
Strategies to Improve the Antitumor Effect of γδ T Cell Immunotherapy for Clinical Application


Abstract
:1. Introduction
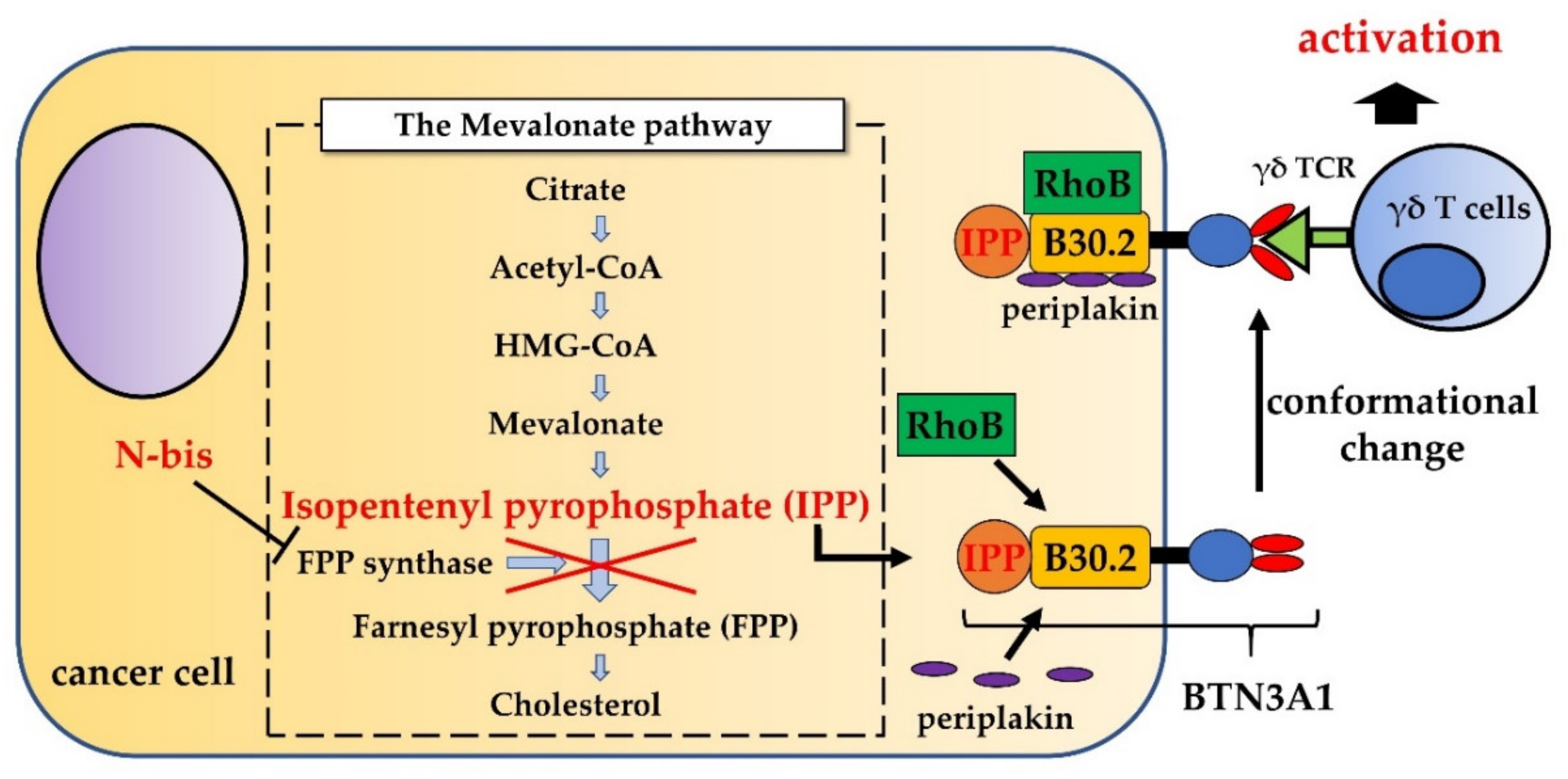
2. Phosphoantigens and Nitrogen-Containing Bisphosphonates Stimulate γδ T Cells
3. Administration of γδ T Cells into a Local Cavity Improves the E/T Ratio to Achieve a Maximum Cytotoxic Effect
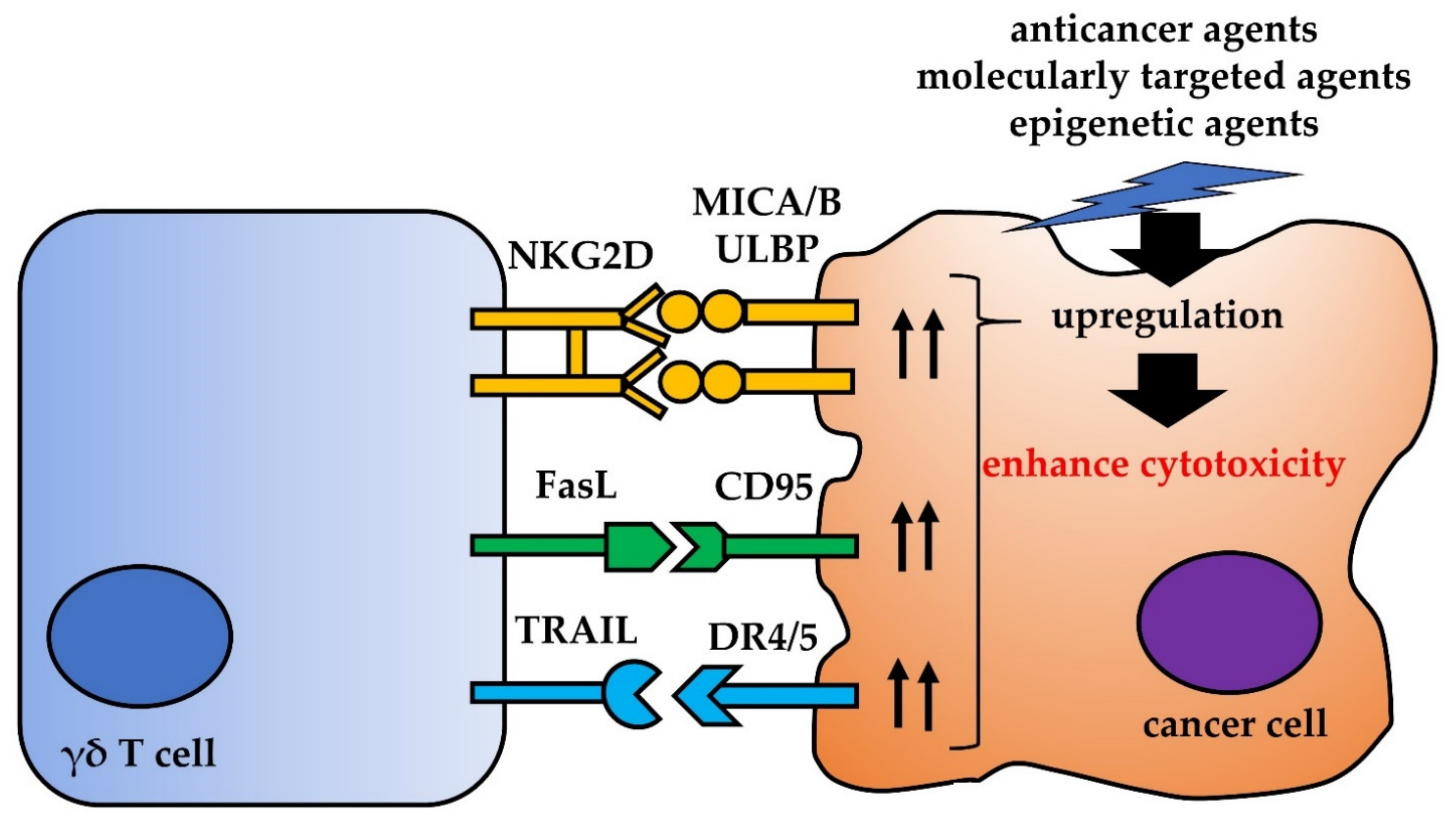
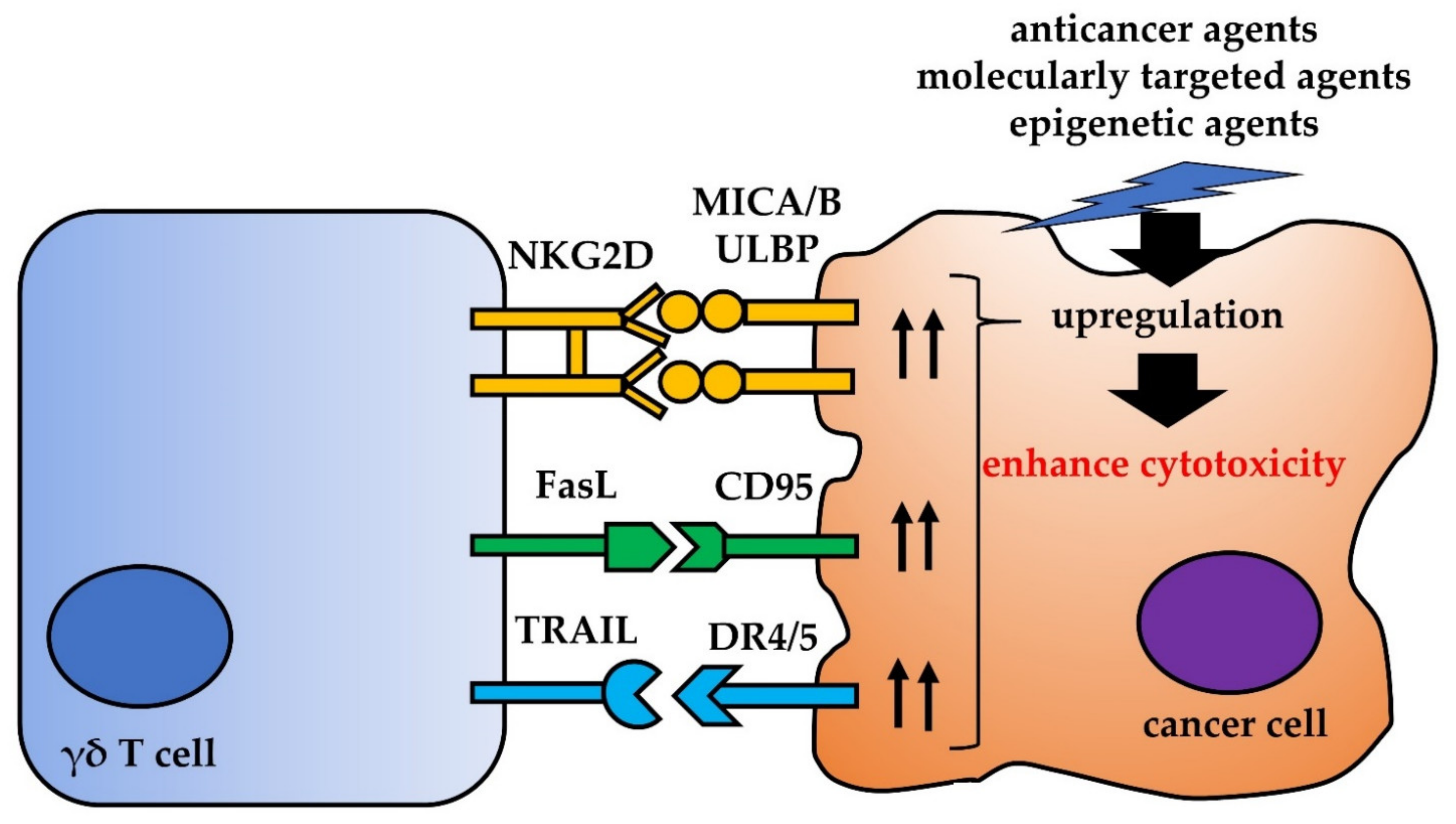
4. Other Interactions between γδ T Cells and Cancer Cells
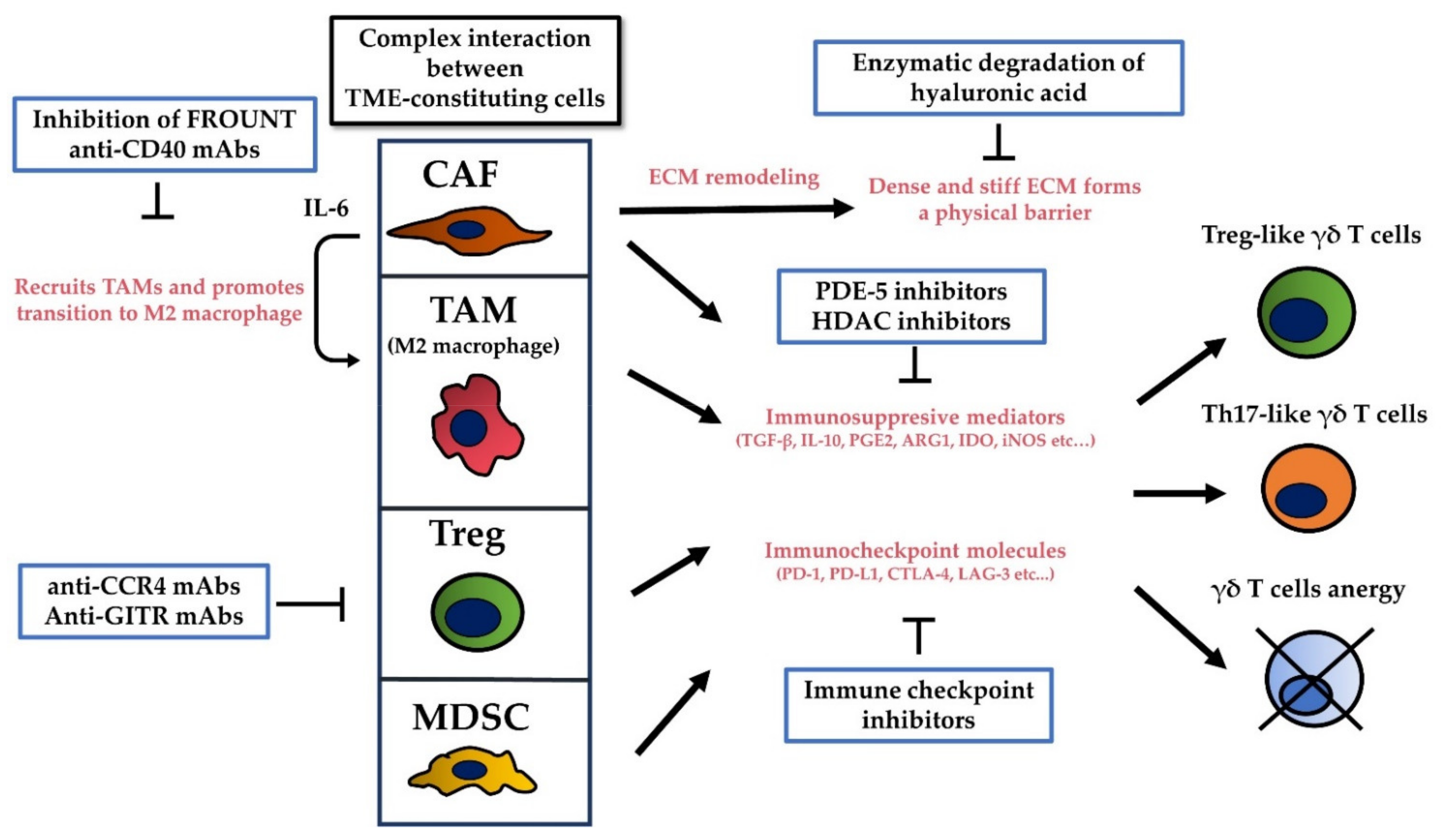
5. The Tumor Microenvironment (TME) Limits the Cytotoxicity of γδ T Cells by Promoting Their Regulatory Functions, by Secreting Immunosuppressive Cytokines, and by Inhibiting Immune Checkpoint Molecules
6. Cancer Stem Cells (CSCs) Could Mediate Resistance to γδ T Cell Immunotherapy
7. Novel Forms of γδ T Cell Therapy Overcome Current Therapeutic Limitations
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Aptsiauri, N.; Cabrera, T.; Garcia-Lora, A.; Lopez-Nevot, M.A.; Ruiz-Cabello, F.; Garrido, F. MHC class I antigens and immune surveillance in transformed cells. Int. Rev. Cytol. 2007, 256, 139–189. [Google Scholar] [CrossRef]
- Saito, H.; Kranz, D.M.; Takagaki, Y.; Hayday, A.C.; Eisen, H.N.; Tonegawa, S. Complete primary structure of a heterodimeric T-cell receptor deduced from cDNA sequences. Nature 1984, 309, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.B.; McLean, J.; Dialynas, D.P.; Strominger, J.L.; Smith, J.A.; Owen, F.L.; Seidman, J.G.; Ip, S.; Rosen, F.; Krangel, M.S. Identification of a putative second T-cell receptor. Nature 1986, 322, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Iwashima, M.; Kaplan, K.B.; Elliott, J.F.; Davis, M.M. A new T-cell receptor gene located within the alpha locus and expressed early in T-cell differentiation. Nature 1987, 327, 677–682. [Google Scholar] [CrossRef]
- Morita, C.T.; Beckman, E.M.; Bukowski, J.F.; Tanaka, Y.; Band, H.; Bloom, B.R.; Golan, D.E.; Brenner, M.B. Direct presentation of nonpeptide prenyl pyrophosphate antigens to human γδ T cells. Immunity 1995, 3, 495–507. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Guo, B.L.; Gehrs, B.C.; Nan, L.; Lopez, R.D. Ex vivo expanded human Vλ9Vδ 2+ λδ-T cells mediate innate antitumor activity against human prostate cancer cells in vitro. J. Urol. 2005, 173, 1552–1556. [Google Scholar] [CrossRef]
- Corvaisier, M.; Moreau-Aubry, A.; Diez, E.; Bennouna, J.; Mosnier, J.F.; Scotet, E.; Bonneville, M.; Jotereau, F. Vγ9Vδ2 T cell response to colon carcinoma cells. J. Immunol. 2005, 175, 5481–5488. [Google Scholar] [CrossRef] [Green Version]
- Viey, E.; Fromont, G.; Escudier, B.; Morel, Y.; Da Rocha, S.; Chouaib, S.; Caignard, A. Phosphostim-activated γδ T cells kill autologous metastatic renal cell carcinoma. J. Immunol. 2005, 174, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Uchida, R.; Ashihara, E.; Sato, K.; Kimura, S.; Kuroda, J.; Takeuchi, M.; Kawata, E.; Taniguchi, K.; Okamoto, M.; Shimura, K.; et al. γδ T cells kill myeloma cells by sensing mevalonate metabolites and ICAM-1 molecules on cell surface. Biochem. Biophys. Res. Commun. 2007, 354, 613–618. [Google Scholar] [CrossRef]
- Toutirais, O.; Cabillic, F.; Le Friec, G.; Salot, S.; Loyer, P.; Le Gallo, M.; Desille, M.; de La Pintière, C.T.; Daniel, P.; Bouet, F.; et al. DNAX accessory molecule-1 (CD226) promotes human hepatocellular carcinoma cell lysis by Vγ9Vδ2 T cells. Eur. J. Immunol. 2009, 39, 1361–1368. [Google Scholar] [CrossRef]
- Ashihara, E.; Munaka, T.; Kimura, S.; Nakagawa, S.; Nakagawa, Y.; Kanai, M.; Hirai, H.; Abe, H.; Miida, T.; Yamato, S.; et al. Isopentenyl pyrophosphate secreted from Zoledronate-stimulated myeloma cells, activates the chemotaxis of γδT cells. Biochem. Biophys. Res. Commun. 2015, 463, 650–655. [Google Scholar] [CrossRef]
