenesis in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e219–e232. [Google Scholar] [CrossRef]
- Gomez-Muñoz, A.; Duffy, P.A.; Martin, A.; O’Brien, L.; Byun, H.S.; Bittman, R.; Brindley, D.N. Short-chain ceramide-1-phosphates are novel stimulators of DNA synthesis and cell division: Antagonism by cell-permeable ceramides. Mol. Pharmacol. 1995, 47, 833–839. [Google Scholar]
- Gatt, S.; Herzl, A.; Barenholz, Y. Hydrolysis of sphingomyelin liposomes by sphingomyelinase. FEBS Lett. 1973, 30, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Kölzer, M.; Arenz, C.; Ferlinz, K.; Werth, N.; Schulze, H.; Klingenstein, R.; Sandhoff, K. Phosphatidylinositol-3,5-Bisphosphate is a potent and selective inhibitor of acid sphingomyelinase. Biol. Chem. 2003, 384, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Testai, F.D.; Landek, M.A.; Goswami, R.; Ahmed, M.; Dawson, G. Acid sphingomyelinase and inhibition by phosphate ion: Role of inhibition by phosphatidyl-myo-inositol 3,4,5-triphosphate in oligodendrocyte cell signaling. J. Neurochem. 2004, 89, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Muñoz, A.; Kong, J.; Salh, B.; Steinbrecher, U.P. Sphingosine-1-phosphate inhibits acid sphingomyelinase and blocks apoptosis in macrophages. FEBS Lett. 2003, 539, 56–60. [Google Scholar] [CrossRef] [Green Version]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Savini, M.; Zhao, Q.; Wang, M.C. Lysosomes: Signaling hubs for metabolic sensing and longevity. Trends Cell Biol. 2019, 29, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Yim, W.W.-Y.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kolter, T.; Sandhoff, K. Principles of lysosomal membrane digestion: Stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev. Cell Dev. Biol. 2005, 21, 81–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruenberg, J. Life in the lumen: The multivesicular endosome. Traffic 2020, 21, 76–93. [Google Scholar] [CrossRef] [Green Version]
- Finkbeiner, S. The autophagy lysosomal pathway and neurodegeneration. Cold Spring Harb. Perspect. Biol. 2020, 12, a033993. [Google Scholar] [CrossRef]
- Herb, M.; Gluschko, A.; Schramm, M. LC3-associated phagocytosis—The highway to hell for phagocytosed microbes. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2020; Volume 101, pp. 68–76. [Google Scholar] [CrossRef]
- Ferguson, S.M. Neuronal lysosomes. Neurosci. Lett. 2019, 697, 1–9. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- Möbius, W.; Herzog, V.; Sandhoff, K.; Schwarzmann, G. Intracellular distribution of a biotin-labeled ganglioside, GM1, by immunoelectron microscopy after endocytosis in fibroblasts. J. Histochem. Cytochem. 1999, 47, 1005–1014. [Google Scholar] [CrossRef] [Green Version]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.-L.; Tanaka, Y.; Saftig, P. At the acidic edge: Emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003, 13, 137–145. [Google Scholar] [CrossRef]
- Gallala, H.; Sandhoff, K. Biological function of the cellular lipid BMP—BMP as a key activator for cholesterol sorting and membrane digestion. Neurochem. Res. 2011, 36, 1594–1600. [Google Scholar] [CrossRef]
- Remmel, N.; Locatelli-Hoops, S.; Breiden, B.; Schwarzmann, G.; Sandhoff, K. Saposin B mobilizes lipids from cholesterol-poor and bis(monoacylglycero)phosphate-rich membranes at acidic pH. Unglycosylated patient variant saposin B lacks lipid-extraction capacity. FEBS J. 2007, 274, 3405–3420. [Google Scholar] [CrossRef]
- Locatelli-Hoops, S.; Remmel, N.; Klingenstein, R.; Breiden, B.; Rossocha, M.; Schoeniger, M.; Koenigs, C.; Saenger, W.; Sandhoff, K. Saposin A mobilizes lipids from low cholesterol and high bis(monoacylglycerol)phosphate-containing membranes: Patient variant Saposin A lacks lipid extraction capacity. J. Biol. Chem. 2006, 281, 32451–32460. [Google Scholar] [CrossRef] [Green Version]
