
Computational Insight on the Interaction of Common Blood Proteins with Gold Nanoparticles


Abstract
:1. Introduction
2. Results
2.1. Interaction Sites of Proteins with AuNP
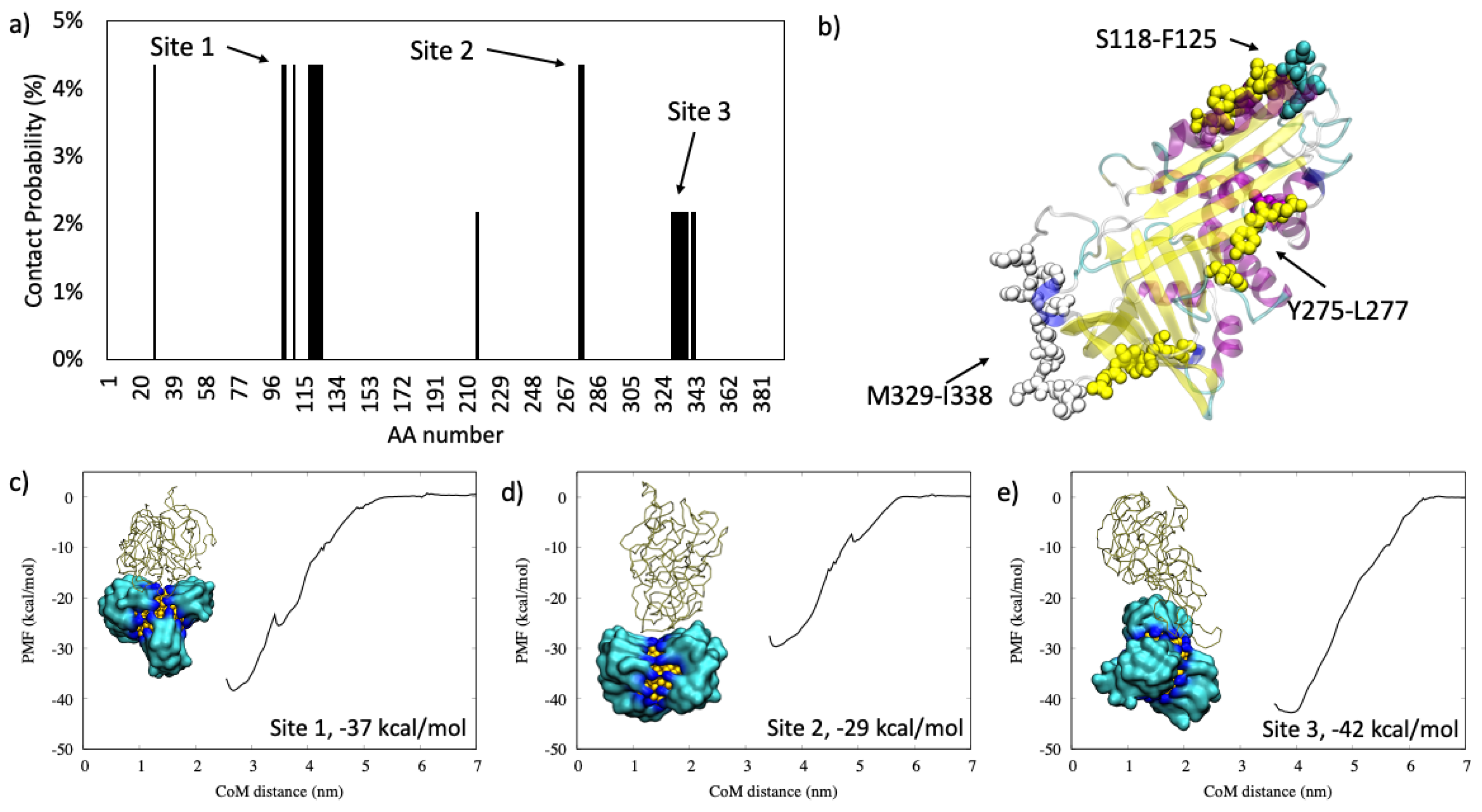
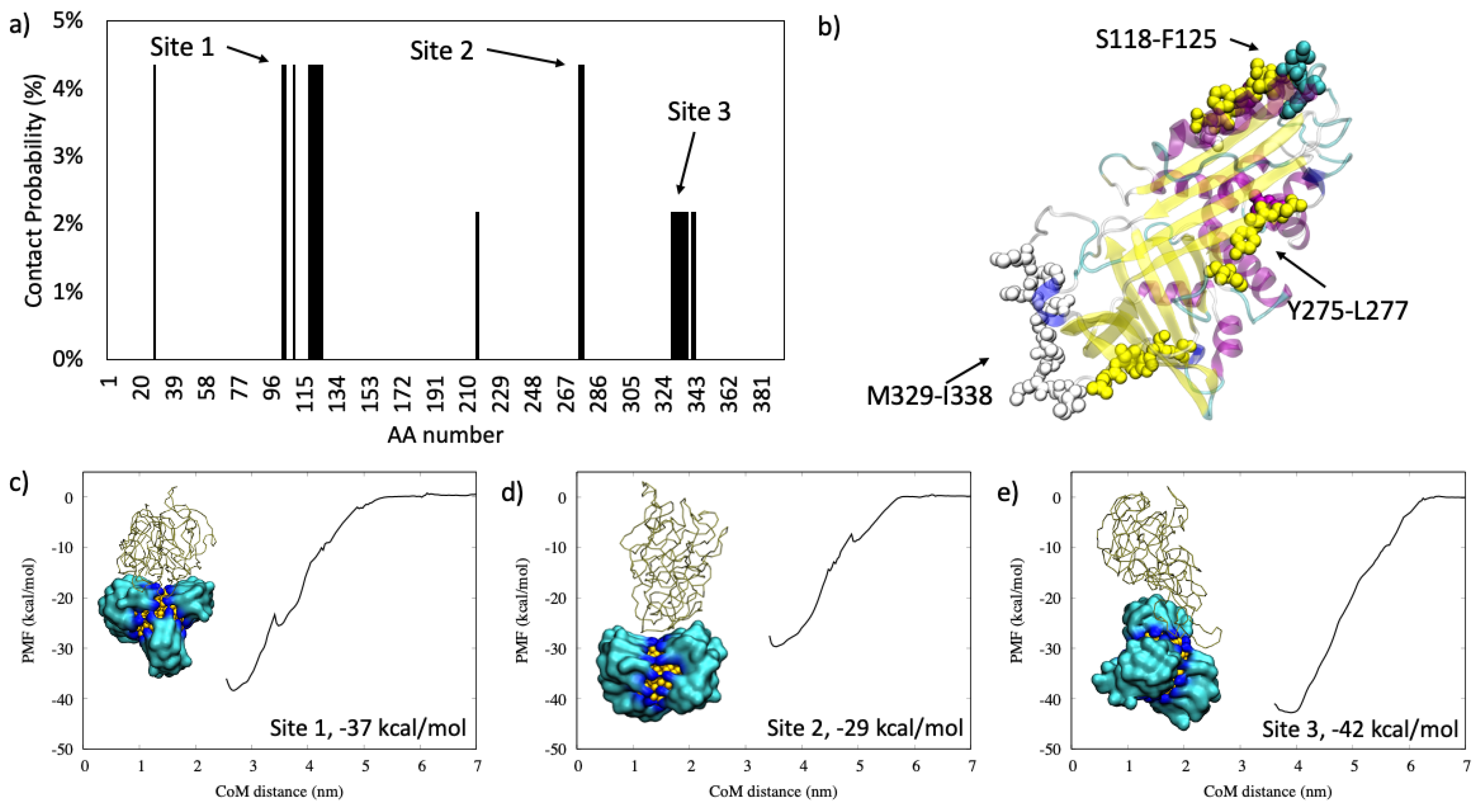
2.1.1. Antiproteinase
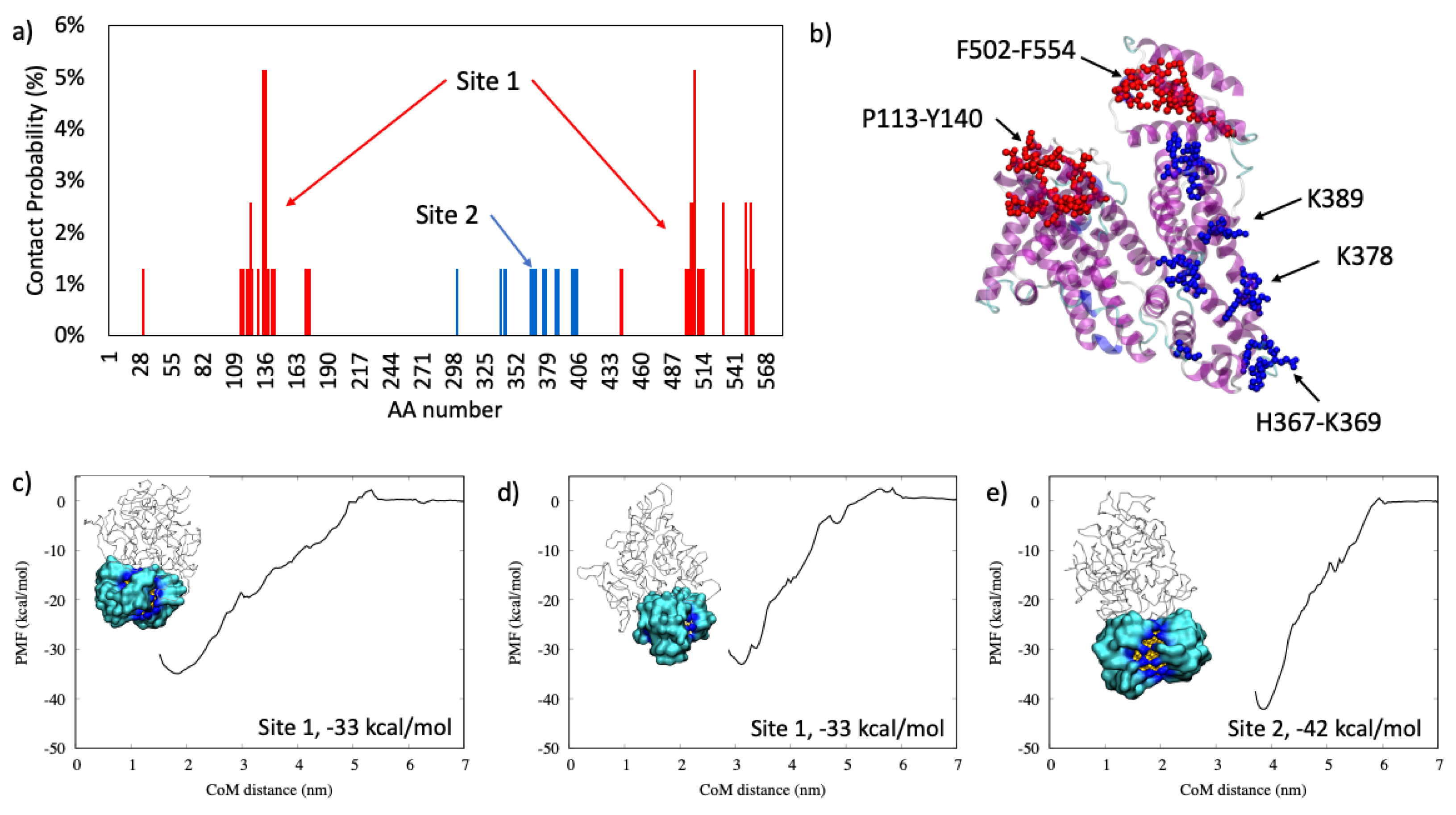
2.1.2. Serum Albumin
2.1.3. Complement C3
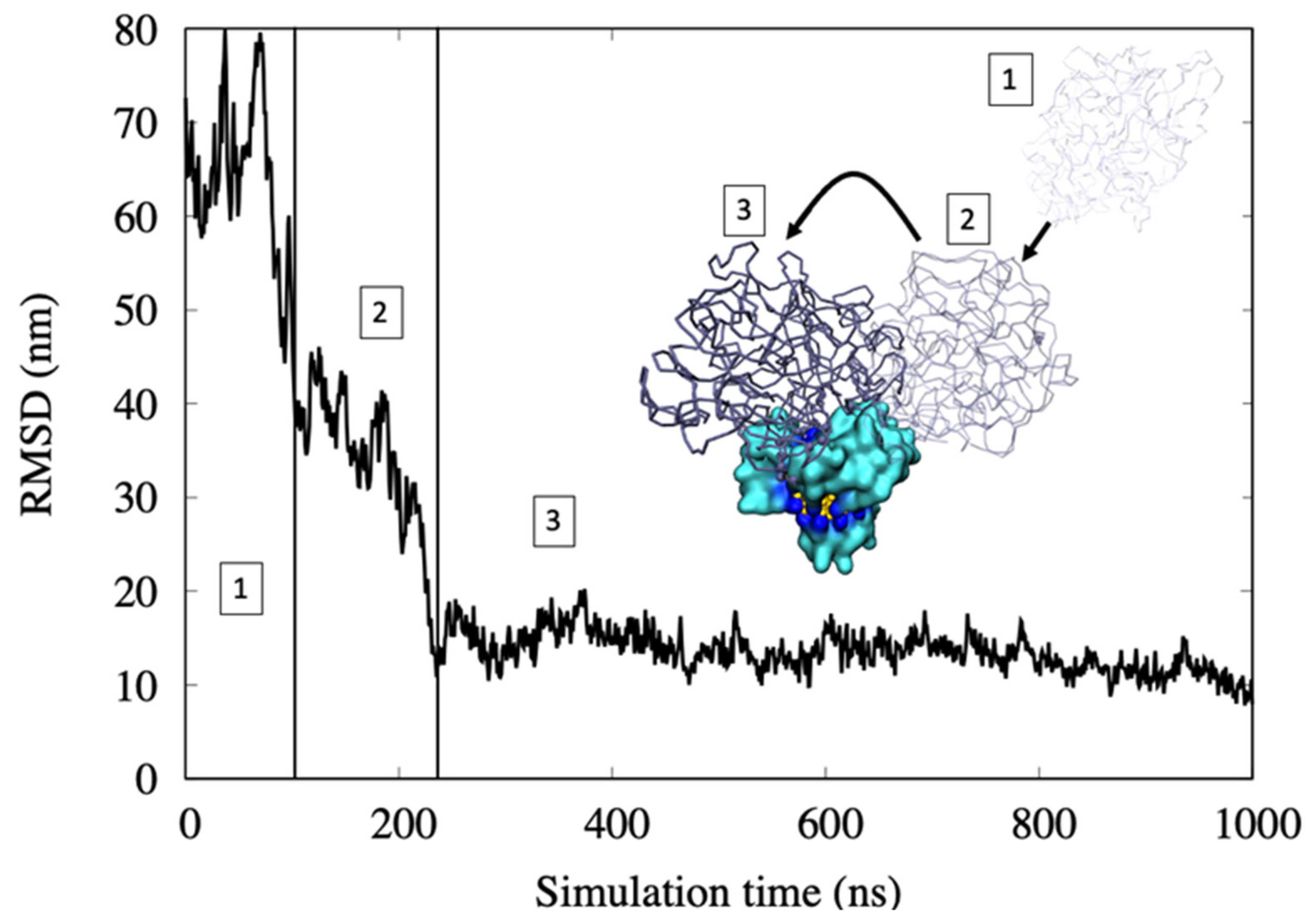
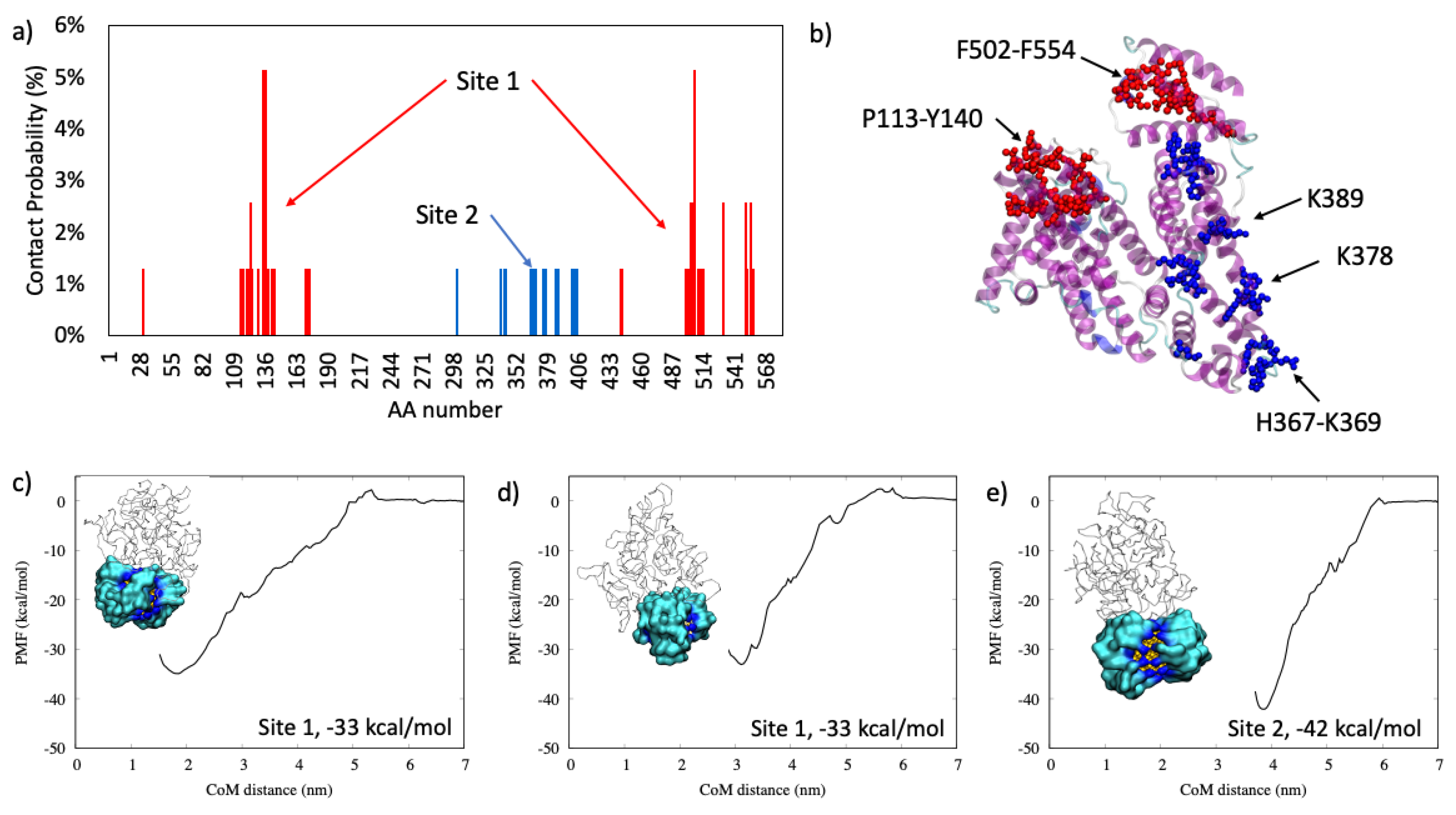
2.1.4. Hemoglobin
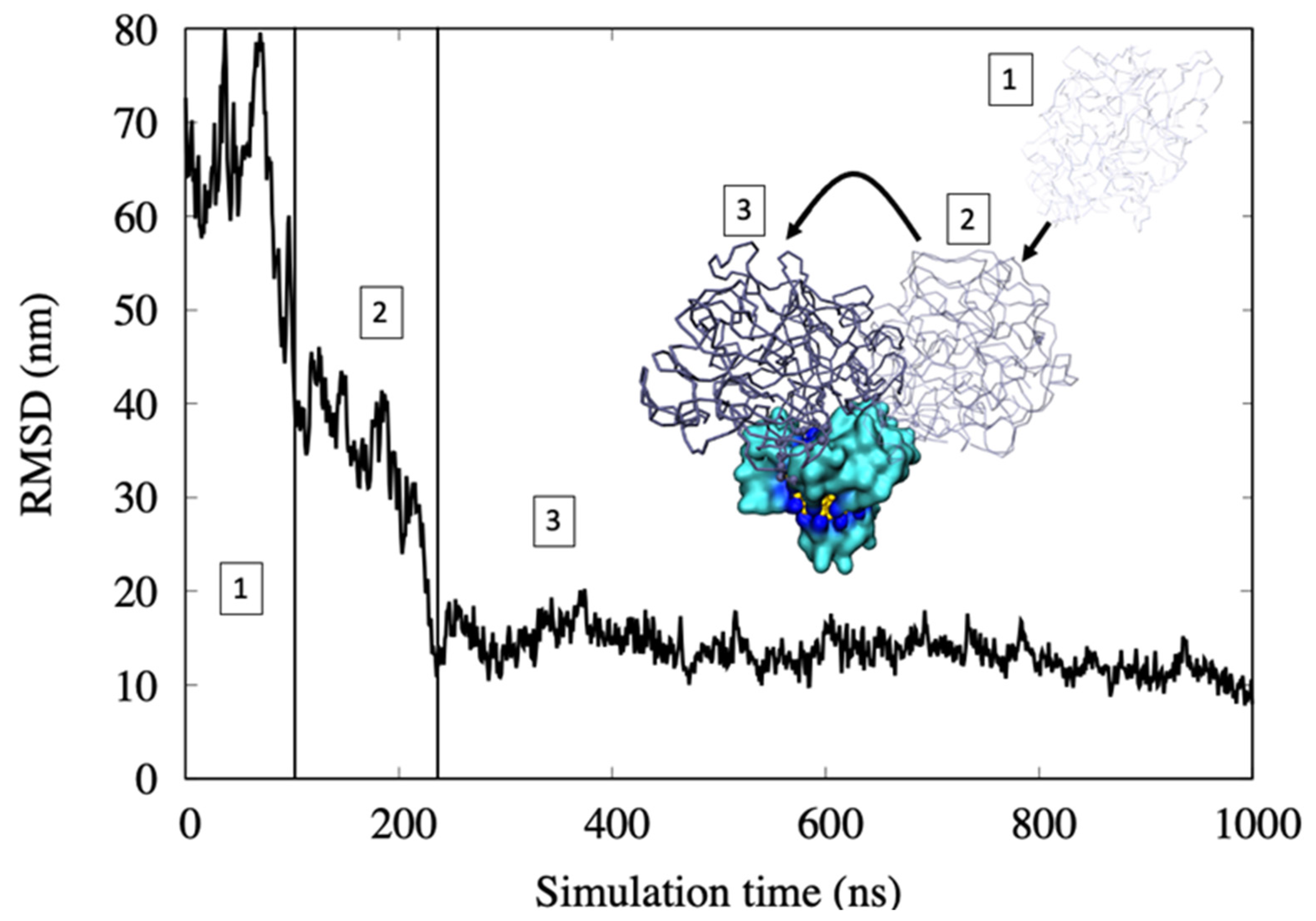
- In the first step, protein motion in water is regulated by a diffusion regime. Here, the RMSD values are high due to the high mobility of the hemoglobin.
- The first binding with the all-MUS AuNP is with the MUS ligands that cap the surface. The RMSD goes down fast due to the lower mobility of the protein. This binding is weak because only a few amino acids interact with the ligand chains.
- The protein rearranges its position over the AuNP and, by being involved in binding several chains of MUS ligands, yields strong hydrophobic interactions. Here, the RMSD reaches a lower and more stable value, indicating a stable binding with the AuNP.
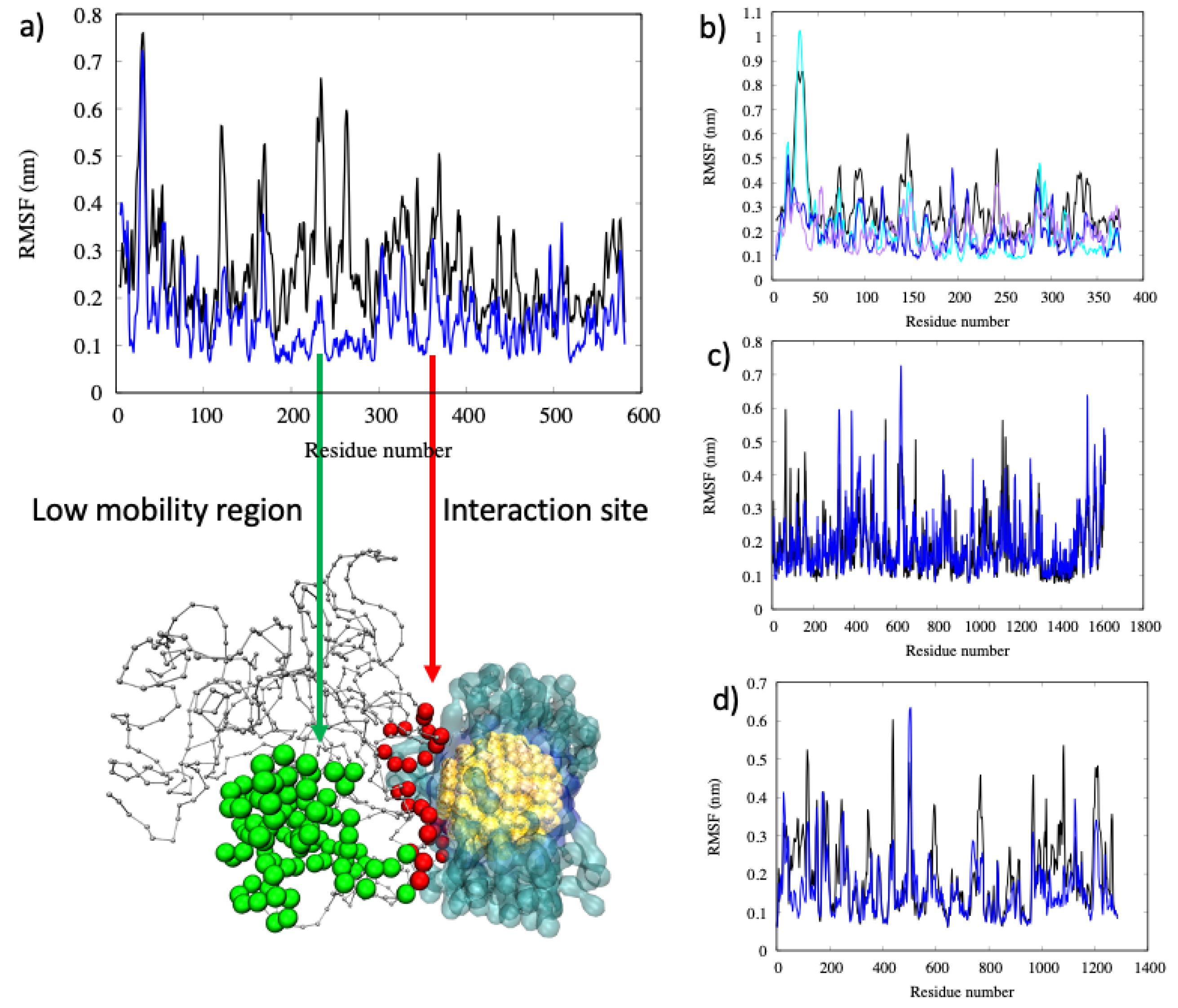
2.2. Conformational Changes
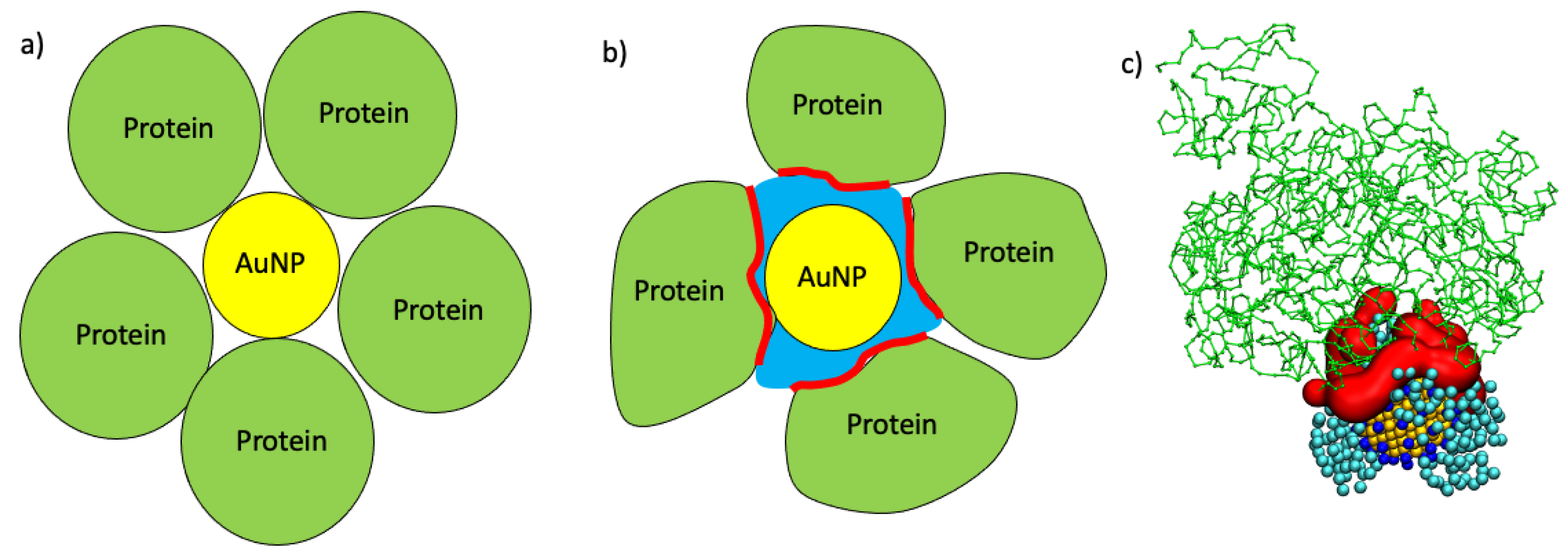