- Hintz, M.; Reichenberg, A.; Altincicek, B.; Bahr, U.; Gschwind, R.M.; Kollas, A.K.; Beck, E.; Wiesner, J.; Eberl, M.; Jomaa, H. Identification of (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate as a major activator for human γδ T cells in Escherichia coli. FEBS Lett. 2001, 509, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Gober, H.J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; De Libero, G. Human T cell receptor γδ cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, K.; Schoel, B.; Gulle, H.; Kaufmann, S.H.; Wagner, H. Primary responses of human T cells to mycobacteria: A frequent set of γ/δ T cells are stimulated by protease-resistant ligands. Eur. J. Immunol. 1990, 20, 1175–1179. [Google Scholar] [CrossRef]
- Constant, P.; Davodeau, F.; Peyrat, M.A.; Poquet, Y.; Puzo, G.; Bonneville, M.; Fournié, J.J. Stimulation of human gamma delta T cells by nonpeptidic mycobacterial ligands. Science 1994, 264, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Harly, C.; Peigné, C.M.; Scotet, E. Molecules and Mechanisms Implicated in the Peculiar Antigenic Activation Process of Human Vγ9Vδ2 T Cells. Front. Immunol. 2015, 5, 657. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.A.; Chen, H.C.; Price, A.J.; Keeble, A.H.; Davey, M.S.; James, L.C.; Eberl, M.; Trowsdale, J. Activation of human γδ T cells by cytosolic interactions of BTN3A1 with soluble phosphoantigens and the cytoskeletal adaptor periplakin. J. Immunol. 2015, 194, 2390–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutin, L.; Scotet, E. Towards Deciphering the Hidden Mechanisms That Contribute to the Antigenic Activation Process of Human Vγ9Vδ2 T Cells. Front. Immunol. 2018, 9, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunakaran, M.M.; Willcox, C.R.; Salim, M.; Paletta, D.; Fichtner, A.S.; Noll, A.; Starick, L.; Nöhren, A.; Begley, C.R.; Berwick, K.A.; et al. Butyrophilin-2A1 Directly Binds Germline-Encoded Regions of the Vγ9Vδ2 TCR and Is Essential for Phosphoantigen Sensing. Immunity 2020, 52, 487–498.e6. [Google Scholar] [CrossRef]
- Cano, C.E.; Pasero, C.; De Gassart, A.; Kerneur, C.; Gabriac, M.; Fullana, M.; Granarolo, E.; Hoet, R.; Scotet, E.; Rafia, C.; et al. BTN2A1, an immune checkpoint targeting Vγ9Vδ2 T cell cytotoxicity against malignant cells. Cell Rep. 2021, 36, 109359. [Google Scholar] [CrossRef]
- Clendening, J.W.; Pandyra, A.; Boutros, P.C.; El Ghamrasni, S.; Khosravi, F.; Trentin, G.A.; Martirosyan, A.; Hakem, A.; Hakem, R.; Jurisica, I.; et al. Dysregulation of the mevalonate pathway promotes transformation. Proc. Natl. Acad. Sci. USA 2010, 107, 15051–15056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruenbacher, G.; Thurnher, M. Mevalonate Metabolism in Cancer Stemness and Trained Immunity. Front. Oncol. 2018, 8, 394. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, K.L.; Guo, K.; Dunford, J.E.; Wu, X.; Knapp, S.; Ebetino, F.H.; Rogers, M.J.; Russell, R.G.; Oppermann, U. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 7829–7834. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Tanaka, Y. γδ T Cell Immunotherapy-A Review. Pharmaceuticals 2015, 8, 40–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieli, F.; Vermijlen, D.; Fulfaro, F.; Caccamo, N.; Meraviglia, S.; Cicero, G.; Roberts, A.; Buccheri, S.; D’Asaro, M.; Gebbia, N.; et al. Targeting human γδ T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res. 2007, 67, 7450–7457. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, M.; Kunzmann, V.; Eckstein, S.; Reimer, P.; Weissinger, F.; Ruediger, T.; Tony, H.P. γδ T cells for immune therapy of patients with lymphoid malignancies. Blood 2003, 102, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Lang, J.M.; Kaikobad, M.R.; Wallace, M.; Staab, M.J.; Horvath, D.L.; Wilding, G.; Liu, G.; Eickhoff, J.C.; McNeel, D.G.; Malkovsky, M. Pilot trial of interleukin-2 and zoledronic acid to augment γδ T cells as treatment for patients with refractory renal cell carcinoma. Cancer Immunol. Immunother. 2011, 60, 1447–1460. [Google Scholar] [CrossRef] [Green Version]
- Meraviglia, S.; Eberl, M.; Vermijlen, D.; Todaro, M.; Buccheri, S.; Cicero, G.; La Mendola, C.; Guggino, G.; D’Asaro, M.; Orlando, V.; et al. In vivo manipulation of Vγ9Vδ2 T cells with zoledronate and low-dose interleukin-2 for immunotherapy of advanced breast cancer patients. Clin. Exp. Immunol. 2010, 161, 290–297. [Google Scholar] [CrossRef]