- Anheuser, S.; Breiden, B.; Sandhoff, K. Ganglioside GM2 catabolism is inhibited by storage compounds of mucopolysaccharidoses and by cationic amphiphilic drugs. Mol. Genet. Metab. 2019, 128, 75–83. [Google Scholar] [CrossRef]
- Kamoshita, S.; Aron, A.M.; Suzuki, K.; Suzuki, K. Infantile Niemann-Pick disease. A chemical study with isolation and characterization of membranous cytoplasmic bodies and myelin. Am. J. Dis. Child. 1969, 117, 379–394. [Google Scholar] [CrossRef]
- Jatzkewitz, H.; Pilz, H.; Sandhoff, K. Quantitative Bestimmungen von Gangliosiden und ihren Neuraminsäurefreien Derivaten bei infantilen, juvenilen und adulten Formen der amaurotischen Idiotie und einer spätinfantilen biochemischen Sonderform. J. Neurochem. 1965, 12, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hammed, M.; Breiden, B.; Adebayo, M.A.; Babalola, J.O.; Schwarzmann, G.; Sandhoff, K. Role of endosomal membrane lipids and NPC2 in cholesterol transfer and membrane fusion. J. Lipid Res. 2010, 51, 1747–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Argüello, M.B.; Basáñez, G.; Goñi, F.M.; Alonso, A. Different effects of enzyme-generated ceramides and diacylglycerols in phospholipid membrane fusion and leakage. J. Biol. Chem. 1996, 271, 26616–26621. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, S.; Martin, B.M.; Kishimoto, Y.; O’Brien, J.S. Saposin D: A sphingomyelinase activator. Biochem. Biophys. Res. Commun. 1988, 156, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Graf, C.G.F.; Schulz, C.; Schmälzlein, M.; Heinlein, C.; Mönnich, M.; Perkams, L.; Püttner, M.; Boos, I.; Hessefort, M.; Lombana Sanchez, J.N.; et al. Synthetic glycoforms reveal carbohydrate-dependent bioactivity of human saposin D. Angew. Chem. Int. Ed. 2017, 56, 5252–5257. [Google Scholar] [CrossRef]
- Schwarzmann, G.; Breiden, B.; Sandhoff, K. Membrane-spanning lipids for an uncompromised monitoring of membrane fusion and intermembrane lipid transfer. J. Lipid Res. 2015, 56, 1861–1879. [Google Scholar] [CrossRef] [Green Version]
- Vaccaro, A.M.; Ciaffoni, F.; Tatti, M.; Salvioli, R.; Barca, A.; Tognozzi, D.; Scerch, C. pH-dependent conformational properties of saposins and their interactions with phospholipid membranes. J. Biol. Chem. 1995, 270, 30576–30580. [Google Scholar] [CrossRef] [Green Version]
- Vaccaro, A.M.; Salvioli, R.; Tatti, M.; Ciaffoni, F. Saposins and their interaction with lipids. Neurochem. Res. 1999, 24, 307–314. [Google Scholar] [CrossRef]
- Vaccaro, A.M.; Tatti, M.; Ciaffoni, F.; Salvioli, R.; Serafino, A.; Barca, A. Saposin-C induces pH-dependent destabilization and fusion of phosphatidylserine-containing vesicles. FEBS Lett. 1994, 349, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Chu, Z. Fusogenic domain and lysines in saposin C. Arch. Biochem. Biophys. 2004, 424, 210–218. [Google Scholar] [CrossRef]
- Kölzer, M.; Ferlinz, K.; Bartelsen, O.; Hoops, S.L.; Lang, F.; Sandhoff, K. Functional characterization of the postulated intramolecular sphingolipid activator protein domain of human acid sphingomyelinase. Biol. Chem. 2004, 385, 1193–1195. [Google Scholar] [CrossRef]
- Kobayashi, T.; Beuchat, M.H.; Chevallier, J.; Makino, A.; Mayran, N.; Escola, J.M.; Lebrand, C.; Cosson, P.; Gruenberg, J. Separation and characterization of late endosomal membrane domains. J. Biol. Chem. 2002, 277, 32157–32164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möbius, W.; van Donselaar, E.; Ohno-Iwashita, Y.; Shimada, Y.; Heijnen, H.F.; Slot, J.W.; Geuze, H.J. Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic 2003, 4, 222–231. [Google Scholar] [CrossRef]
- Kölzer, M.; Werth, N.; Sandhoff, K. Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Lett. 2004, 559, 96–98. [Google Scholar] [CrossRef] [Green Version]