2.3. Maximum AuNP Adsorption Capacity
3. Discussion
4. Materials and Methods
5. Analysis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Abadeer, N.S.; Murphy, C.J. Recent Progress in Cancer Thermal Therapy Using Gold Nanoparticles. J. Phys. Chem. C 2016, 120, 4691–4716. [Google Scholar] [CrossRef]
- Chen, G.; Roy, I.; Yang, C.; Prasad, P.N. Nanochemistry and Nanomedicine for Nanoparticle-Based Diagnostics and Therapy. Chem. Rev. 2016, 116, 2826–2885. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Yan, B. Nano Products in Daily Life: To Know What We Do Not Know. Natl. Sci. Rev. 2016, 3, 414–415. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, P.; Han, G.; De, M.; Kim, C.K.; Rotello, V.M. Gold Nanoparticles in Delivery Applications. Adv. Drug Deliv. Rev. 2008, 60, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Tian, J.; Zhao, Y.; Chen, C.; Zhou, R.; Chai, Z. Towards Understanding of Nanoparticle–Protein Corona. Arch. Toxicol. 2015, 89, 519–539. [Google Scholar] [CrossRef] [PubMed]
- Del Pino, P.; Pelaz, B.; Zhang, Q.; Maffre, P.; Nienhaus, G.U.; Parak, W.J. Protein Corona Formation around Nanoparticles – from the Past to the Future. Mater. Horizons. 2014, 1, 301. [Google Scholar] [CrossRef]
