
Tideglusib, a Non-ATP Competitive Inhibitor of GSK-3β as a Drug Candidate for the Treatment of Amyotrophic Lateral Sclerosis

 ,
, 
 and
and 
Abstract
1. Introduction
2. Results
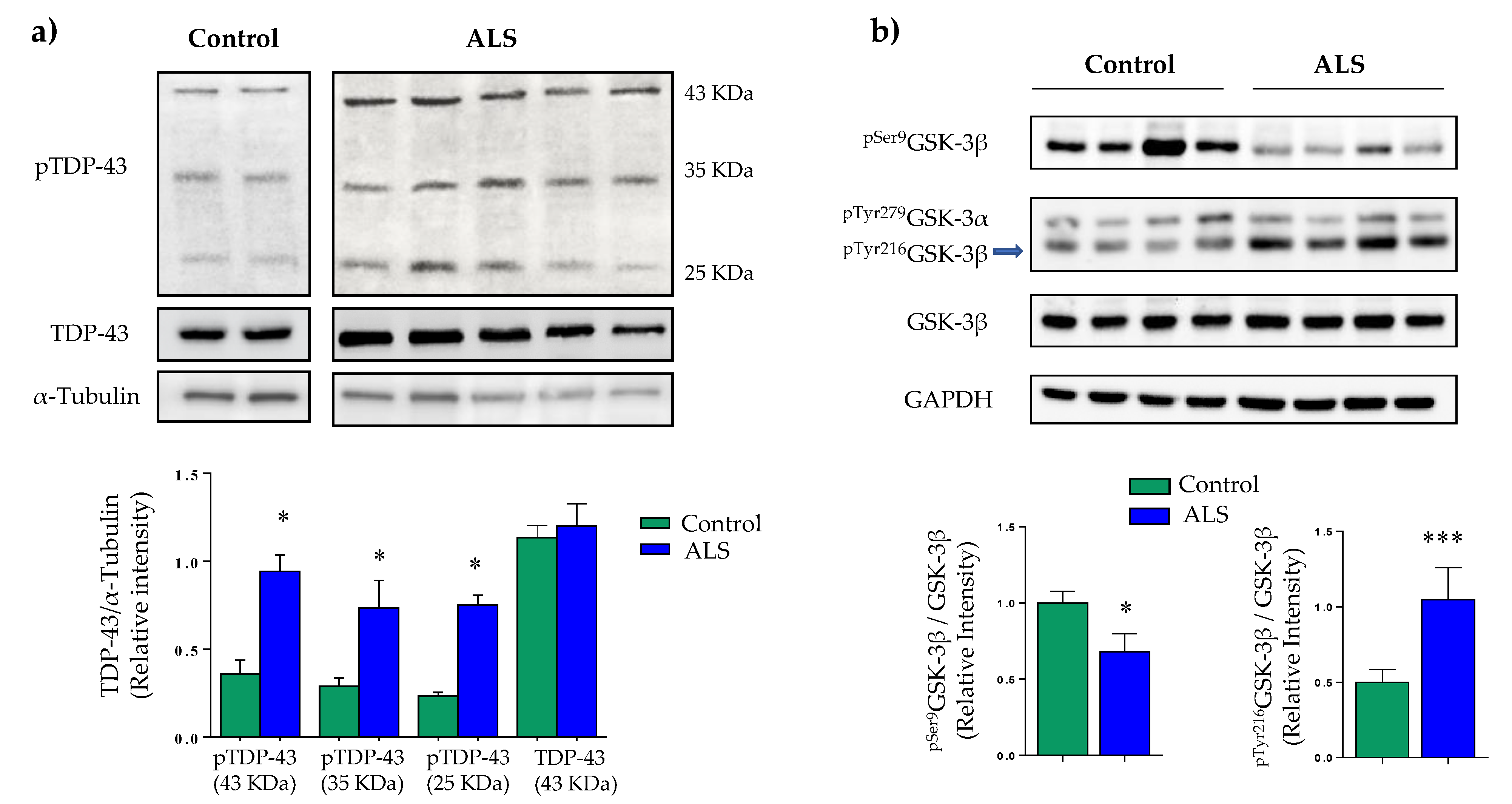
2.1. Characterization of Immortalized Lymphocytes from Sporadic ALS Patients
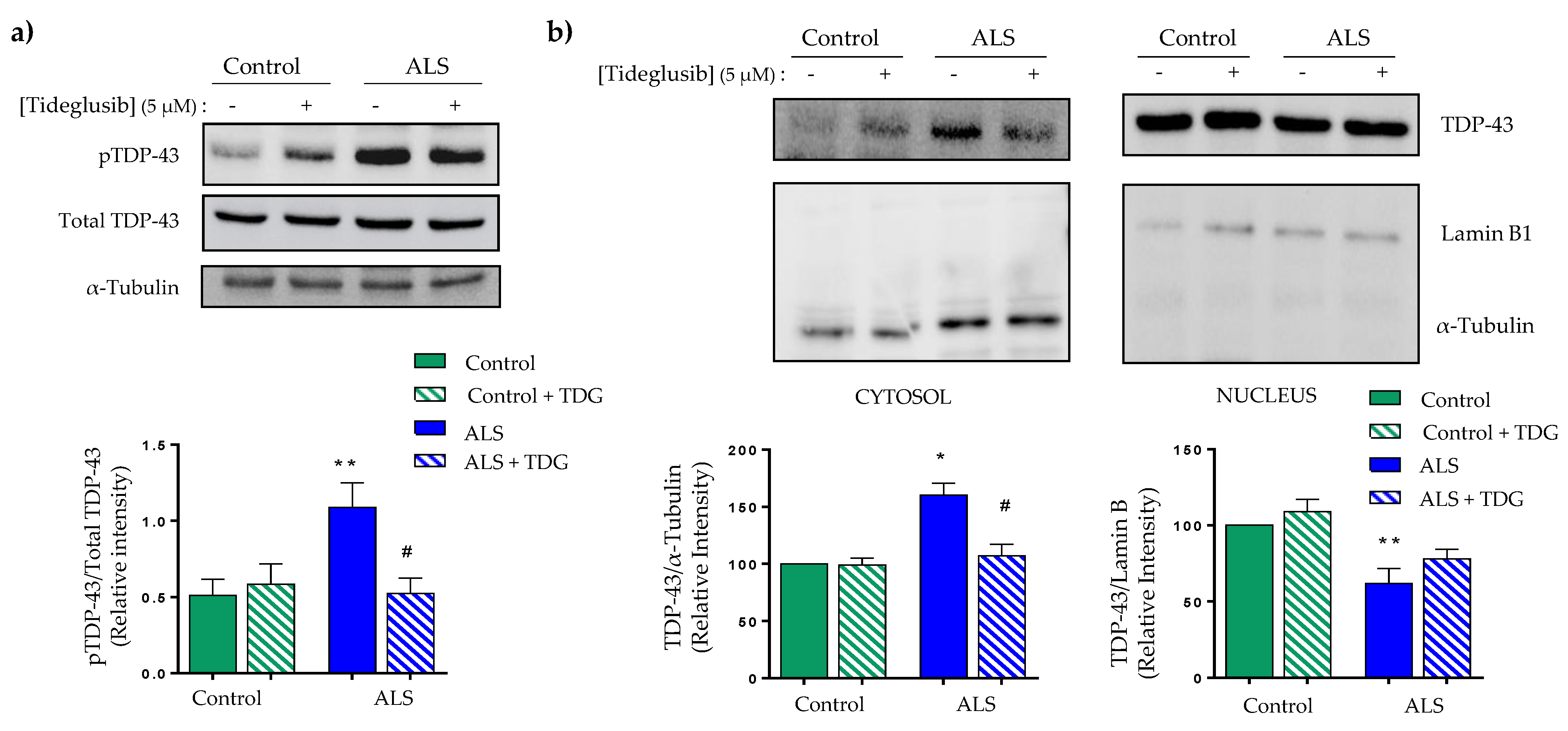
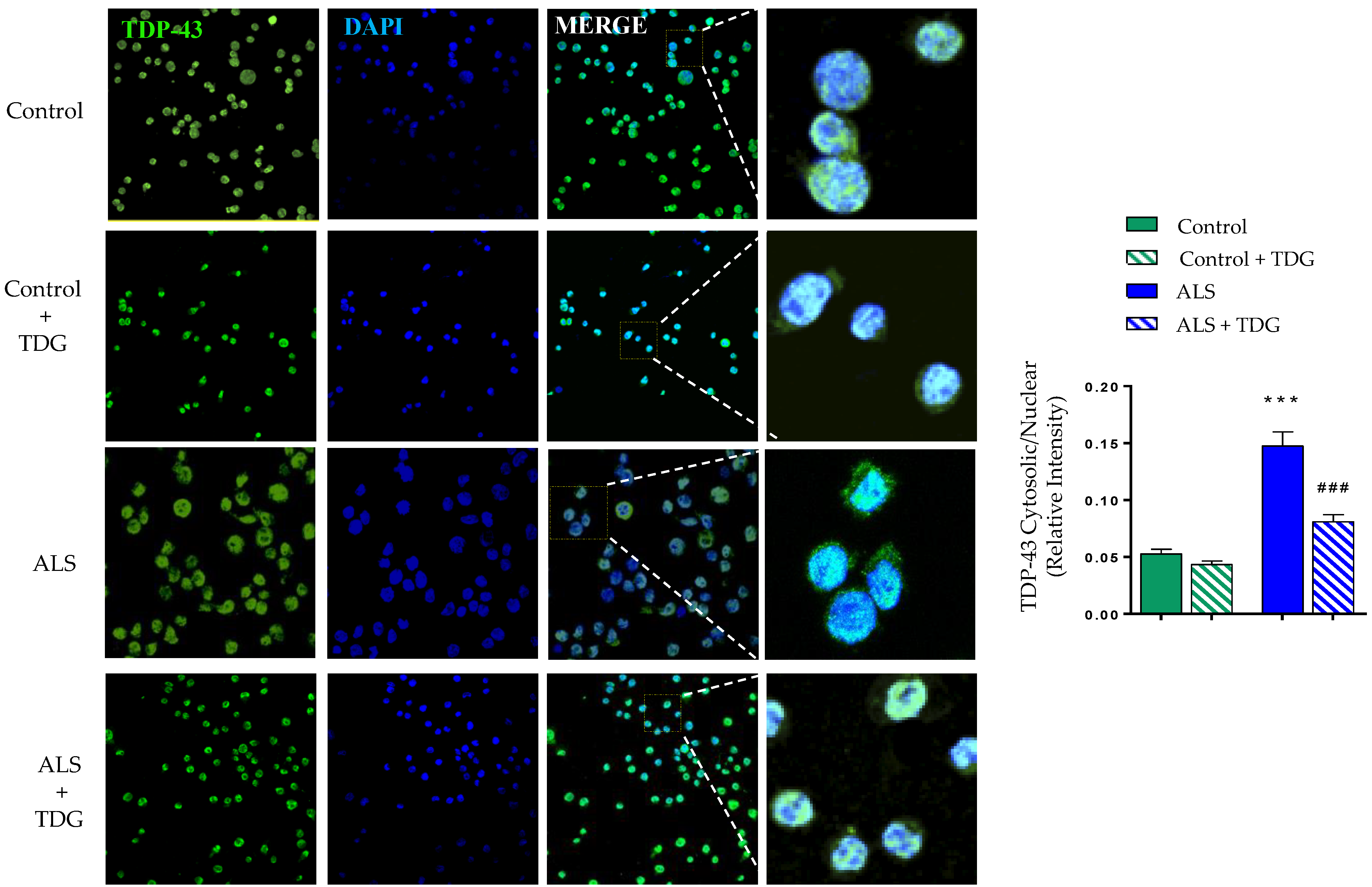
2.2. Tideglusib Restored TDP-43 Homeostasis in Immortalized Lymphocytes from Sporadic ALS Patients
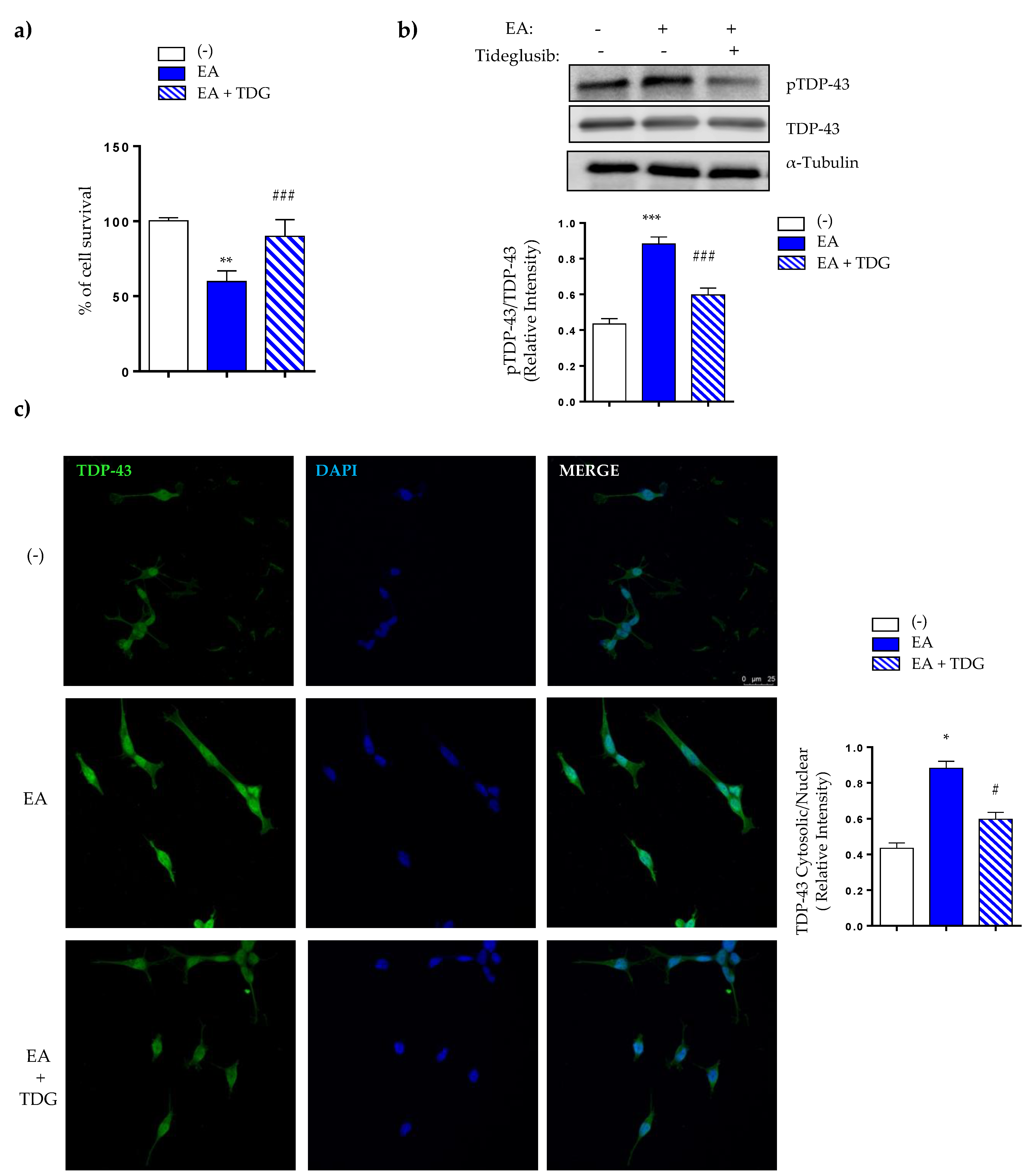
2.3. Effects of Tideglusib on TDP-43 Phosphorylation and Viability in Ethacrynic Acid-Treated Neuroblastoma SH-SY5Y Cells
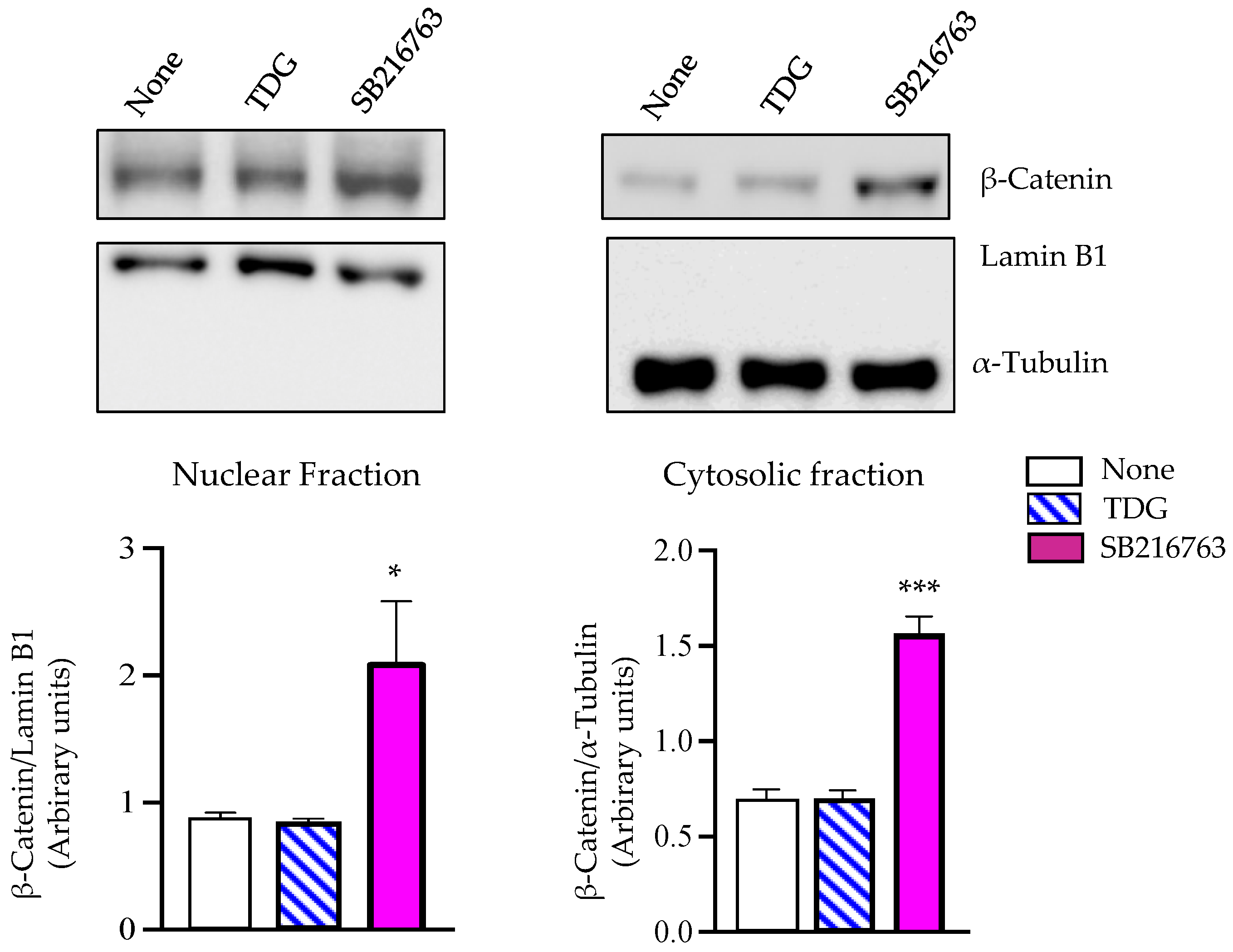
2.4. Tideglusib Does Not Induce Nuclear ß-Catenin Accumulation
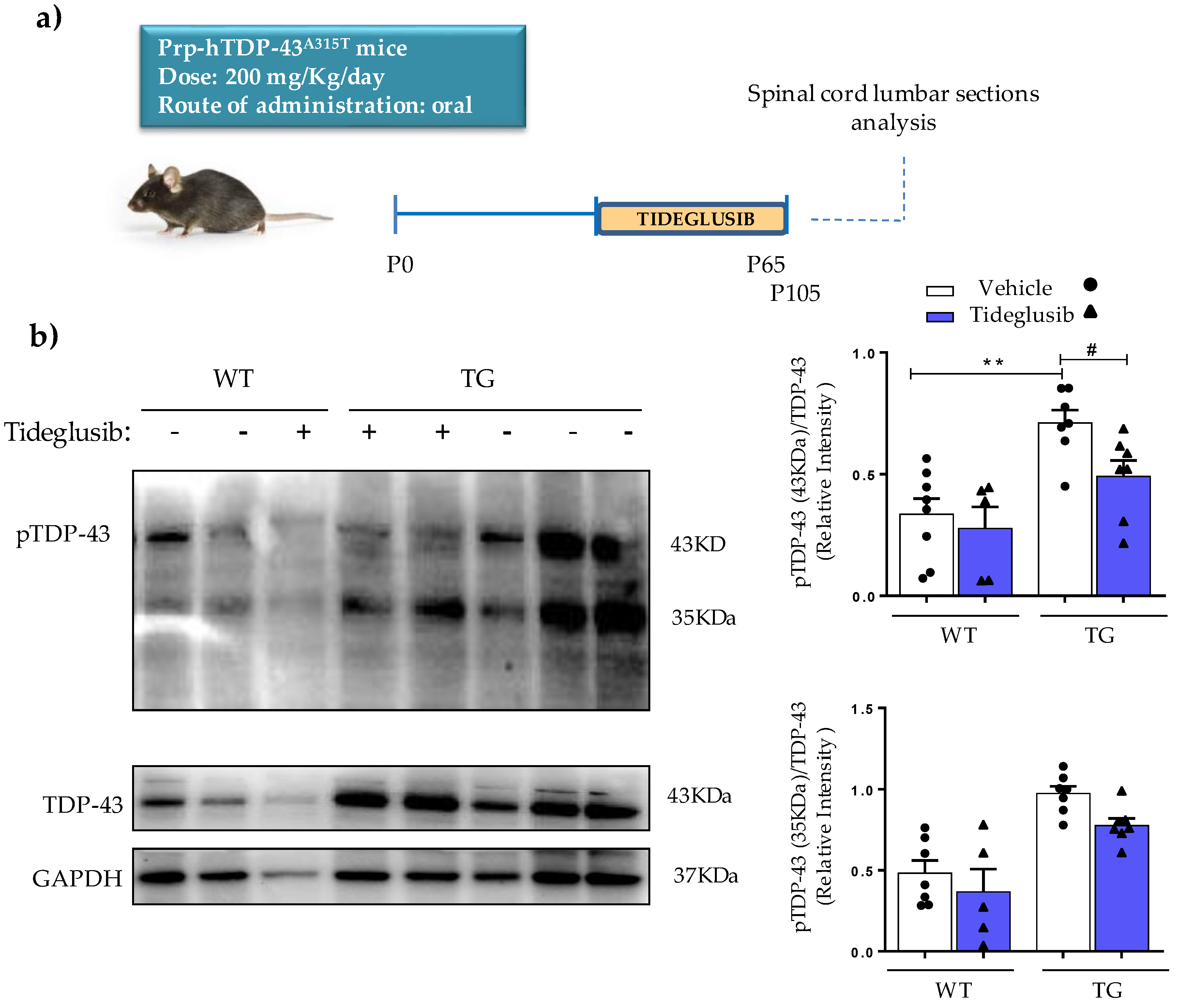
2.5. Tideglusib Counteracts TDP-43 Hyperphosphorylation in Prp-hTDP-43 (A315T) Mice
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Animal Procedures
4.3. Immunoblotting Analysis
4.4. Immunofluorescence
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scotter, E.L.; Chen, H.J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Palomo, V.; Tosat-Bitrian, C.; Nozal, V.; Nagaraj, S.; Martin-Requero, A.; Martinez, A. TDP-43: A Key Therapeutic Target beyond Amyotrophic Lateral Sclerosis. ACS Chem. Neurosci. 2019, 10, 1183–1196. [Google Scholar] [CrossRef]
- Neumann, M.; Kwong, L.K.; Lee, E.B.; Kremmer, E.; Flatley, A.; Xu, Y.; Forman, M.S.; Troost, D.; Kretzschmar, H.A.; Trojanowski, J.Q.; et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009, 117, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Neukomm, L.J.; Brown, R.H., Jr.; Freeman, M.R. Age-Dependent