- Cockburn, C.L.; Green, R.S.; Damle, S.R.; Martin, R.K.; Ghahrai, N.N.; Colonne, P.M.; Fullerton, M.S.; Conrad, D.H.; Chalfant, C.E.; Voth, D.E.; et al. Functional inhibition of acid sphingomyelinase disrupts infection by intracellular bacterial pathogens. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Elojeimy, S.; Holman, D.H.; Liu, X.; El-Zawahry, A.; Villani, M.; Cheng, J.C.; Mahdy, A.; Zeidan, Y.; Bielwaska, A.; Hannun, Y.A.; et al. New insights on the use of desipramine as an inhibitor for acid ceramidase. FEBS Lett. 2006, 580, 4751–4756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellegaard, A.M.; Bach, P.; Jäättelä, M. Targeting cancer lysosomes with good old cationic amphiphilic drugs. Rev. Physiol. Biochem. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Berger, A.; Rosenthal, D.; Spiegel, S. Sphingosylphosphocholine, a signaling molecule which accumulates in Niemann-Pick disease type A, stimulates DNA-binding activity of the transcription activator protein AP-1. Proc. Natl. Acad. Sci. USA 1995, 92, 5885–5889. [Google Scholar] [CrossRef] [Green Version]
- Rouser, G.; Kritchevsky, G.; Knudson, A.G., Jr.; Simon, G. Accumulation of a glycerolphospholipid in classical niemann-pick disease. Lipids 1968, 3, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Biochemical studies in Niemann-Pick disease I. Major sphingolipids of liver and spleen. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1983, 750, 178–184. [Google Scholar] [CrossRef]
- Newcomb, B.; Rhein, C.; Mileva, I.; Ahmad, R.; Clarke, C.J.; Snider, J.; Obeid, L.M.; Hannun, Y.A. Identification of an acid sphingomyelinase ceramide kinase pathway in the regulation of the chemokine CCL5. J. Lipid Res. 2018, 59, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presa, N.; Gomez-Larrauri, A.; Dominguez-Herrera, A.; Trueba, M.; Gomez-Muñoz, A. Novel signaling aspects of ceramide 1-phosphate. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2020, 1865, 158630. [Google Scholar] [CrossRef] [PubMed]
- Dadsena, S.; Bockelmann, S.; Mina, J.G.M.; Hassan, D.G.; Korneev, S.; Razzera, G.; Jahn, H.; Niekamp, P.; Müller, D.; Schneider, M.; et al. Ceramides bind VDAC2 to trigger mitochondrial apoptosis. Nat. Commun. 2019, 10, 1832. [Google Scholar] [CrossRef]
- Leonetti, D.; Estéphan, H.; Ripoche, N.; Dubois, N.; Aguesse, A.; Gouard, S.; Brossard, L.; Chiavassa, S.; Corre, I.; Pecqueur, C.; et al. Secretion of acid sphingomyelinase and ceramide by endothelial cells contributes to radiation-induced intestinal toxicity. Cancer Res. 2020, 80, 2651–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketteler, J.; Wittka, A.; Leonetti, D.; Roy, V.V.; Estephan, H.; Maier, P.; Reis, H.; Herskind, C.; Jendrossek, V.; Paris, F.; et al. Caveolin-1 regulates the ASMase/ceramide-mediated radiation response of endothelial cells in the context of tumor-stroma interactions. Cell Death Dis. 2020, 11, 228. [Google Scholar] [CrossRef] [Green Version]
- Henry, B.; Ziobro, R.; Becker, K.; Kolesnick, R.; Gulbins, E. Acid Sphingomyelinase. In Sphingolipids: Basic Science and Drug Development, 215th ed.; Gulbins, E., Petrache, I., Eds.; Springer: Vienna, Austria, 2013; pp. 77–88. [Google Scholar]
- Rana, S.; Espinosa-Diez, C.; Ruhl, R.; Chatterjee, N.; Hudson, C.; Fraile-Bethencourt, E.; Agarwal, A.; Khou, S.; Thomas, C.R., Jr.; Anand, S. Differential regulation of microRNA-15a by radiation affects angiogenesis and tumor growth via modulation of acid sphingomyelinase. Sci. Rep. 2020, 10, 5581. [Google Scholar] [CrossRef] [Green Version]
- Heneweer, C.; Penate Medina, T.; Tower, R.; Kalthoff, H.; Kolesnick, R.; Larson, S.; Penate Medina, O. Acid-sphingomyelinase triggered fluorescently labeled sphingomyelin containing liposomes in tumor diagnosis after radiation-induced stress. Int. J. Mol. Sci. 2021, 22, 3864. [Google Scholar] [CrossRef]
- Kolesnick, R.; Fuks, Z. Radiation and ceramide-induced apoptosis. Oncogene 2003, 22, 5897–5906. [Google Scholar] [CrossRef] [Green Version]