- Deyev, S.; Proshkina, G.; Ryabova, A.; Tavanti, F.; Menziani, M.C.; Eidelshtein, G.; Avishai, G.; Kotlyar, A. Synthesis, Characterization, and Selective Delivery of DARPin–Gold Nanoparticle Conjugates to Cancer Cells. Bioconjugate Chem. 2017, 28, 2569–2574. [Google Scholar] [CrossRef]
- Fleischer, C.C.; Payne, C.K. Nanoparticle–Cell Interactions: Molecular Structure of the Protein Corona and Cellular Outcomes. Acc. Chem. Res. 2014, 47, 2651–2659. [Google Scholar] [CrossRef] [PubMed]
- Tavanti, F.; Pedone, A.; Menziani, M.C. A Closer Look into the Ubiquitin Corona on Gold Nanoparticles by Computational Studies. New J. Chem. 2015, 39, 2474–2482. [Google Scholar] [CrossRef]
- Calzolai, L.; Franchini, F.; Gilliland, D.; Rossi, F. Protein−Nanoparticle Interaction: Identification of the Ubiquitin−Gold Nanoparticle Interaction Site. Nano Lett. 2010, 10, 3101–3105. [Google Scholar] [CrossRef] [PubMed]
- Tavanti, F.; Pedone, A.; Menziani, M.C. Competitive Binding of Proteins to Gold Nanoparticles Disclosed by Molecular Dynamics Simulations. J. Phys. Chem. C 2015, 119, 22172–22180. [Google Scholar] [CrossRef]
- Brancolini, G.; Corazza, A.; Vuano, M.; Fogolari, F.; Mimmi, M.C.; Bellotti, V.; Stoppini, M.; Corni, S.; Esposito, G. Probing the Influence of Citrate-Capped Gold Nanoparticles on an Amyloidogenic Protein. ACS Nano 2015, 9, 2600–2613. [Google Scholar] [CrossRef] [PubMed]
