
Pathogenic Mechanisms of Hypertrophic Cardiomyopathy beyond Sarcomere Dysfunction



{kind=link}
Abstract
:1. Introduction
2. Genetic and Non-Genetic Risk Modifiers
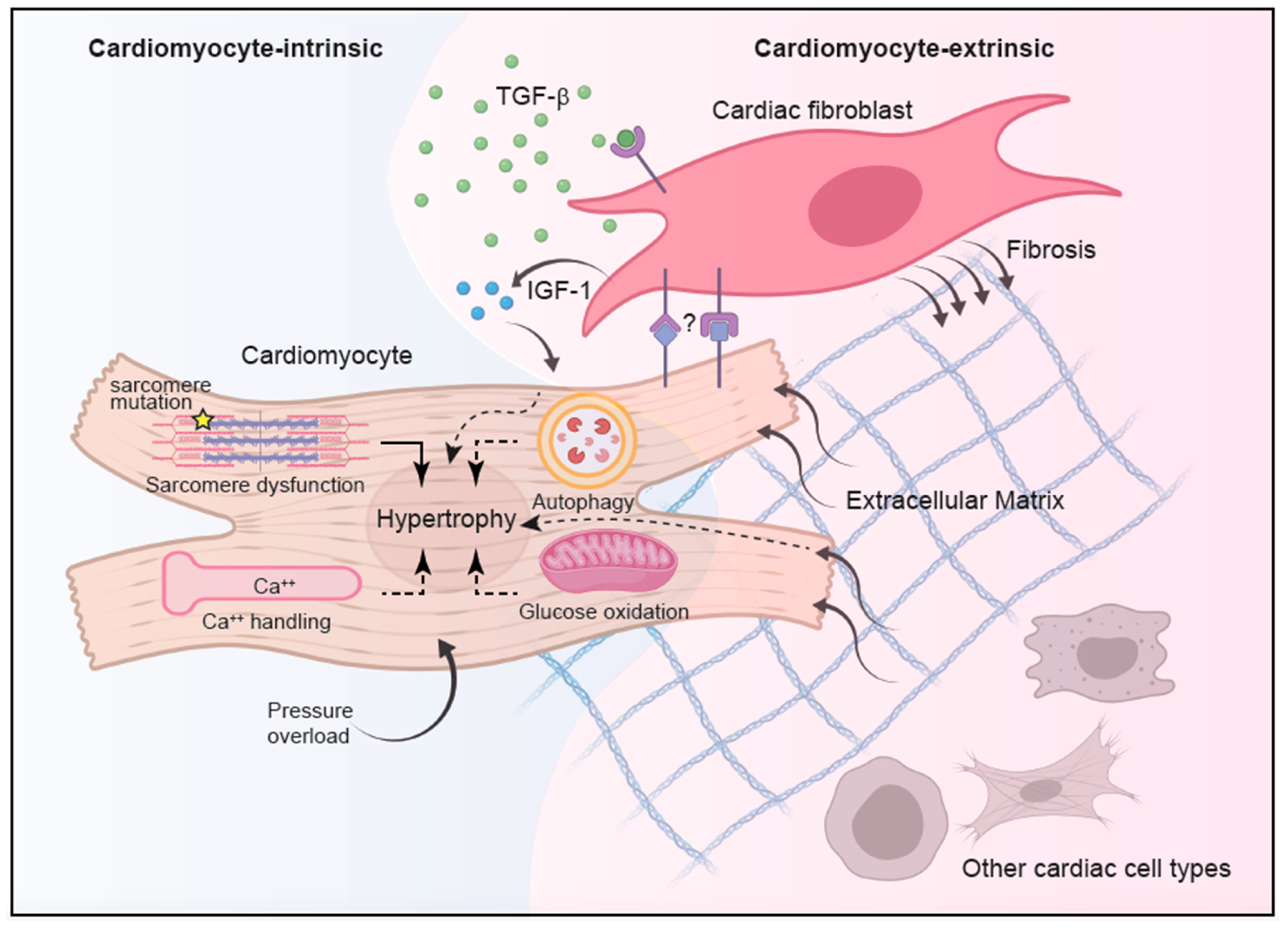
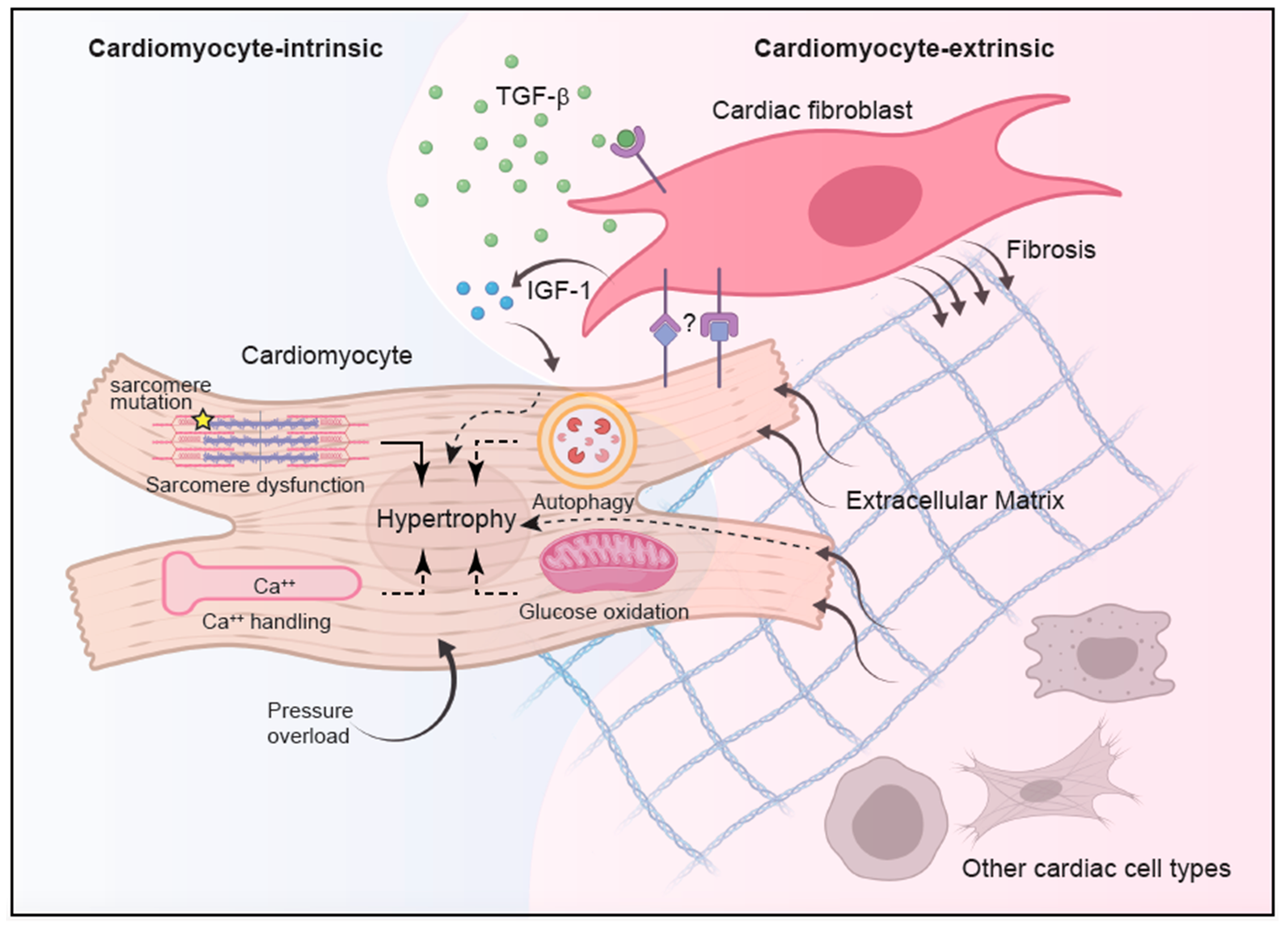
3. Cell-Intrinsic Mechanisms of Cardiomyocyte Hypertrophy
3.1. Calcium Signaling
3.2. RAS Pathway
3.3. Autophagy
3.4. Glucose Metabolism
3.5. Non-Sarcomere Structural Proteins
4. Cell-Extrinsic Mechanisms of HCM
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HCM | Hypertrophic Cardiomyopathy |
| GWAS | Genome Wide Association Study |
| DCM | Dilated Cardiomyopathy |
| LV | Left Ventricle |
| CaMKII | Calcium-calmodulin Dependent Protein Kinase II |
| SERCA2A | Sarcoendoplasmic Reticulum Calcium Transport ATPase |
| CRYAB | αB-Crystallin |
| NFAT | Nuclear Factor of Activated T-cells |
| TAC | Transverse Aortic Constriction |
| MPC | Mitochondrial Pyruvate Carrier |
| TTN | Titin |
| PKD | Protein Kinase D |
| LMNA | Lamin A/C |
| ECM | Extracellular Matrix |
| TGF-β | Transforming Growth Factor-β |
References
- Bonow, R.O. Hypertrophic cardiomyopathy: Past, present... and future. Trends Cardiovasc. Med. 2015, 25, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Ommen, S.R.; Semsarian, C.; Spirito, P.; Olivotto, I.; Maron, M.S. Hypertrophic cardiomyopathy: Present and future, with translation into contemporary cardiovascular medicine. J. Am. Coll. Cardiol. 2014, 64, 83–99. [Google Scholar] [CrossRef] [Green Version]
- Seidman, C.E.; Seidman, J.G. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: A personal history. Circ. Res. 2011, 108, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Thierfelder, L.; Watkins, H.; MacRae, C.; Lamas, R.; McKenna, W.; Vosberg, H.P.; Seidman, J.G.; Seidman, C.E. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 1994, 77, 701–712. [Google Scholar] [CrossRef]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Watkins, H.; MacRae, C.; Thierfelder, L.; Chou, Y.H.; Frenneaux, M.; McKenna, W.; Seidman, J.G.; Seidman, C.E. A disease locus for familial hypertrophic cardiomyopathy maps to chromosome 1q3. Nat. Genet. 1993, 3, 333–337. [Google Scholar] [CrossRef]
- Akhtar, M.; Elliott, P. The genetics of hypertrophic cardiomyopathy. Glob. Cardiol. Sci. Pract. 2018, 2018, 36. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Maron, M.S.; Maron, B.A.; Loscalzo, J. Moving beyond the Sarcomere to Explain Heterogeneity in Hypertrophic Cardiomyopathy: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 1978–1986. [Google Scholar] [CrossRef]