- Peña, L.A.; Fuks, Z.; Kolesnick, R.N. Radiation-induced apoptosis of endothelial cells in the murine central nervous system: Protection by fibroblast growth factor and sphingomyelinase deficiency. Cancer Res. 2000, 60, 321–327. [Google Scholar] [PubMed]
- Contreras, F.X.; Villar, A.V.; Alonso, A.; Kolesnick, R.N.; Goñi, F.M. Sphingomyelinase activity causes transbilayer lipid translocation in model and cell membranes. J. Biol. Chem. 2003, 278, 37169–37174. [Google Scholar] [CrossRef] [Green Version]
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 2010, 189, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Rhein, C.; Zoicas, I.; Marx, L.M.; Zeitler, S.; Hepp, T.; von Zimmermann, C.; Mühle, C.; Richter-Schmidinger, T.; Lenz, B.; Erim, Y.; et al. mRNA expression of SMPD1 encoding acid sphingomyelinase decreases upon antidepressant treatment. Int. J. Mol. Sci. 2021, 22, 5700. [Google Scholar] [CrossRef]
- Zhang, L.; Dai, J.; Zeng, Z.; Jia, Y. Nitric oxide induces HepG2 cell death via extracellular signal-regulated protein kinase activation by regulating acid sphingomyelinase. Mol. Biol. Rep. 2020, 47, 8353–8359. [Google Scholar] [CrossRef] [PubMed]
- Vadlamudi, Y.; Muthu, K.; Kumar, M.S. Structural exploration of acid sphingomyelinase at different physiological pH through molecular dynamics and docking studies. RSC Adv. 2016, 6, 74859–74873. [Google Scholar] [CrossRef]
- O’Sullivan, M.J.; Lindsay, A.J. The endosomal recycling pathway-at the crossroads of the cell. Int. J. Mol. Sci. 2020, 21, 6074. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Claus, R.A. Keep your friends close, but your enemies closer: Role of acid sphingomyelinase during infection and host response. Front. Med. 2020, 7, 616500. [Google Scholar] [CrossRef]
- Schissel, S.L.; Jiang, X.; Tweedie-Hardman, J.; Jeong, T.; Camejo, E.H.; Najib, J.; Rapp, J.H.; Williams, K.J.; Tabas, I. Secretory sphingomyelinase, a product of the acid sphingomyelinase gene, can hydrolyze atherogenic lipoproteins at neutral pH. Implications for atherosclerotic lesion development. J. Biol. Chem. 1998, 273, 2738–2746. [Google Scholar] [CrossRef] [Green Version]
- Mizrachi, A.; Ben-Aharon, I.; Li, H.; Bar-Joseph, H.; Bodden, C.; Hikri, E.; Popovtzer, A.; Shalgi, R.; Haimovitz-Friedman, A. Chemotherapy-induced acute vascular injury involves intracellular generation of ROS via activation of the acid sphingomyelinase pathway. Cell. Signal. 2021, 82, 109969. [Google Scholar] [CrossRef]
- Thayyullathil, F.; Cheratta, A.R.; Alakkal, A.; Subburayan, K.; Pallichankandy, S.; Hannun, Y.A.; Galadari, S. Acid sphingomyelinase-dependent autophagic degradation of GPX4 is critical for the execution of ferroptosis. Cell Death Dis. 2021, 12, 26. [Google Scholar] [CrossRef]
- Luong, T.T.D.; Tuffaha, R.; Schuchardt, M.; Moser, B.; Schelski, N.; Boehme, B.; Gollmann-Tepekoylu, C.; Schramm, C.; Holfeld, J.; Pieske, B.; et al. Acid sphingomyelinase promotes SGK1-dependent vascular calcification. Clin. Sci. 2021, 135, 515–534. [Google Scholar] [CrossRef]
- Pinkert, T.; Furkert, D.; Korte, T.; Herrmann, A.; Arenz, C. Amplification of a FRET Probe by lipid–water partition for the detection of acid sphingomyelinase in live cells. Angew Chem. Int. Ed. 2017, 56, 2790–2794. [Google Scholar] [CrossRef]
- Kappe, C.; Mohamed, Z.H.; Naser, E.; Carpinteiro, A.; Arenz, C. A novel visible range FRET probe for monitoring acid sphingomyelinase activity in living cells. Chemistry 2020, 26, 5780–5783. [Google Scholar] [CrossRef]
- Mohamed, Z.H.; Rhein, C.; Schmid, B.; Tripal, P.; Kornhuber, J.; Arenz, C. Synthesis and characterization of a new two photon excitable acid sphingomyelinase FRET probe. Bioorganic Med. Chem. 2021, 44, 116303. [Google Scholar] [CrossRef] [PubMed]