TDP-43-Mediated Motor Neuron Degeneration Requires GSK3, hat-trick, and xmas-2. Curr. Biol. 2015, 25, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Castro, A.; Dorronsoro, I.; Alonso, M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med. Res. Rev. 2002, 22, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Zhang, H.; Wagey, R.; Krieger, C.; Pelech, S.L. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2003, 85, 432–442. [Google Scholar] [CrossRef]
- Yang, W.; Leystra-Lantz, C.; Strong, M.J. Upregulation of GSK3beta expression in frontal and temporal cortex in ALS with cognitive impairment (ALSci). Brain Res. 2008, 1196, 131–139. [Google Scholar] [CrossRef]
- Choi, H.J.; Cha, S.J.; Lee, J.W.; Kim, H.J.; Kim, K. Recent Advances on the Role of GSK3beta in the Pathogenesis of Amyotrophic Lateral Sclerosis. Brain Sci. 2020, 10, 675. [Google Scholar] [CrossRef]
- Kihira, T.; Suzuki, A.; Kondo, T.; Wakayama, I.; Yoshida, S.; Hasegawa, K.; Garruto, R.M. Immunohistochemical expression of IGF-I and GSK in the spinal cord of Kii and Guamanian ALS patients. Neuropathology 2009, 29, 548–558. [Google Scholar] [CrossRef]
- González-Muñoz, M.I.; Rodríguez-Mahillo, A.; Gil, C.; Morán, Y.; Moneo, I.; Martínez, A.; Mora, J.S. Glycogen Synthase Kinase-3β Expression and Phosphorylation in Peripheral Blood Mononuclear Cells of Patients with Amyotrophic Lateral Sclerosis. JAMMR 2013, 4, 263–271. [Google Scholar] [CrossRef]
- Moujalled, D.; James, J.L.; Parker, S.J.; Lidgerwood, G.E.; Duncan, C.; Meyerowitz, J.; Nonaka, T.; Hasegawa, M.; Kanninen, K.M.; Grubman, A.; et al. Kinase Inhibitor Screening Identifies Cyclin-Dependent Kinases and Glycogen Synthase Kinase 3 as Potential Modulators of TDP-43 Cytosolic Accumulation during Cell Stress. PLoS ONE 2013, 8, e67433. [Google Scholar] [CrossRef]