- Maron, B.J. Clinical Course and Management of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Gruner, C.; Ivanov, J.; Care, M.; Williams, L.; Moravsky, G.; Yang, H.; Laczay, B.; Siminovitch, K.; Woo, A.; Rakowski, H. Toronto hypertrophic cardiomyopathy genotype score for prediction of a positive genotype in hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingles, J.; Burns, C.; Bagnall, R.D.; Lam, L.; Yeates, L.; Sarina, T.; Puranik, R.; Briffa, T.; Atherton, J.J.; Driscoll, T.; et al. Nonfamilial Hypertrophic Cardiomyopathy: Prevalence, Natural History, and Clinical Implications. Circ. Cardiovasc. Genet. 2017, 10, e001620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.V.; et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2019, 12, e002460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manrai, A.K.; Funke, B.H.; Rehm, H.L.; Olesen, M.S.; Maron, B.A.; Szolovits, P.; Margulies, D.M.; Loscalzo, J.; Kohane, I.S. Genetic Misdiagnoses and the Potential for Health Disparities. N. Engl. J. Med. 2016, 375, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, N.; Michels, M.; Rowin, E.J.; Semsarian, C.; Girolami, F.; Tomberli, B.; Cecchi, F.; Maron, M.S.; Olivotto, I.; Maron, B.J. Clinical Course and Significance of Hypertrophic Cardiomyopathy Without Left Ventricular Hypertrophy. Circulation 2019, 139, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef]
- Walsh, R.; Mazzarotto, F.; Whiffin, N.; Buchan, R.; Midwinter, W.; Wilk, A.; Li, N.; Felkin, L.; Ingold, N.; Govind, R.; et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: The case of hypertrophic cardiomyopathy. Genome Med. 2019, 11, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Task Force, M.; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Tadros, R.; Francis, C.; Xu, X.; Vermeer, A.M.C.; Harper, A.R.; Huurman, R.; Kelu Bisabu, K.; Walsh, R.; Hoorntje, E.T.; Te Rijdt, W.P.; et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat. Genet. 2021, 53, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef]
- Neubauer, S.; Kolm, P.; Ho, C.Y.; Kwong, R.Y.; Desai, M.Y.; Dolman, S.F.; Appelbaum, E.; Desvigne-Nickens, P.; DiMarco, J.P.; Friedrich, M.G.; et al. Distinct Subgroups in Hypertrophic Cardiomyopathy in the NHLBI HCM Registry. J. Am. Coll. Cardiol. 2019, 74, 2333–2345. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H. Time to Think Differently About Sarcomere-Negative Hypertrophic Cardiomyopathy. Circulation 2021, 143, 2415–2417. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Luedde, M.; Katus, H.A. Mechanisms of disease: Hypertrophic cardiomyopathy. Nat. Rev. Cardiol. 2011, 9, 91–100. [Google Scholar] [CrossRef]
- Viola, H.M.; Hool, L.C. Impaired calcium handling and mitochondrial metabolic dysfunction as early markers of hypertrophic cardiomyopathy. Arch. Biochem. Biophys. 2019, 665, 166–174. [Google Scholar] [CrossRef]