- Prause, K.; Naseri, G.; Schumacher, F.; Kappe, C.; Kleuser, B.; Arenz, C. A photocaged inhibitor of acid sphingomyelinase. Chem. Commun. 2020, 56, 14885–14888. [Google Scholar] [CrossRef] [PubMed]
- Holme, M.N.; Rana, S.; Barriga, H.M.G.; Kauscher, U.; Brooks, N.J.; Stevens, M.M. A robust liposomal platform for direct colorimetric detection of sphingomyelinase enzyme and inhibitors. ACS Nano 2018, 12, 8197–8207. [Google Scholar] [CrossRef] [Green Version]
- Arenz, C. Small molecule inhibitors of acid sphingomyelinase. Cell Physiol. Biochem. 2010, 26, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kornhuber, J.; Tripal, P.; Reichel, M.; Terfloth, L.; Bleich, S.; Wiltfang, J.; Gulbins, E. Identification of new functional inhibitors of acid sphingomyelinase using a structure-property-activity relation model. J. Med. Chem. 2007, 51, 219–237. [Google Scholar] [CrossRef]
- Kornhuber, J.; Muehlbacher, M.; Trapp, S.; Pechmann, S.; Friedl, A.; Reichel, M.; Muhle, C.; Terfloth, L.; Groemer, T.W.; Spitzer, G.M.; et al. Identification of novel functional inhibitors of acid sphingomyelinase. PLoS ONE 2011, 6, e23852. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Nong, K.; Gu, Q.; Dong, J.; Wang, J. Discovery of N-hydroxy-3-alkoxybenzamides as direct acid sphingomyelinase inhibitors using a ligand-based pharmacophore model. Eur. J. Med. Chem. 2018, 151, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Yu, J.; Nong, K.; Wang, Y.; Niu, A.; Chen, W.; Dong, J.; Wang, J. Discovery of potent, selective, and direct acid sphingomyelinase inhibitors with antidepressant activity. J. Med. Chem. 2020, 63, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.M.; Henkels, K.M.; Rehl, K.M.; Liang, H.; Zhou, Y.; Gutterman, J.U.; Cho, K.J. Avicin G is a potent sphingomyelinase inhibitor and blocks oncogenic K- and H-Ras signaling. Sci. Rep. 2020, 10, 9120. [Google Scholar] [CrossRef] [PubMed]
- Naser, E.; Kadow, S.; Schumacher, F.; Mohamed, Z.H.; Kappe, C.; Hessler, G.; Pollmeier, B.; Kleuser, B.; Arenz, C.; Becker, K.A.; et al. Characterization of the small molecule ARC39, a direct and specific inhibitor of acid sphingomyelinase in vitro. J. Lipid Res. 2020, 61, 896–910. [Google Scholar] [CrossRef] [Green Version]
- Böll, S.; Ziemann, S.; Ohl, K.; Klemm, P.; Rieg, A.D.; Gulbins, E.; Becker, K.A.; Kamler, M.; Wagner, N.; Uhlig, S.; et al. Acid sphingomyelinase regulates T(H) 2 cytokine release and bronchial asthma. Allergy 2020, 75, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Justice, M.J.; Bronova, I.; Schweitzer, K.S.; Poirier, C.; Blum, J.S.; Berdyshev, E.V.; Petrache, I. Inhibition of acid sphingomyelinase disrupts LYNUS signaling and triggers autophagy. J. Lipid Res. 2018, 59, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Gulbins, A.; Schumacher, F.; Becker, K.A.; Wilker, B.; Soddemann, M.; Boldrin, F.; Müller, C.P.; Edwards, M.J.; Goodman, M.; Caldwell, C.C.; et al. Antidepressants act by inducing autophagy controlled by sphingomyelin-ceramide. Mol. Psychiatry 2018, 23, 2324–2346. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jin, S.-J.; Su, J.; Li, X.-X.; Xu, M. Acid sphingomyelinase down-regulation alleviates vascular endothelial insulin resistance in diabetic rats. Basic Clin. Pharmacol. Toxicol. 2018, 123, 645–659. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Huang, S.; Duan, W.; Liu, Q.; Lei, M. Inhibition of acid sphingomyelinase activity ameliorates endothelial dysfunction in db/db mice. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Li, Y.; Syn, W.K.; Wang, Z.; Lopes-Virella, M.F.; Lyons, T.J.; Huang, Y. Amitriptyline inhibits nonalcoholic steatohepatitis and atherosclerosis induced by high-fat diet and LPS through modulation of sphingolipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E131–E144. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Voltin, J.R.; Gooz, M.A.; Li, L.; Lemasters, J.J.; Gudz, T.I. Acid sphingomyelinase promotes mitochondrial dysfunction due to glutamate-induced regulated necrosis. J. Lipid Res. 2018, 59, 312–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Jin, H.K.; Park, M.H.; Kim, B.R.; Lee, P.H.; Nakauchi, H.; Carter, J.E.; He, X.; Schuchman, E.H.; Bae, J.S. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J. Exp. Med. 2014, 211, 1551–1570. [Google Scholar] [CrossRef] [Green Version]