- Yang, Y.M.; Gupta, S.K.; Kim, K.J.; Powers, B.E.; Cerqueira, A.; Wainger, B.J.; Ngo, H.D.; Rosowski, K.A.; Schein, P.A.; Ackeifi, C.A.; et al. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell 2013, 12, 713–726. [Google Scholar] [CrossRef]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef]
- Ambegaokar, S.S.; Jackson, G.R. Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum. Mol. Genet. 2011, 20, 4947–4977. [Google Scholar] [CrossRef]
- Hu, J.H.; Chernoff, K.; Pelech, S.; Krieger, C. Protein kinase and protein phosphatase expression in the central nervous system of G93A mSOD over-expressing mice. J. Neurochem. 2003, 85, 422–431. [Google Scholar] [CrossRef]
- Koh, S.H.; Kim, Y.; Kim, H.Y.; Hwang, S.; Lee, C.H.; Kim, S.H. Inhibition of glycogen synthase kinase-3 suppresses the onset of symptoms and disease progression of G93A-SOD1 mouse model of ALS. Exp. Neurol. 2007, 205, 336–346. [Google Scholar] [CrossRef]
- de Munck, E.; Munoz-Saez, E.; Miguel, B.G.; Solas, M.T.; Martinez, A.; Arahuetes, R.M. Morphometric and neurochemical alterations found in l-BMAA treated rats. Environ. Toxicol. Pharmacol. 2015, 39, 1232–1245. [Google Scholar] [CrossRef] [PubMed]
- de Munck, E.; Munoz-Saez, E.; Miguel, B.G.; Solas, M.T.; Ojeda, I.; Martinez, A.; Gil, C.; Arahuetes, R.M. beta-N-methylamino-l-alanine causes neurological and pathological phenotypes mimicking Amyotrophic Lateral Sclerosis (ALS): The first step towards an experimental model for sporadic ALS. Environ. Toxicol. Pharmacol. 