- Bennouna, J.; Levy, V.; Sicard, H.; Senellart, H.; Audrain, M.; Hiret, S.; Rolland, F.; Bruzzoni-Giovanelli, H.; Rimbert, M.; Galéa, C.; et al. Phase I study of bromohydrin pyrophosphate (BrHPP, IPH 1101), a Vγ9Vδ2 T lymphocyte agonist in patients with solid tumors. Cancer Immunol. Immunother. 2010, 59, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Bennouna, J.; Bompas, E.; Neidhardt, E.M.; Rolland, F.; Philip, I.; Galéa, C.; Salot, S.; Saiagh, S.; Audrain, M.; Rimbert, M.; et al. Phase-I study of Innacell γδ, an autologous cell-therapy product highly enriched in γ9δ2 T lymphocytes, in combination with IL-2, in patients with metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2008, 57, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Kunzmann, V.; Smetak, M.; Kimmel, B.; Weigang-Koehler, K.; Goebeler, M.; Birkmann, J.; Becker, J.; Schmidt-Wolf, I.G.; Einsele, H.; Wilhelm, M. Tumor-promoting versus tumor-antagonizing roles of γδ T cells in cancer immunotherapy: Results from a prospective phase I/II trial. J. Immunother. 2012, 35, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Tanaka, Y.; Yagi, J.; Osaka, Y.; Nakazawa, H.; Uchiyama, T.; Minato, N.; Toma, H. Safety profile and anti-tumor effects of adoptive immunotherapy using gamma-delta T cells against advanced renal cell carcinoma: A pilot study. Cancer Immunol. Immunother. 2007, 56, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Tanaka, Y.; Yagi, J.; Minato, N.; Tanabe, K. Phase I/II study of adoptive transfer of γδ T cells in combination with zoledronic acid and IL-2 to patients with advanced renal cell carcinoma. Cancer Immunol. Immunother. 2011, 60, 1075–1084. [Google Scholar] [CrossRef]
- Nicol, A.J.; Tokuyama, H.; Mattarollo, S.R.; Hagi, T.; Suzuki, K.; Yokokawa, K.; Nieda, M. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br. J. Cancer 2011, 105, 778–786. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Muto, M.; Nieda, M.; Nakagawa, Y.; Nicol, A.; Kaneko, T.; Goto, S.; Yokokawa, K.; Suzuki, K. Clinical and immunological evaluation of zoledronate-activated Vγ9γδ T-cell-based immunotherapy for patients with multiple myeloma. Exp. Hematol. 2009, 37, 956–968. [Google Scholar] [CrossRef]
- Nakajima, J.; Murakawa, T.; Fukami, T.; Goto, S.; Kaneko, T.; Yoshida, Y.; Takamoto, S.; Kakimi, K. A phase I study of adoptive immunotherapy for recurrent non-small-cell lung cancer patients with autologous γδ T cells. Eur. J. Cardiothorac. Surg. 2010, 37, 1191–1197. [Google Scholar] [CrossRef] [Green Version]
- Wada, I.; Matsushita, H.; Noji, S.; Mori, K.; Yamashita, H.; Nomura, S.; Shimizu, N.; Seto, Y.; Kakimi, K. Intraperitoneal injection of in vitro expanded Vγ9Vδ2 T cells together with zoledronate for the treatment of malignant ascites due to gastric cancer. Cancer Med. 2014, 3, 362–375. [Google Scholar] [CrossRef]
- Sakamoto, M.; Nakajima, J.; Murakawa, T.; Fukami, T.; Yoshida, Y.; Murayama, T.; Takamoto, S.; Matsushita, H.; Kakimi, K. Adoptive immunotherapy for advanced non-small cell lung cancer using zoledronate-expanded γδTcells: A phase I clinical study. J. Immunother. 2011, 34, 202–211. [Google Scholar] [CrossRef]
- Noguchi, A.; Kaneko, T.; Kamigaki, T.; Fujimoto, K.; Ozawa, M.; Saito, M.; Ariyoshi, N.; Goto, S. Zoledronate-activated Vγ9γδ T cell-based immunotherapy is feasible and restores the impairment of γδ T cells in patients with solid tumors. Cytotherapy 2011, 13, 92–97. [Google Scholar] [CrossRef]
- Izumi, T.; Kondo, M.; Takahashi, T.; Fujieda, N.; Kondo, A.; Tamura, N.; Murakawa, T.; Nakajima, J.; Matsushita, H.; Kakimi, K. Ex vivo characterization of γδ T-cell repertoire in patients after adoptive transfer of Vγ9Vδ2 T cells expressing the interleukin-2 receptor β-chain and the common γ-chain. Cytotherapy 2013, 15, 481–491. [Google Scholar] [CrossRef]