- Brancolini, G.; Maschio, M.C.; Cantarutti, C.; Corazza, A.; Fogolari, F.; Bellotti, V.; Corni, S.; Esposito, G. Citrate Stabilized Gold Nanoparticles Interfere with Amyloid Fibril Formation: D76N and ΔN6 Β2-Microglobulin Variants. Nanoscale 2018, 10, 4793–4806. [Google Scholar] [CrossRef] [Green Version]
- Brancolini, G.; Kokh, D.B.; Calzolai, L.; Wade, R.C.; Corni, S. Docking of Ubiquitin to Gold Nanoparticles. ACS Nano. 2012, 6, 9863–9878. [Google Scholar] [CrossRef]
- Lin, W.; Insley, T.; Tuttle, M.D.; Zhu, L.; Berthold, D.A.; Král, P.; Rienstra, C.M.; Murphy, C.J. Control of Protein Orientation on Gold Nanoparticles. J. Phys. Chem. C 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Tieleman, D.P.; Liang, Q. Modulating Interactions between Ligand-Coated Nanoparticles and Phase-Separated Lipid Bilayers by Varying the Ligand Density and the Surface Charge. Nanoscale. 2018, 10, 2481–2491. [Google Scholar] [CrossRef] [PubMed]
- Salassi, S.; Simonelli, F.; Bochicchio, D.; Ferrando, R.; Rossi, G. Au Nanoparticles in Lipid Bilayers: A Comparison between Atomistic and Coarse-Grained Models. J. Phys. Chem. C 2017, 121, 10927–10935. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.; Andreozzi, P.; Dal Magro, R.; Fiordaliso, F.; Corbelli, A.; Talamini, L.; Chinello, C.; Raimondo, F.; Magni, F.; Tringali, M.; et al. Evolution of Nanoparticle Protein Corona across the Blood–Brain Barrier. ACS Nano. 2018, 12, 7292–7300. [Google Scholar] [CrossRef]
- Cagno, V.; Andreozzi, P.; D’Alicarnasso, M.; Jacob Silva, P.; Mueller, M.; Galloux, M.; Le Goffic, R.; Jones, S.T.; Vallino, M.; Hodek, J.; et al. Broad-Spectrum Non-Toxic Antiviral Nanoparticles with a Virucidal Inhibition Mechanism. Nat. Mater. 2018, 17, 195–203. [Google Scholar] [CrossRef]
- Gasbarri, M.; V’kovski, P.; Torriani, G.; Thiel, V.; Stellacci, F.; Tapparel, C.; Cagno, V. SARS-CoV-2 Inhibition by Sulfonated Compounds. Microorganisms 2020, 8, 1894. [Google Scholar] [CrossRef]
- Yu, Q.; Zhao, L.; Guo, C.; Yan, B.; Su, G. Regulating Protein Corona Formation and Dynamic Protein Exchange by Controlling Nanoparticle Hydrophobicity. Front. Bioeng. Biotechnol. 2020, 8, 210. [Google Scholar] [CrossRef] [Green Version]
- Chew, A.K.; Dallin, B.C.; Van Lehn, R.C. The Interplay of Ligand Properties and Core Size Dictates the Hydrophobicity of Monolayer-Protected Gold Nanoparticles. ACS Nano. 2021, 15, 4534–4545. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Radic, S.; Chen, R.; Chen, P.; Geitner, N.K.; Brown, J.M.; Ke, P.C. Direct Observation of a Single Nanoparticle–Ubiquitin Corona Formation. Nanoscale 2013, 5, 9162–9169. [Google Scholar] [CrossRef] [Green Version]
- Tavanti, F.; Pedone, A.; Menziani, M.C. Multiscale Molecular Dynamics Simulation of Multiple Protein Adsorption on Gold Nanoparticles. Int. J. Mol. Sci. 2019, 20, 3539. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Vangala, K.; Vo, T.; Zhang, D.; Fitzkee, N.C. A Three-Step Model for Protein–Gold Nanoparticle Adsorption. J. Phys. Chem. C 2014, 118, 8134–8142. [Google Scholar] [CrossRef]
- Dell’Orco, D.; Lundqvist, M.; Oslakovic, C.; Cedervall, T.; Linse, S. Modeling the Time Evolution of the Nanoparticle-Protein Corona in a Body Fluid. PLoS ONE 2010, 5, e10949. [Google Scholar] [CrossRef] [Green Version]
- Tavanti, F.; Pedone, A.; Matteini, P.; Menziani, M.C. Computational Insight into the Interaction of Cytochrome C with Wet and PVP-Coated Ag Surfaces. J. Phys. Chem. B 2017, 121, 9532–9540. [Google Scholar] [CrossRef]
- Tavanti, F.; Pedone, A.; Menziani, M.C. Disclosing the Interaction of Gold Nanoparticles with Aβ(1–40) Monomers through Replica Exchange Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 22, 26. [Google Scholar] [CrossRef]