- Faict, S.; Oudaert, I.; D’Auria, L.; Dehairs, J.; Maes, K.; Vlummens, P.; De Veirman, K.; De Bruyne, E.; Fostier, K.; Vande Broek, I.; et al. The transfer of sphingomyelinase contributes to drug resistance in multiple myeloma. Cancers 2019, 11, 1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellkofer, R.; Sandhoff, K. Halothane increases membrane fluidity and stimulates sphingomyelin degradation by membrane-bound neutral sphingomyelinase of synaptosomal plasma membranes from calf brain already at clinical concentrations. J. Neurochem. 1980, 34, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Sandhoff, K.; Pallmann, B. Membrane-bound neuraminidase from calf brain: Regulation of oligosialoganglioside degradation by membrane fluidity and membrane components. Proc. Natl. Acad. Sci. USA 1978, 75, 122–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinoff, A.; Herrmann, N.; Lanctôt, K.L. Ceramides and depression: A systematic review. J. Affect. Disord 2017, 213, 35–43. [Google Scholar] [CrossRef]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 301, 805–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulbins, E.; Palmada, M.; Reichel, M.; Lüth, A.; Böhmer, C.; Amato, D.; Müller, C.P.; Tischbirek, C.H.; Groemer, T.W.; Tabatabai, G.; et al. Acid sphingomyelinase-ceramide system mediates effects of antidepressant drugs. Nat. Med. 2013, 19, 934–938. [Google Scholar] [CrossRef] [Green Version]
- Niziolek, G.M.; Hoehn, R.S.; Seitz, A.P.; Jernigan, P.L.; Makley, A.T.; Gulbins, E.; Edwards, M.J.; Goodman, M.D. The role of acid sphingomyelinase inhibition in repetitive mild traumatic brain injury. J. Surg. Res. 2020, 259, 296–304. [Google Scholar] [CrossRef]
- Jaddoa, E.; Masania, J.; Masiero, E.; Sgamma, T.; Arroo, R.; Sillence, D.; Zetterström, T. Effect of antidepressant drugs on the brain sphingolipid system. J. Psychopharmacol. 2020, 34, 716–725. [Google Scholar] [CrossRef]
- Zoicas, I.; Schumacher, F.; Kleuser, B.; Reichel, M.; Gulbins, E.; Fejtova, A.; Kornhuber, J.; Rhein, C. The forebrain-specific overexpression of acid sphingomyelinase induces depressive-like symptoms in mice. Cells 2020, 9, 1244. [Google Scholar] [CrossRef]
- Zoicas, I.; Mühle, C.; Schmidtner, A.K.; Gulbins, E.; Neumann, I.D.; Kornhuber, J. Anxiety and depression are related to higher activity of sphingolipid metabolizing enzymes in the rat brain. Cells 2020, 9, 1239. [Google Scholar] [CrossRef]
- Huston, J.P.; Kornhuber, J.; Mühle, C.; Japtok, L.; Komorowski, M.; Mattern, C.; Reichel, M.; Gulbins, E.; Kleuser, B.; Topic, B.; et al. A sphingolipid mechanism for behavioral extinction. J. Neurochem. 2016, 137, 589–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.H.; Jin, H.K.; Bae, J.-S. Potential therapeutic target for aging and age-related neurodegenerative diseases: The role of acid sphingomyelinase. Exp. Mol. Med. 2020, 52, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Taniguchi, K.; Lee, H.M.; Lee, M.Y.; Bandu, R.; Komura, K.; Lee, K.Y.; Akao, Y.; Kim, K.P. Comparative lipidomics of 5-Fluorouracil-sensitive and -resistant colorectal cancer cells reveals altered sphingomyelin and ceramide controlled by acid sphingomyelinase (SMPD1). Sci. Rep. 2020, 10, 6124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, R.O. Enzyme replacement for lysosomal diseases. Annu. Rev. Med. 2006, 57, 283–296. [Google Scholar] [CrossRef]