- Yoshida, Y.; Nakajima, J.; Wada, H.; Kakimi, K. γδ T-cell immunotherapy for lung cancer. Surg. Today 2011, 41, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, A.J.; Jauhiainen, M.; Mönkkönen, H.; Rogers, M.J.; Mönkkönen, J.; Thompson, K. Peripheral blood monocytes are responsible for γδ T cell activation induced by zoledronic acid through accumulation of IPP/DMAPP. Br. J. Haematol. 2009, 144, 245–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieli, F.; Gebbia, N.; Poccia, F.; Caccamo, N.; Montesano, C.; Fulfaro, F.; Arcara, C.; Valerio, M.R.; Meraviglia, S.; Di Sano, C.; et al. Induction of γδ T-lymphocyte effector functions by bisphosphonate zoledronic acid in cancer patients in vivo. Blood 2003, 102, 2310–2311. [Google Scholar] [CrossRef]
- Bryant, N.L.; Gillespie, G.Y.; Lopez, R.D.; Markert, J.M.; Cloud, G.A.; Langford, C.P.; Arnouk, H.; Su, Y.; Haines, H.L.; Suarez-Cuervo, C.; et al. Preclinical evaluation of ex vivo expanded/activated γδ T cells for immunotherapy of glioblastoma multiforme. J. Neurooncol. 2011, 101, 179–188. [Google Scholar] [CrossRef]
- Yuasa, T.; Sato, K.; Ashihara, E.; Takeuchi, M.; Maita, S.; Tsuchiya, N.; Habuchi, T.; Maekawa, T.; Kimura, S. Intravesical administration of γδ T cells successfully prevents the growth of bladder cancer in the murine model. Cancer Immunol. Immunother. 2009, 58, 493–502. [Google Scholar] [CrossRef]
- Shimizu, T.; Tomogane, M.; Miyashita, M.; Ukimura, O.; Ashihara, E. Low dose gemcitabine increases the cytotoxicity of human Vγ9Vδ2 T cells in bladder cancer cells in vitro and in an orthotopic xenograft model. Oncoimmunology 2018, 7, e1424671. [Google Scholar] [CrossRef] [PubMed]
- Rincon-Orozco, B.; Kunzmann, V.; Wrobel, P.; Kabelitz, D.; Steinle, A.; Herrmann, T. Activation of Vγ9Vδ2 T cells by NKG2D. J. Immunol. 2005, 175, 2144–2151. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Das, H.; Groh, V.; Kuijl, C.; Sugita, M.; Morita, C.T.; Spies, T.; Bukowski, J.F. MICA engagement by human Vγ2Vδ2 T cells enhances their antigen-dependent effector function. Immunity 2001, 15, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Groh, V.; Steinle, A.; Bauer, S.; Spies, T. Recognition of stress-induced MHC molecules by intestinal epithelial γδ T cells. Science 1998, 279, 1737–1740. [Google Scholar] [CrossRef] [PubMed]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Chalupny, N.J.; Sutherland, C.L.; Lawrence, W.A.; Rein-Weston, A.; Cosman, D. ULBP4 is a novel ligand for human NKG2D. Biochem. Biophys. Commun. 2003, 305, 129–135. [Google Scholar] [CrossRef]
- Chen, G.; Emens, L.A. Chemoimmunotherapy: Reengineering tumor immunity. Cancer Immunol. Immunother. 2013, 62, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef]
- Gasser, S.; Raulet, D. The DNA damage response, immunity and cancer. Semin. Cancer Biol. 2006, 16, 344–347. [Google Scholar] [CrossRef]
- Todaro, M.; Orlando, V.; Cicero, G.; Caccamo, N.; Meraviglia, S.; Stassi, G.; Dieli, F. Chemotherapy sensitizes colon cancer initiating cells to Vγ9Vδ2 T cell-mediated cytotoxicity. PLoS ONE 2013, 8, e65145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, L.S., Jr.; Bowersock, J.; Dasgupta, A.; Gillespie, G.Y.; Su, Y.; Johnson, A.; Spencer, H.T. Engineered drug resistant γδ T cells kill glioblastoma cell lines during a chemotherapy challenge: A strategy for combining chemo- and immunotherapy. PLoS ONE 2013, 8, e51805. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, Y.; Li, Y.; Guo, K.; He, Y. Role of sorafenib and sunitinib in the induction of expressions of NKG2D ligands in nasopharyngeal carcinoma with high expression of ABCG2. J. Cancer Res. Clin. Oncol. 2011, 137, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.H.; Kim, M.J.; Kim, S.J.; Park, S.J.; Chung, J.S.; Bae, J.H.; Kang, C.D. EGFR inhibitors enhanced the susceptibility to NK cell-mediated lysis of lung cancer cells. J. Immunother. 2011, 34, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Story, J.Y.; Zoine, J.T.; Burnham, R.E.; Hamilton, J.; Spencer, H.T.; Doering, C.B.; Raikar, S.S. Bortezomib enhances cytotoxicity of ex vivo-expanded gamma delta T cells against acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Cytotherapy 2021, 23, 12–24. [Google Scholar] [CrossRef]