- Coppini, R.; Ferrantini, C.; Mugelli, A.; Poggesi, C.; Cerbai, E. Altered Ca(2+) and Na(+) Homeostasis in Human Hypertrophic Cardiomyopathy: Implications for Arrhythmogenesis. Front. Physiol. 2018, 9, 1391. [Google Scholar] [CrossRef] [PubMed]
- Helms, A.S.; Alvarado, F.J.; Yob, J.; Tang, V.T.; Pagani, F.; Russell, M.W.; Valdivia, H.H.; Day, S.M. Genotype-Dependent and -Independent Calcium Signaling Dysregulation in Human Hypertrophic Cardiomyopathy. Circulation 2016, 134, 1738–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molkentin, J.D.; Lu, J.R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.U.N.; Canseco, D.C.; Xiao, F.; Nakada, Y.; Li, S.; Lam, N.T.; Muralidhar, S.A.; Savla, J.J.; Hill, J.A.; Le, V.; et al. A calcineurin-Hoxb13 axis regulates growth mode of mammalian cardiomyocytes. Nature 2020, 582, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Kumarapeli, A.R.; Su, H.; Huang, W.; Tang, M.; Zheng, H.; Horak, K.M.; Li, M.; Wang, X. Alpha B-crystallin suppresses pressure overload cardiac hypertrophy. Circ. Res. 2008, 103, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Osinska, H.; Klevitsky, R.; Gerdes, A.M.; Nieman, M.; Lorenz, J.; Hewett, T.; Robbins, J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ. Res. 2001, 89, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Rowin, E.J.; Arkun, K.; Rastegar, H.; Larson, A.M.; Maron, M.S.; Chin, M.T. Adult Monozygotic Twins With Hypertrophic Cardiomyopathy and Identical Disease Expression and Clinical Course. Am. J. Cardiol. 2020, 127, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.C.; Lepri, F.; Baban, A.; Dentici, M.L.; Versacci, P.; Capolino, R.; Ferese, R.; De Luca, A.; Tartaglia, M.; Marino, B.; et al. RASopathies: Clinical Diagnosis in the First Year of Life. Mol. Syndromol. 2011, 1, 282–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monda, E.; Rubino, M.; Lioncino, M.; Di Fraia, F.; Pacileo, R.; Verrillo, F.; Cirillo, A.; Caiazza, M.; Fusco, A.; Esposito, A.; et al. Hypertrophic Cardiomyopathy in Children: Pathophysiology, Diagnosis, and Treatment of Non-sarcomeric Causes. Front. Pediatr. 2021, 9, 632293. [Google Scholar] [CrossRef]
- Tidyman, W.E.; Rauen, K.A. The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 2009, 19, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moak, J.P.; Kaski, J.P. Hypertrophic cardiomyopathy in children. Heart 2012, 98, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Hickey, E.J.; Mehta, R.; Elmi, M.; Asoh, K.; McCrindle, B.W.; Williams, W.G.; Manlhiot, C.; Benson, L. Survival implications: Hypertrophic cardiomyopathy in Noonan syndrome. Congenit. Heart Dis. 2011, 6, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.N.; Schneider, M.D. Sizing up the heart: Development redux in disease. Genes Dev. 2003, 17, 1937–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaglia, M.; Gelb, B.D. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: Phenotypic spectrum and molecular mechanisms. Ann. N. Y. Acad. Sci. 2010, 1214, 99–121. [Google Scholar] [CrossRef] [Green Version]