2013, 36, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Boll, M.C.; Bayliss, L.; Vargas-Canas, S.; Burgos, J.; Montes, S.; Penaloza-Solano, G.; Rios, C.; Alcaraz-Zubeldia, M. Clinical and biological changes under treatment with lithium carbonate and valproic acid in sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 2014, 340, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Morales-Garcia, J.A.; Luna-Medina, R.; Alonso-Gil, S.; Sanz-Sancristobal, M.; Palomo, V.; Gil, C.; Santos, A.; Martinez, A.; Perez-Castillo, A. Glycogen synthase kinase 3 inhibition promotes adult hippocampal neurogenesis in vitro and in vivo. ACS Chem. Neurosci. 2012, 3, 963–971. [Google Scholar] [CrossRef]
- Luna-Medina, R.; Cortes-Canteli, M.; Sanchez-Galiano, S.; Morales-Garcia, J.A.; Martinez, A.; Santos, A.; Perez-Castillo, A. NP031112, a thiadiazolidinone compound, prevents inflammation and neurodegeneration under excitotoxic conditions: Potential therapeutic role in brain disorders. J. Neurosci. 2007, 27, 5766–5776. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Tolosa, E.; Litvan, I.; Hoglinger, G.U.; Burn, D.; Lees, A.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; Del Ser, T.; Investigators, T. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disord. 2014, 29, 470–478. [Google Scholar] [CrossRef]
- Anagnostou, E.; Bennett, T.A.; Thorpe, K.; Nicolson, R. 5.16 A Phase 2 Randomized, Placebo-Controlled Trial of Tideglusib, an Orally Administered GSK-3 Beta Inhibitor, in the Treatment of Adolescents with ASD. J. Am. Acad. Child. Adolesc. 2018, 57, S232. [Google Scholar] [CrossRef]