- Skov, S.; Pedersen, M.T.; Andresen, L.; Straten, P.T.; Woetmann, A.; Odum, N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005, 65, 11136–11145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braza, M.S.; Klein, B. Anti-tumour immunotherapy with Vγ9Vδ2 T lymphocytes: From the bench to the bedside. Br. J. Haematol. 2013, 160, 123–132. [Google Scholar] [CrossRef]
- Nagata, S. Early work on the function of CD95, an interview with Shige Nagata. Cell Death Differ. 2004, 11 (Suppl. S1), S23–S27. [Google Scholar] [CrossRef] [Green Version]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef]
- Wu, G.S.; Burns, T.F.; McDonald, E.R., 3rd; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.D.; Zhou, J.Y.; Muschel, R.; et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 1997, 17, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Ni, J.; Wei, Y.F.; Yu, G.; Gentz, R.; Dixit, V.M. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 1997, 277, 815–818. [Google Scholar] [CrossRef]
- Degli-Esposti, M.A.; Dougall, W.C.; Smolak, P.J.; Waugh, J.Y.; Smith, C.A.; Goodwin, R.G. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Emery, J.G.; McDonnell, P.; Burke, M.B.; Deen, K.C.; Lyn, S.; Silverman, C.; Dul, E.; Appelbaum, E.R.; Eichman, C.; DiPrinzio, R.; et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J. Biol. Chem. 1998, 273, 14363–14367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S. TRAIL: A sword for killing tumors. Curr. Med. Chem. 2010, 17, 3309–3317. [Google Scholar] [CrossRef]
- Shankar, S.; Chen, X.; Srivastava, R.K. Effects of sequential treatments with chemotherapeutic drugs followed by TRAIL on prostate cancer in vitro and in vivo. Prostate 2005, 62, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Mattarollo, S.R.; Kenna, T.; Nieda, M.; Nicol, A.J. Chemotherapy pretreatment sensitizes solid tumor-derived cell lines to Vα24+ NKT cell-mediated cytotoxicity. Int. J. Cancer 2006, 119, 1630–1637. [Google Scholar] [CrossRef]
- Wesch, D.; Glatzel, A.; Kabelitz, D. Differentiation of resting human peripheral blood gamma delta T cells toward Th1- or Th2-phenotype. Cell. Immunol. 2001, 212, 110–117. [Google Scholar] [CrossRef]
- Vermijlen, D.; Ellis, P.; Langford, C.; Klein, A.; Engel, R.; Willimann, K.; Jomaa, H.; Hayday, A.C.; Eberl, M. Distinct cytokine-driven responses of activated blood γδ T cells: Insights into unconventional T cell pleiotropy. J. Immunol. 2007, 178, 4304–4314. [Google Scholar] [CrossRef]
- Bansal, R.R.; Mackay, C.R.; Moser, B.; Eberl, M. IL-21 enhances the potential of human γδ T cells to provide B-cell help. Eur. J. Immunol. 2012, 42, 110–119. [Google Scholar] [CrossRef]
- Casetti, R.; Agrati, C.; Wallace, M.; Sacchi, A.; Martini, F.; Martino, A.; Rinaldi, A.; Malkovsky, M. Cutting edge: TGF-β1 and IL-15 Induce FOXP3+ γδ regulatory T cells in the presence of antigen stimulation. J. Immunol. 2009, 183, 3574–3577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Presti, E.; Toia, F.; Oieni, S.; Buccheri, S.; Turdo, A.; Mangiapane, L.R.; Campisi, G.; Caputo, V.; Todaro, M.; Stassi, G.; et al. Squamous Cell Tumors Recruit γδ T Cells Producing either IL17 or IFNγ Depending on the Tumor Stage. Cancer Immunol. Res. 2017, 5, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, N.; La Mendola, C.; Orlando, V.; Meraviglia, S.; Todaro, M.; Stassi, G.; Sireci, G.; Fournié, J.J.; Dieli, F. Differentiation, phenotype, and function of interleukin-17-producing human Vγ9Vδ2 T cells. Blood 2011, 118, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, R.S.; Shah, S.U.; Shrikhande, S.V.; Goel, M.; Dikshit, R.P.; Chiplunkar, S.V. IL17 producing γδT cells induce angiogenesis and are associated with poor survival in gallbladder cancer patients. Int. J. Cancer 2016, 139, 869–881. [Google Scholar] [CrossRef]
- Wakita, D.; Sumida, K.; Iwakura, Y.; Nishikawa, H.; Ohkuri, T.; Chamoto, K.; Kitamura, H.; Nishimura, T. Tumor-infiltrating IL-17-producing γδ T cells support the progression of tumor by promoting angiogenesis. Eur. J. Immunol. 2010, 40, 1927–1937. [Google Scholar] [CrossRef]