- Pandit, B.; Sarkozy, A.; Pennacchio, L.A.; Carta, C.; Oishi, K.; Martinelli, S.; Pogna, E.A.; Schackwitz, W.; Ustaszewska, A.; Landstrom, A.; et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 2007, 39, 1007–1012. [Google Scholar] [CrossRef]
- Zenker, M.; Buheitel, G.; Rauch, R.; Koenig, R.; Bosse, K.; Kress, W.; Tietze, H.U.; Doerr, H.G.; Hofbeck, M.; Singer, H.; et al. Genotype-phenotype correlations in Noonan syndrome. J. Pediatr. 2004, 144, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Simpson, J.; Hong, J.H.; Kim, K.H.; Thavarajah, N.K.; Backx, P.H.; Neel, B.G.; Araki, T. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J. Clin. Investig. 2011, 121, 1009–1025. [Google Scholar] [CrossRef] [Green Version]
- Marin, T.M.; Keith, K.; Davies, B.; Conner, D.A.; Guha, P.; Kalaitzidis, D.; Wu, X.; Lauriol, J.; Wang, B.; Bauer, M.; et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J. Clin. Investig. 2011, 121, 1026–1043. [Google Scholar] [CrossRef] [Green Version]
- Dalin, M.G.; Zou, Z.; Scharin-Tang, M.; Safari, R.; Karlsson, C.; Bergo, M.O. Myocardial KRAS(G12D) expression does not cause cardiomyopathy in mice. Cardiovasc. Res. 2014, 101, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Shimada, Y.J.; Raita, Y.; Liang, L.W.; Maurer, M.S.; Hasegawa, K.; Fifer, M.A.; Reilly, M.P. Comprehensive Proteomics Profiling Reveals Circulating Biomarkers of Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2021, 14, e007849. [Google Scholar] [CrossRef]
- Singh, S.R.; Zech, A.T.L.; Geertz, B.; Reischmann-Dusener, S.; Osinska, H.; Prondzynski, M.; Kramer, E.; Meng, Q.; Redwood, C.; van der Velden, J.; et al. Activation of Autophagy Ameliorates Cardiomyopathy in Mybpc3-Targeted Knockin Mice. Circ. Heart Fail. 2017, 10, e004140. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, U.; Fohr, J.; Wilhelm, W.; Dammrich, J. Short-term inhibition of cardiac cellular autophagy by isoproterenol. J. Mol. Cell Cardiol. 1987, 19, 1179–1184. [Google Scholar] [CrossRef]
- Dammrich, J.; Pfeifer, U. Cardiac hypertrophy in rats after supravalvular aortic constriction. II. Inhibition of cellular autophagy in hypertrophying cardiomyocytes. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1983, 43, 287–307. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Dennemarker, J.; Lohmuller, T.; Muller, S.; Aguilar, S.V.; Tobin, D.J.; Peters, C.; Reinheckel, T. Impaired turnover of autophagolysosomes in cathepsin L deficiency. Biol. Chem. 2010, 391, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Ouzounian, M.; de Couto, G.; Chen, M.; Yan, R.; Fukuoka, M.; Li, G.; Moon, M.; Liu, Y.; Gramolini, A.; et al. Cathepsin-L ameliorates cardiac hypertrophy through activation of the autophagy-lysosomal dependent protein processing pathways. J. Am. Heart Assoc. 2013, 2, e000191. [Google Scholar] [CrossRef] [Green Version]