- Horrigan, J.; Gomes, T.B.; Snape, M.; Nikolenko, N.; McMorn, A.; Evans, S.; Yaroshinsky, A.; Della Pasqua, O.; Oosterholt, S.; Lochmuller, H. A Phase 2 Study of AMO-02 (Tideglusib) in Congenital and Childhood-Onset Myotonic Dystrophy Type 1 (DM1). Pediatr. Neurol. 2020, 112, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Posa, D.; Martinez-Gonzalez, L.; Bartolome, F.; Nagaraj, S.; Porras, G.; Martinez, A.; Martin-Requero, A. Recapitulation of Pathological TDP-43 Features in Immortalized Lymphocytes from Sporadic ALS Patients. Mol. Neurobiol. 2019, 56, 2424–2432. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, L.; Rodriguez-Cueto, C.; Cabezudo, D.; Bartolome, F.; Andres-Benito, P.; Ferrer, I.; Gil, C.; Martin-Requero, A.; Fernandez-Ruiz, J.; Martinez, A.; et al. Motor neuron preservation and decrease of in vivo TDP-43 phosphorylation by protein CK-1delta kinase inhibitor treatment. Sci. Rep. 2020, 10, 4449. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, Y.; Katsuno, M.; Takagi, S.; Ishigaki, S.; Niwa, J.; Hasegawa, M.; Tanaka, F.; Sobue, G. Oxidative stress induced by glutathione depletion reproduces pathological modifications of TDP-43 linked to TDP-43 proteinopathies. Neurobiol. Dis. 2012, 45, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Alquezar, C.; Salado, I.G.; de la Encarnacion, A.; Perez, D.I.; Moreno, F.; Gil, C.; de Munain, A.L.; Martinez, A.; Martin-Requero, A. Targeting TDP-43 phosphorylation by Casein Kinase-1delta inhibitors: A novel strategy for the treatment of frontotemporal dementia. Mol. Neurodegener. 2016, 11, 36. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; Maestro, R.; et al. Multifaceted roles of GSK-3 and Wnt/beta-catenin in hematopoiesis and leukemogenesis: Opportunities for therapeutic intervention. Leukemia 2014, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Xiao, G.; Hu, J. Regulation of Wnt/beta-catenin signaling by posttranslational modifications. Cell. Biosci. 2014, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef] [PubMed]
- del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andres, M.V.; Gomez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; Leon, T. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: A pilot study. J. Alzheimers Dis. 2013, 33, 205–215. [Google Scholar] [CrossRef]
- Vaca, G.; Martinez-Gonzalez, L.; Fernandez, A.; Rojas-Prats, E.; Porras, G.; Cuevas, E.P.; Gil, C.; Martinez, A.; Martin-Requero, A. Therapeutic potential of novel Cell Division Cycle Kinase 7 inhibitors on TDP-43-related pathogenesis such as Frontotemporal Lobar Degeneration (FTLD) and amyotrophic lateral sclerosis (ALS). J. Neurochem. 2021, 156, 379–390. [Google Scholar] [CrossRef]
- Janssens, J.; Van Broeckhoven, C. Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD-ALS spectrum disorders. Hum. Mol. Genet. 2013, 22, R77–R87. [Google Scholar] [CrossRef]
- Geser, F.; Lee, V.M.; Trojanowski, J.Q. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: A spectrum of TDP-43 proteinopathies. Neuropathology 2010, 30, 103–112. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001, 359 Pt 1, 1–16. [Google Scholar] [CrossRef]
- Palomo, V.; Perez, D.I.; Roca, C.; Anderson, C.; Rodriguez-Muela, N.; Perez, C.; Morales-Garcia, J.A.; Reyes, J.A.; Campillo, N.E.; Perez-Castillo, A.M.; et al. Subtly Modulating Glycogen Synthase Kinase 3 beta: Allosteric Inhibitor Development and Their Potential for the Treatment of Chronic Diseases. J. Med. Chem. 2017, 60, 4983–5001. [Google Scholar] [CrossRef]