- Yi, Y.; He, H.W.; Wang, J.X.; Cai, X.Y.; Li, Y.W.; Zhou, J.; Cheng, Y.F.; Jin, J.J.; Fan, J.; Qiu, S.J. The functional impairment of HCC-infiltrating γδ T cells, partially mediated by regulatory T cells in a TGFβ- and IL-10-dependent manner. J. Hepatol. 2013, 58, 977–983. [Google Scholar] [CrossRef]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, J. Tumor stroma as targets for cancer therapy. Pharmacol. Ther. 2013, 137, 200–215. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Nazareth, M.R.; Broderick, L.; Simpson-Abelson, M.R.; Kelleher, R.J., Jr.; Yokota, S.J.; Bankert, R.B. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J. Immunol. 2007, 178, 5552–5562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, A.; Tumino, N.; Sabatini, A.; Cimini, E.; Casetti, R.; Bordoni, V.; Grassi, G.; Agrati, C. Myeloid-Derived Suppressor Cells Specifically Suppress IFN-γ Production and Antitumor Cytotoxic Activity of Vδ2 T Cells. Front. Immunol. 2018, 9, 1271. [Google Scholar] [CrossRef] [Green Version]
- Blattner, C.; Fleming, V.; Weber, R.; Himmelhan, B.; Altevogt, P.; Gebhardt, C.; Schulze, T.J.; Razon, H.; Hawila, E.; Wildbaum, G.; et al. CCR5+ Myeloid-Derived Suppressor Cells Are Enriched and Activated in Melanoma Lesions. Cancer Res. 2018, 78, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.C.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.S.; Linehan, D.C. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J. Immunol. 2009, 182, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Velázquez, M.; Jiao, X.; De La Fuente, M.; Pestell, T.G.; Ertel, A.; Lisanti, M.P.; Pestell, R.G. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012, 72, 3839–3850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujio, K.; Yamamoto, K.; Okamura, T. Overview of LAG-3-Expressing, IL-10-Producing Regulatory T Cells. Curr. Top. Microbiol. Immunol. 2017, 410, 29–45. [Google Scholar] [CrossRef]
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, N.S.; Dranoff, G.; Brogdon, J.L. Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liakou, C.I.; Kamat, A.; Tang, D.N.; Chen, H.; Sun, J.; Troncoso, P.; Logothetis, C.; Sharma, P. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 14987–14992. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; Butler, M.; Oble, D.A.; Seiden, M.V.; Haluska, F.G.; Kruse, A.; Macrae, S.; Nelson, M.; Canning, C.; Lowy, I.; et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 3005–3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Faget, J.; Biota, C.; Bachelot, T.; Gobert, M.; Treilleux, I.; Goutagny, N.; Durand, I.; Léon-Goddard, S.; Blay, J.Y.; Caux, C.; et al. Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res. 2011, 71, 6143–6152. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef] [Green Version]
- Ko, K.; Yamazaki, S.; Nakamura, K.; Nishioka, T.; Hirota, K.; Yamaguchi, T.; Shimizu, J.; Nomura, T.; Chiba, T.; Sakaguchi, S. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J. Exp. Med. 2005, 202, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Tanaka, T.; Yamamoto, Y.; Akasaki, Y.; Sasaki, H. Dual role of macrophage in tumor immunity. Immunotherapy 2018, 10, 899–909. [Google Scholar] [CrossRef]
- Terashima, Y.; Onai, N.; Murai, M.; Enomoto, M.; Poonpiriya, V.; Hamada, T.; Motomura, K.; Suwa, M.; Ezaki, T.; Haga, T.; et al. Pivotal function for cytoplasmic protein FROUNT in CCR2-mediated monocyte chemotaxis. Nat. Immunol. 2005, 6, 827–835. [Google Scholar] [CrossRef]
- Toda, E.; Terashima, Y.; Sato, T.; Hirose, K.; Kanegasaki, S.; Matsushima, K. FROUNT is a common regulator of CCR2 and CCR5 signaling to control directional migration. J. Immunol. 2009, 183, 6387–6394. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.M.; Back, T.C.; Scarzello, A.J.; Subleski, J.J.; Hall, V.L.; Stauffer, J.K.; Chen, X.; Micic, D.; Alderson, K.; Murphy, W.J.; et al. Successful immunotherapy with IL-2/anti-CD40 induces the chemokine-mediated mitigation of an immunosuppressive tumor microenvironment. Proc. Natl. Acad. Sci. USA 2009, 106, 19455–19460. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Flippot, R.; Escudier, B.; Albiges, L. Immune Checkpoint Inhibitors: Toward New Paradigms in Renal Cell Carcinoma. Drugs 2018, 78, 1443–1457. [Google Scholar] [CrossRef]