- Roe, N.D.; Xu, X.; Kandadi, M.R.; Hu, N.; Pang, J.; Weiser-Evans, M.C.; Ren, J. Targeted deletion of PTEN in cardiomyocytes renders cardiac contractile dysfunction through interruption of Pink1-AMPK signaling and autophagy. Biochim. Biophys. Acta 2015, 1852, 290–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onay-Besikci, A. Regulation of cardiac energy metabolism in newborn. Mol. Cell Biochem. 2006, 287, 1. [Google Scholar] [CrossRef] [PubMed]
- Nascimben, L.; Ingwall, J.S.; Lorell, B.H.; Pinz, I.; Schultz, V.; Tornheim, K.; Tian, R. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension 2004, 44, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, H.S.; Brownsey, R.W.; Kulpa, J.E.; Allard, M.F. Glycolysis and pyruvate oxidation in cardiac hypertrophy--why so unbalanced? Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2003, 135, 499–513. [Google Scholar] [CrossRef]
- Kundu, B.K.; Zhong, M.; Sen, S.; Davogustto, G.; Keller, S.R.; Taegtmeyer, H. Remodeling of glucose metabolism precedes pressure overload-induced left ventricular hypertrophy: Review of a hypothesis. Cardiology 2015, 130, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donthi, R.V.; Ye, G.; Wu, C.; McClain, D.A.; Lange, A.J.; Epstein, P.N. Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J. Biol. Chem. 2004, 279, 48085–48090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xu, J.; Wang, Q.; Brainard, R.E.; Watson, L.J.; Jones, S.P.; Epstein, P.N. Reduced cardiac fructose 2,6 bisphosphate increases hypertrophy and decreases glycolysis following aortic constriction. PLoS ONE 2013, 8, e53951. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Caggiano, M.; Kamynina, A.; Francois, A.A.; Prysyazhna, O.; Eykyn, T.R.; Krasemann, S.; Crespo-Leiro, M.G.; Vieites, M.G.; Bianchi, K.; Morales, V.; et al. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat. Metab. 2020, 2, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Tadamura, E.; Tamaki, N.; Matsumori, A.; Magata, Y.; Yonekura, Y.; Nohara, R.; Sasayama, S.; Yoshibayashi, M.; Kamiya, T.; Konishi, J. Myocardial metabolic changes in hypertrophic cardiomyopathy. J. Nucl. Med. 1996, 37, 572–577. [Google Scholar]