- Agosta, F.; Al-Chalabi, A.; Filippi, M.; Hardiman, O.; Kaji, R.; Meininger, V.; Nakano, I.; Shaw, P.; Shefner, J.; van den Berg, L.H.; et al. The El Escorial criteria: Strengths and weaknesses. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2015, 16, 1–7. [Google Scholar] [CrossRef]
- Koistinen, P. Human peripheral blood and bone marrow cell separation using density gradient centrifugation on Lymphoprep and Percoll in haematological diseases. Scand. J. Clin. Lab. Invest. 1987, 47, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.M. Tetrazolium (MTT) assay for cellular viability and activity. Methods Mol. Biol. 1998, 79, 179–183. [Google Scholar] [PubMed]
- Coughlan, K.S.; Halang, L.; Woods, I.; Prehn, J.H. A high-fat jelly diet restores bioenergetic balance and extends lifespan in the presence of motor dysfunction and lumbar spinal cord motor neuron loss in TDP-43A315T mutant C57BL6/J mice. Dis. Model. Mech. 2016, 9, 1029–1037. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Source | Catalog Number | Dilution |
|---|---|---|---|
| Lamin B1 | Santa Cruz Biotechnologies | sc-6217 | 1:1000 |
| α-Tubulin | Sigma | L7543 | 1:1000 |
| pTDP-43 (Ser409/410) | Proteintech | 22309-1-AP | 1:500 |
| TDP-43 | Proteintech | 10782-2-AP | 1:1000 |
| GAPDH | Santa Cruz Biotechnologies | sc-25778 | 1:500 |
| p-GSK-3β(Ser9) | Santa Cruz Biotechnologies | sc-11757) | 1:500 |
| p-GSK3β(Y216) | Santa Cruz Biotechnologies | sc-81496 | 1:500 |
| GSK-3β | Santa Cruz Biotechnologies | sc-9166 | 1:500 |