- Iwasaki, M.; Tanaka, Y.; Kobayashi, H.; Murata-Hirai, K.; Miyabe, H.; Sugie, T.; Toi, M.; Minato, N. Expression and function of PD-1 in human γδ T cells that recognize phosphoantigens. Eur. J. Immunol. 2011, 41, 345–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, C.; Gravelle, P.; Decaup, E.; Bordenave, J.; Poupot, M.; Tosolini, M.; Franchini, D.M.; Laurent, C.; Morin, R.; Lagarde, J.M.; et al. Boosting γδ T cell-mediated antibody-dependent cellular cytotoxicity by PD-1 blockade in follicular lymphoma. Oncoimmunology 2018, 8, 1554175. [Google Scholar] [CrossRef]
- Tomogane, M.; Sano, Y.; Shimizu, D.; Shimizu, T.; Miyashita, M.; Toda, Y.; Hosogi, S.; Tanaka, Y.; Kimura, S.; Ashihara, E. Human Vγ9Vδ2 T cells exert anti-tumor activity independently of PD-L1 expression in tumor cells. Biochem. Biophys. Res. Commun. 2021, 573, 132–139. [Google Scholar] [CrossRef]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [Green Version]
- Ojo, D.; Lin, X.; Wong, N.; Gu, Y.; Tang, D. Prostate Cancer Stem-like Cells Contribute to the Development of Castration-Resistant Prostate Cancer. Cancers 2015, 7, 2290–2308. [Google Scholar] [CrossRef] [PubMed]
- Duru, N.; Fan, M.; Candas, D.; Menaa, C.; Liu, H.C.; Nantajit, D.; Wen, Y.; Xiao, K.; Eldridge, A.; Chromy, B.A.; et al. HER2-associated radioresistance of breast cancer stem cells isolated from HER2-negative breast cancer cells. Clin. Cancer Res. 2012, 18, 6634–6647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, C.W.; Kim, L.G.; Kong, H.H.; Low, L.Y.; Iacopetta, B.; Soong, R.; Salto-Tellez, M. CD133 expression predicts for non-response to chemotherapy in colorectal cancer. Mod. Pathol. 2010, 23, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Jinushi, M. Role of cancer stem cell-associated inflammation in creating pro-inflammatory tumorigenic microenvironments. Oncoimmunology 2014, 3, e28862. [Google Scholar] [CrossRef] [Green Version]
- Schatton, T.; Schütte, U.; Frank, N.Y.; Zhan, Q.; Hoerning, A.; Robles, S.C.; Zhou, J.; Hodi, F.S.; Spagnoli, G.C.; Murphy, G.F.; et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 2010, 70, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res. 2010, 16, 800–813. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Shin, J.H.; Longmire, M.; Wang, H.; Kohrt, H.E.; Chang, H.Y.; Sunwoo, J.B. CD44+ Cells in Head and Neck Squamous Cell Carcinoma Suppress T-Cell-Mediated Immunity by Selective Constitutive and Inducible Expression of PD-L1. Clin. Cancer Res. 2016, 22, 3571–3581. [Google Scholar] [CrossRef] [Green Version]
- Miyashita, M.; Tomogane, M.; Nakamura, Y.; Shimizu, T.; Fujihara, A.; Ukimura, O.; Ashihara, E. Sphere-derived Prostate Cancer Stem Cells Are Resistant to γδ T Cell Cytotoxicity. Anticancer Res. 2020, 40, 5481–5487. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Kontermann, R.E.; Brinkmann, U. Tumor-antigen-binding bispecific antibodies for cancer treatment. Semin. Oncol. 2014, 41, 653–660. [Google Scholar] [CrossRef] [Green Version]
- Hoh, A.; Dewerth, A.; Vogt, F.; Wenz, J.; Baeuerle, P.A.; Warmann, S.W.; Fuchs, J.; Armeanu-Ebinger, S. The activity of γδ T cells against paediatric liver tumour cells and spheroids in cell culture. Liver Int. 2013, 33, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Oberg, H.H.; Peipp, M.; Kellner, C.; Sebens, S.; Krause, S.; Petrick, D.; Adam-Klages, S.; Röcken, C.; Becker, T.; Vogel, I.; et al. Novel bispecific antibodies increase γδ T-cell cytotoxicity against pancreatic cancer cells. Cancer Res. 2014, 74, 1349–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberg, H.H.; Kellner, C.; Gonnermann, D.; Peipp, M.; Peters, C.; Sebens, S.; Kabelitz, D.; Wesch, D. γδ T cell activation by bispecific antibodies. Cell. Immunol. 2015, 296, 41–49. [Google Scholar] [CrossRef]