- Ishida, Y.; Nagata, S.; Uehara, T.; Yasumura, Y.; Fukuchi, K.; Miyatake, K. Clinical analysis of myocardial perfusion and metabolism in patients with hypertrophic cardiomyopathy by single photon emission tomography and positron emission tomography. J. Cardiol. 2001, 37 (Suppl. 1), 121–128. [Google Scholar] [PubMed]
- Granzier, H.L.; Labeit, S. The giant protein titin: A major player in myocardial mechanics, signaling, and disease. Circ. Res. 2004, 94, 284–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, M.; Takahashi, M.; Sakamoto, T.; Hiroe, M.; Marumo, F.; Kimura, A. Structural analysis of the titin gene in hypertrophic cardiomyopathy: Identification of a novel disease gene. Biochem. Biophys. Res. Commun. 1999, 262, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, H.; Wu, G.; Luo, X.; Zhang, C.; Zou, Y.; Wang, H.; Hui, R.; Wang, J.; Song, L. Titin-Truncating Variants Increase the Risk of Cardiovascular Death in Patients With Hypertrophic Cardiomyopathy. Can. J. Cardiol. 2017, 33, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Zekavati, A.; Syrris, P.; Hubank, M.; Giambartolomei, C.; Dalageorgou, C.; Jenkins, S.; McKenna, W.; Uk10k, C.; Plagnol, V.; et al. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J. Med. Genet. 2013, 50, 228–239. [Google Scholar] [CrossRef]
- Herwig, M.; Kolijn, D.; Lodi, M.; Holper, S.; Kovacs, A.; Papp, Z.; Jaquet, K.; Haldenwang, P.; Dos Remedios, C.; Reusch, P.H.; et al. Modulation of Titin-Based Stiffness in Hypertrophic Cardiomyopathy via Protein Kinase D. Front. Physiol. 2020, 11, 240. [Google Scholar] [CrossRef]
- Fielitz, J.; Kim, M.S.; Shelton, J.M.; Qi, X.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc. Natl. Acad. Sci. USA 2008, 105, 3059–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broers, J.L.; Peeters, E.A.; Kuijpers, H.J.; Endert, J.; Bouten, C.V.; Oomens, C.W.; Baaijens, F.P.; Ramaekers, F.C. Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: Implications for the development of laminopathies. Hum. Mol. Genet. 2004, 13, 2567–2580. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Adams, W.J.; Alford, P.W.; McCain, M.L.; Feinberg, A.W.; Sheehy, S.P.; Goss, J.A.; Parker, K.K. Cytoskeletal prestress regulates nuclear shape and stiffness in cardiac myocytes. Exp. Biol. Med. 2015, 240, 1543–1554. [Google Scholar] [CrossRef]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cupesi, M.; Yoshioka, J.; Gannon, J.; Kudinova, A.; Stewart, C.L.; Lammerding, J. Attenuated hypertrophic response to pressure overload in a lamin A/C haploinsufficiency mouse. J. Mol. Cell Cardiol. 2010, 48, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Araujo-Vilar, D.; Lado-Abeal, J.; Palos-Paz, F.; Lattanzi, G.; Bandin, M.A.; Bellido, D.; Dominguez-Gerpe, L.; Calvo, C.; Perez, O.; Ramazanova, A.; et al. A novel phenotypic expression associated with a new mutation in LMNA gene, characterized by partial lipodystrophy, insulin resistance, aortic stenosis and hypertrophic cardiomyopathy. Clin. Endocrinol. 