| β-Catenin | Santa Cruz Biotechnologies | sc-7199 | 1:500 |
| Control (N = 5) | ALS (N = 5) | |
|---|---|---|
| Gender (M/F) | (2/3) | (2/3) |
| Family history | No | No |
| Age (±SD) | 62 ± 7 | 65 ± 1 |
| Site of onset | ||
| Bulbar | NA | 4 |
| Limb | NA | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-González, L.; Gonzalo-Consuegra, C.; Gómez-Almería, M.; Porras, G.; de Lago, E.; Martín-Requero, Á.; Martínez, A. Tideglusib, a Non-ATP Competitive Inhibitor of GSK-3β as a Drug Candidate for the Treatment of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 8975. https://doi.org/10.3390/ijms22168975
Martínez-González L, Gonzalo-Consuegra C, Gómez-Almería M, Porras G, de Lago E, Martín-Requero Á, Martínez A. Tideglusib, a Non-ATP Competitive Inhibitor of GSK-3β as a Drug Candidate for the Treatment of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2021; 22(16):8975. https://doi.org/10.3390/ijms22168975
Chicago/Turabian StyleMartínez-González, Loreto, Claudia Gonzalo-Consuegra, Marta Gómez-Almería, Gracia Porras, Eva de Lago, Ángeles Martín-Requero, and Ana Martínez. 2021. "Tideglusib, a Non-ATP Competitive Inhibitor of GSK-3β as a Drug Candidate for the Treatment of Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 22, no. 16: 8975. https://doi.org/10.3390/ijms22168975
APA StyleMartínez-González, L., Gonzalo-Consuegra, C., Gómez-Almería, M., Porras, G., de Lago, E., Martín-Requero, Á., & Martínez, A. (2021). Tideglusib, a Non-ATP Competitive Inhibitor of GSK-3β as a Drug Candidate for the Treatment of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 22(16), 8975. https://doi.org/10.3390/ijms22168975