- Diaz, G.A.; Jones, S.A.; Scarpa, M.; Mengel, K.E.; Giugliani, R.; Guffon, N.; Batsu, I.; Fraser, P.A.; Li, J.; Zhang, Q.; et al. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genet. Med. 2021, 1–8. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Diaz, G.A.; Lachmann, R.H.; Jouvin, M.H.; Nandy, I.; Ji, A.J.; Puga, A.C. Olipudase alfa for treatment of acid sphingomyelinase deficiency (ASMD): Safety and efficacy in adults treated for 30 months. J. Inherit. Metab. Dis. 2018, 41, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Wasserstein, M.P.; Jones, S.A.; Soran, H.; Diaz, G.A.; Lippa, N.; Thurberg, B.L.; Culm-Merdek, K.; Shamiyeh, E.; Inguilizian, H.; Cox, G.F.; et al. Successful within-patient dose escalation of olipudase alfa in acid sphingomyelinase deficiency. Mol. Genet. Metab. 2015, 116, 88–97. [Google Scholar] [CrossRef]
- Aldosari, M.H.; de Vries, R.P.; Rodriguez, L.R.; Hesen, N.A.; Beztsinna, N.; van Kuilenburg, A.B.P.; Hollak, C.E.M.; Schellekens, H.; Mastrobattista, E. Liposome-targeted recombinant human acid sphingomyelinase: Production, formulation, and in vitro evaluation. Eur. J. Pharm. Biopharm. 2019, 137, 185–195. [Google Scholar] [CrossRef]
- Simonis, A.; Schubert-Unkmeir, A. The role of acid sphingomyelinase and modulation of sphingolipid metabolism in bacterial infection. Biol. Chem. 2018, 399, 1135–1146. [Google Scholar] [CrossRef]
- Wu, Y.; Gulbins, E.; Grassmé, H. The function of sphingomyelinases in mycobacterial infections. Biol. Chem. 2018, 399, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, C.; Peng, H.; Swaidan, A.; Riehle, A.; Pollmeier, B.; Zhang, Y.; Gulbins, E.; Grassmé, H. Acid sphingomyelinase contributes to the control of mycobacterial infection via a signaling cascade leading from reactive oxygen species to cathepsin D. Cells 2020, 9, 2406. [Google Scholar] [CrossRef]
- Naimi, W.A.; Gumpf, J.J.; Cockburn, C.L.; Camus, S.; Chalfant, C.E.; Li, P.L.; Carlyon, J.A. Functional inhibition or genetic deletion of acid sphingomyelinase bacteriostatically inhibits Anaplasma phagocytophilum infection in vivo. Pathog. Dis. 2021, 79. [Google Scholar] [CrossRef] [PubMed]
- Meiners, J.; Palmieri, V.; Klopfleisch, R.; Ebel, J.F.; Japtok, L.; Schumacher, F.; Yusuf, A.M.; Becker, K.A.; Zöller, J.; Hose, M.; et al. Intestinal acid sphingomyelinase protects from severe pathogen-driven colitis. Front. Immunol. 2019, 10, 1386. [Google Scholar] [CrossRef] [PubMed]
- Guzman, G.; Niekamp, P.; Tafesse, F.G. The squeaky yeast gets greased: The roles of host lipids in the clearance of pathogenic fungi. J. Fungi 2020, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Guo, S.; Pang, W.; Zhao, Z. Crosstalk between acid sphingomyelinase and inflammasome signaling and their emerging roles in tissue injury and fibrosis. Front. Cell Dev. Biol. 2020, 7, 378. [Google Scholar] [CrossRef] [Green Version]
- White, J.M.; Whittaker, G.R. Fusion of enveloped viruses in endosomes. Traffic 2016, 17, 593–614. [Google Scholar] [CrossRef] [Green Version]
- Kühnl, A.; Musiol, A.; Heitzig, N.; Johnson, D.E.; Ehrhardt, C.; Grewal, T.; Gerke, V.; Ludwig, S.; Rescher, U. Late endosomal/lysosomal cholesterol accumulation is a host cell-protective mechanism inhibiting endosomal escape of influenza A virus. MBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Schoer, J.K.; Gallegos, A.M.; McIntosh, A.L.; Starodub, O.; Kier, A.B.; Billheimer, J.T.; Schroeder, F. Lysosomal membrane cholesterol dynamics. Biochemistry 2000, 39, 7662–7677. [Google Scholar] [CrossRef]
- Shoemaker, C.J.; Schornberg, K.L.; Delos, S.E.; Scully, C.; Pajouhesh, H.; Olinger, G.G.; Johansen, L.M.; White, J.M. Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PLoS ONE 2013, 8, e56265. [Google Scholar] [CrossRef]
- Wang, J.; Pendurthi, U.R.; Yi, G.; Rao, L.V.M. SARS-CoV-2 infection induces the activation of tissue factor-mediated coagulation by activation of acid sphingomyelinase. Blood 2021. [Google Scholar] [CrossRef] [PubMed]