2008, 69, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Brown, S.C.; Nihoyannopoulos, P.; Poulton, J.; Kinali, M.; Richard, P.; Piercy, R.J.; Messina, S.; Sewry, C.; Burke, M.M.; et al. Extreme variability of skeletal and cardiac muscle involvement in patients with mutations in exon 11 of the lamin A/C gene. Muscle Nerve 2005, 31, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Fuseler, J.W.; Price, R.L.; Borg, T.K.; Baudino, T.A. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1883–H1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, N.R.; Chaffin, M.; Fleming, S.J.; Hall, A.W.; Parsons, V.A.; Bedi, K.C., Jr.; Akkad, A.D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation 2020, 142, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Yu, P.; Li, D.; Li, Z.; Liao, Y.; Wang, Y.; Zhou, B.; Wang, L. Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy. Circulation 2020, 141, 1704–1719. [Google Scholar] [CrossRef]
- Li, R.K.; Li, G.; Mickle, D.A.; Weisel, R.D.; Merante, F.; Luss, H.; Rao, V.; Christakis, G.T.; Williams, W.G. Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation 1997, 96, 874–881. [Google Scholar] [CrossRef]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Kai, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 2002, 106, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Wang, R.S.; Shevtsov, S.; Drakos, S.G.; Arons, E.; Wever-Pinzon, O.; Huggins, G.S.; Samokhin, A.O.; Oldham, W.M.; Aguib, Y.; et al. Individualized interactomes for network-based precision medicine in hypertrophic cardiomyopathy with implications for other clinical pathophenotypes. Nat. Commun. 2021, 12, 873. [Google Scholar] [CrossRef] [PubMed]
- Cartledge, J.E.; Kane, C.; Dias, P.; Tesfom, M.; Clarke, L.; McKee, B.; Al Ayoubi, S.; Chester, A.; Yacoub, M.H.; Camelliti, P.; et al. Functional crosstalk between cardiac fibroblasts and adult cardiomyocytes by soluble mediators. Cardiovasc. Res. 2015, 105, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Ceccato, T.L.; Starbuck, R.B.; Hall, J.K.; Walker, C.J.; Brown, T.E.; Killgore, J.P.; Anseth, K.S.; Leinwand, L.A. Defining the Cardiac Fibroblast Secretome in a Fibrotic Microenvironment. J. Am. Heart Assoc. 2020, 9, e017025. [Google Scholar] [CrossRef] [PubMed]