- Oberg, H.H.; Kellner, C.; Gonnermann, D.; Sebens, S.; Bauerschlag, D.; Gramatzki, M.; Kabelitz, D.; Peipp, M.; Wesch, D. Tribody [(HER2)2xCD16] Is More Effective Than Trastuzumab in Enhancing γδ T Cell and Natural Killer Cell Cytotoxicity Against HER2-Expressing Cancer Cells. Front. Immunol. 2018, 9, 814. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Grupp, S.A.; Porter, D.L.; June, C.H. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014, 123, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Deniger, D.C.; Switzer, K.; Mi, T.; Maiti, S.; Hurton, L.; Singh, H.; Huls, H.; Olivares, S.; Lee, D.A.; Champlin, R.E.; et al. Bispecific T-cells expressing polyclonal repertoire of endogenous γδ T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol. Ther. 2013, 21, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.; Abramowski, P.; Wisidagamage Don, N.D.; Flutter, B.; Capsomidis, A.; Cheung, G.W.; Gustafsson, K.; Anderson, J. Avoidance of On-Target Off-Tumor Activation Using a Co-stimulation-Only Chimeric Antigen Receptor. Mol. Ther. 2017, 25, 1234–1247. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Author | Year | Tumor | Interventions | Phase | Ref. or Clinical Trials. Gov Identifier |
|---|---|---|---|---|---|
| Wilhelm et al. | 2003 | MM, NHL | Pam + IL-2 (in vivo) | Pilot study | [27] |
| Kobayashi et al. | 2006 | RCC | Ex-vivo γδ T cell + IL-2 | Pilot study | [33] |
| Kobayashi et al. | 2007 | RCC | Ex-vivo γδ T cell + ZOL + IL-2 | I/II | [34] |
| Dieli et al. | 2007 | Prostate cancer | ZOL/ZOL + IL-2 (in vivo) | I | [26] |
| Bennouna et al. | 2008 | RCC | BrHPP + IL-2 (in vivo) | I | [31] |
| Abe et al. | 2009 | MM | Ex-vivo γδ T cell + ZOL + IL-2 | Pilot study | [36] |
| Meraviglia et al. | 2010 | Breast cancer | ZOL + IL-2 (in vivo) | I | [29] |
| Bennouna et al. | 2010 | Solid cancer | BrHPP + IL-2 (in vivo) | I | [30] |
| Nakajima et al. | 2010 | NSCLC | Ex-vivo γδ T cell + ZOL + IL-2 | I | [37] |
| Lang et al. | 2011 | RCC | ZOL + IL-2 (in vivo) | Pilot study | [28] |
| Nicol et al. | 2011 | Solid cancer | Ex-vivo γδ T cell + ZOL | I | [35] |
| Sakamoto et al. | 2011 | NSCLC | Ex-vivo γδ T cell + ZOL + IL-2 | I | [39] |
| Noguchi et al. | 2011 | Solid cancer | Ex-vivo γδ T cell | Pilot study | [40] |
| Kanzmann et al. | 2012 | RCC, MM, AML | ZOL + IL-2 (in vivo) | I/II | [32] |
| Izumi et al. | 2013 | Colorectal cancer | Ex-vivo γδ T cell | Pilot study | [41] |
| Wada et al. | 2014 | Gastric cancer | Ex-vivo γδ T cell + ZOL (intraperitoneal injection) | Pilot study | [38] |
| Kakimi et al. | 2014 | NSCLC | Ex-vivo γδ T cell | I | [42] |
| Ghigo et al. | 2020 | Solid cancerHematopoietic/Lymphoid cancer | ICT01 (anti-BTN3A mAbs)/ICT01 plus pembrolizumab | I | NCT04243499 |
| Clinical Trials. Gov Identifier | Interventions | Cancers | Phase |
|---|---|---|---|
| NCT02656147 | Anti-CD19-CAR-γδ T cell | Leukemia and lymphoma | I |
| NCT04107142 | NKG2DL-targeting CAR-γδ T cell | Solid cancer | I |
| NCT04702841 | CAR-γδ T cell | Relapsed and refractory CD7 positive T cell-derived malignant tumor | I |
| NCT04796441 | CAR-γδ T cell | AML | Not Applicable |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyashita, M.; Shimizu, T.; Ashihara, E.; Ukimura, O. Strategies to Improve the Antitumor Effect of γδ T Cell Immunotherapy for Clinical Application. Int. J. Mol. Sci. 2021, 22, 8910. https://doi.org/10.3390/ijms22168910
Miyashita M, Shimizu T, Ashihara E, Ukimura O. Strategies to Improve the Antitumor Effect of γδ T Cell Immunotherapy for Clinical Application. International Journal of Molecular Sciences. 2021; 22(16):8910. https://doi.org/10.3390/ijms22168910
Chicago/Turabian StyleMiyashita, Masatsugu, Teruki Shimizu, Eishi Ashihara, and Osamu Ukimura. 2021. "Strategies to Improve the Antitumor Effect of γδ T Cell Immunotherapy for Clinical Application" International Journal of Molecular Sciences 22, no. 16: 8910. https://doi.org/10.3390/ijms22168910
APA StyleMiyashita, M., Shimizu, T., Ashihara, E., & Ukimura, O. (2021). Strategies to Improve the Antitumor Effect of γδ T Cell Immunotherapy for Clinical Application. International Journal of Molecular Sciences, 22(16), 8910. https://doi.org/10.3390/ijms22168910