- Carpinteiro, A.; Edwards, M.J.; Hoffmann, M.; Kochs, G.; Gripp, B.; Weigang, S.; Adams, C.; Carpinteiro, E.; Gulbins, A.; Keitsch, S.; et al. Pharmacological inhibition of acid sphingomyelinase prevents uptake of SARS-CoV-2 by epithelial cells. Cell Rep. Med. 2020, 1, 100142. [Google Scholar] [CrossRef] [PubMed]
- Schloer, S.; Brunotte, L.; Goretzko, J.; Mecate-Zambrano, A.; Korthals, N.; Gerke, V.; Ludwig, S.; Rescher, U. Targeting the endolysosomal host-SARS-CoV-2 interface by clinically licensed functional inhibitors of acid sphingomyelinase (FIASMA) including the antidepressant fluoxetine. Emerg. Microbes Infect. 2020, 9, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Loas, G.; Le Corre, P. Update on functional inhibitors of acid sphingomyelinase (FIASMAs) in SARS-CoV-2 infection. Pharmaceuticals 2021, 14, 691. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.E.; Adhikary, S.; Kolokoltsov, A.A.; Davey, R.A. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J. Virol. 2012, 86, 7473–7483. [Google Scholar] [CrossRef] [Green Version]
- Salata, C.; Calistri, A.; Parolin, C.; Baritussio, A.; Palù, G. Antiviral activity of cationic amphiphilic drugs. Expert Rev. Anti Infect. Ther. 2017, 15, 483–492. [Google Scholar] [CrossRef]
- Hoertel, N.; Sánchez-Rico, M.; Vernet, R.; Beeker, N.; Jannot, A.S.; Neuraz, A.; Salamanca, E.; Paris, N.; Daniel, C.; Gramfort, A.; et al. Association between antidepressant use and reduced risk of intubation or death in hospitalized patients with COVID-19: Results from an observational study. Mol. Psychiatry 2021, 1–14. [Google Scholar] [CrossRef]
- Hoertel, N.; Sánchez-Rico, M.; Gulbins, E.; Kornhuber, J.; Carpinteiro, A.; Lenze, E.J.; Reiersen, A.M.; Abellán, M.; De La Muela, P.; Vernet, R.; et al. Association between FIASMAs and reduced risk of intubation or death in individuals hospitalized for severe COVID-19: An observational multicenter study. Clin. Pharmacol. Ther. 2021. [Google Scholar] [CrossRef]
- Luquain-Costaz, C.; Rabia, M.; Hullin-Matsuda, F.; Delton, I. Bis(monoacylglycero)phosphate, an important actor in the host endocytic machinery hijacked by SARS-CoV-2 and related viruses. Biochimie 2020. [Google Scholar] [CrossRef]
- Martín-Acebes, M.A.; Gabandé-Rodríguez, E.; García-Cabrero, A.M.; Sánchez, M.P.; Ledesma, M.D.; Sobrino, F.; Saiz, J.C. Host sphingomyelin increases West Nile virus infection in vivo. J. Lipid Res. 2016, 57, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, M.J.; Becker, K.A.; Gripp, B.; Hoffmann, M.; Keitsch, S.; Wilker, B.; Soddemann, M.; Gulbins, A.; Carpinteiro, E.; Patel, S.H.; et al. Sphingosine prevents binding of SARS-CoV-2 spike to its cellular receptor ACE2. J. Biol. Chem. 2020, 295, 15174–15182. [Google Scholar] [CrossRef] [PubMed]
- Walkley, S.U.; Vanier, M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breiden, B.; Sandhoff, K. Acid Sphingomyelinase, a Lysosomal and Secretory Phospholipase C, Is Key for Cellular Phospholipid Catabolism. Int. J. Mol. Sci. 2021, 22, 9001. https://doi.org/10.3390/ijms22169001
Breiden B, Sandhoff K. Acid Sphingomyelinase, a Lysosomal and Secretory Phospholipase C, Is Key for Cellular Phospholipid Catabolism. International Journal of Molecular Sciences. 2021; 22(16):9001. https://doi.org/10.3390/ijms22169001
Chicago/Turabian StyleBreiden, Bernadette, and Konrad Sandhoff. 2021. "Acid Sphingomyelinase, a Lysosomal and Secretory Phospholipase C, Is Key for Cellular Phospholipid Catabolism" International Journal of Molecular Sciences 22, no. 16: 9001. https://doi.org/10.3390/ijms22169001
APA StyleBreiden, B., & Sandhoff, K. (2021). Acid Sphingomyelinase, a Lysosomal and Secretory Phospholipase C, Is Key for Cellular Phospholipid Catabolism. International Journal of Molecular Sciences, 22(16), 9001. https://doi.org/10.3390/ijms22169001