- Sewanan, L.R.; Schwan, J.; Kluger, J.; Park, J.; Jacoby, D.L.; Qyang, Y.; Campbell, S.G. Extracellular Matrix From Hypertrophic Myocardium Provokes Impaired Twitch Dynamics in Healthy Cardiomyocytes. JACC Basic Transl. Sci. 2019, 4, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Konstandin, M.H.; Volkers, M.; Collins, B.; Quijada, P.; Quintana, M.; De La Torre, A.; Ormachea, L.; Din, S.; Gude, N.; Toko, H.; et al. Fibronectin contributes to pathological cardiac hypertrophy but not physiological growth. Basic Res. Cardiol. 2013, 108, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valiente-Alandi, I.; Potter, S.J.; Salvador, A.M.; Schafer, A.E.; Schips, T.; Carrillo-Salinas, F.; Gibson, A.M.; Nieman, M.L.; Perkins, C.; Sargent, M.A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, A.; Lenco, J.; Tambor, V.; Rehulkova, H.; Pudil, R.; Stulik, J. Plasma concentration of fibronectin is decreased in patients with hypertrophic cardiomyopathy. Clin. Chim. Acta 2016, 463, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Moretti, F.A.; Chauhan, A.K.; Iaconcig, A.; Porro, F.; Baralle, F.E.; Muro, A.F. A major fraction of fibronectin present in the extracellular matrix of tissues is plasma-derived. J. Biol. Chem. 2007, 282, 28057–28062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitaoka, H.; Kubo, T.; Okawa, M.; Hayato, K.; Yamasaki, N.; Matsumura, Y.; Doi, Y.L. Impact of metalloproteinases on left ventricular remodeling and heart failure events in patients with hypertrophic cardiomyopathy. Circ. J. 2010, 74, 1191–1196. [Google Scholar] [CrossRef] [Green Version]
- Roldan, V.; Marin, F.; Gimeno, J.R.; Ruiz-Espejo, F.; Gonzalez, J.; Feliu, E.; Garcia-Honrubia, A.; Saura, D.; de la Morena, G.; Valdes, M.; et al. Matrix metalloproteinases and tissue remodeling in hypertrophic cardiomyopathy. Am. Heart J. 2008, 156, 85–91. [Google Scholar] [CrossRef]
- Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Larson, A.; Chin, M.T. A method for cryopreservation and single nucleus RNA-sequencing of normal adult human interventricular septum heart tissue reveals cellular diversity and function. BMC Med Genomics. 2021, 14, 161. [Google Scholar] [CrossRef] [PubMed]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chou, C.; Chin, M.T. Pathogenic Mechanisms of Hypertrophic Cardiomyopathy beyond Sarcomere Dysfunction. Int. J. Mol. Sci. 2021, 22, 8933. https://doi.org/10.3390/ijms22168933
Chou C, Chin MT. Pathogenic Mechanisms of Hypertrophic Cardiomyopathy beyond Sarcomere Dysfunction. International Journal of Molecular Sciences. 2021; 22(16):8933. https://doi.org/10.3390/ijms22168933
Chicago/Turabian StyleChou, Chun, and Michael T. Chin. 2021. "Pathogenic Mechanisms of Hypertrophic Cardiomyopathy beyond Sarcomere Dysfunction" International Journal of Molecular Sciences 22, no. 16: 8933. https://doi.org/10.3390/ijms22168933
APA StyleChou, C., & Chin, M. T. (2021). Pathogenic Mechanisms of Hypertrophic Cardiomyopathy beyond Sarcomere Dysfunction. International Journal of Molecular Sciences, 22(16), 8933. https://doi.org/10.3390/ijms22168933