- Deng, Z.J.; Liang, M.; Monteiro, M.; Toth, I.; Minchin, R.F. Nanoparticle-Induced Unfolding of Fibrinogen Promotes Mac-1 Receptor Activation and Inflammation. Nat Nano 2011, 6, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Hazarika, Z.; Jha, A.N. Computational Analysis of the Silver Nanoparticle–Human Serum Albumin Complex. ACS Omega. 2020, 5, 170–178. [Google Scholar] [CrossRef]
- Khan, S.; Gupta, A.; Verma, N.C.; Nandi, C.K. Kinetics of Protein Adsorption on Gold Nanoparticle with Variable Protein Structure and Nanoparticle Size. J. Chem. Phys. 2015, 143, 164709. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.L.; Ramyar, K.X.; Tzekou, A.; Ricklin, D.; McWhorter, W.J.; Lambris, J.D.; Geisbrecht, B.V. Molecular Basis for Complement Recognition and Inhibition Determined by Crystallographic Studies of the Staphylococcal Complement Inhibitor (SCIN) Bound to C3c and C3b. J. Mol. Biol. 2010, 402, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fermi, G.; Perutz, M.F.; Shaanan, B.; Fourme, R. The Crystal Structure of Human Deoxyhaemoglobin at 1.74 Å Resolution. J. Mol. Biol. 1984, 175, 159–174. [Google Scholar] [CrossRef]
- Elliott, P.R.; Pei, X.Y.; Dafforn, T.R.; Lomas, D.A. Topography of a 2.0 Å Structure of A1-Antitrypsin Reveals Targets for Rational Drug Design to Prevent Conformational Disease. Protein Sci. 2000, 9, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal Structure of Human Serum Albumin at 2.5 Å Resolution. Protein Eng. Des. Sel. 1999, 12, 439–446. [Google Scholar] [CrossRef]
- Janssen, B.J.C.; Huizinga, E.G.; Raaijmakers, H.C.A.; Roos, A.; Daha, M.R.; Nilsson-Ekdahl, K.; Nilsson, B.; Gros, P. Structures of Complement Component C3 Provide Insights into the Function and Evolution of Immunity. Nature 2005, 437, 505–511. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A General Purpose Force Field for Coarse-Grained Molecular Dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef]
- de Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tavanti, F.; Pedone, A.; Menziani, M.C.; Alexander-Katz, A. Computational Insights into the Binding of Monolayer-Capped Gold Nanoparticles onto Amyloid-β Fibrils. ACS Chem. Neurosci. 2020, 11, 3153–3160. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S.; de Groot, B.L.; van der Spoel, D. G_wham—A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 2010, 6, 3713–3720. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]


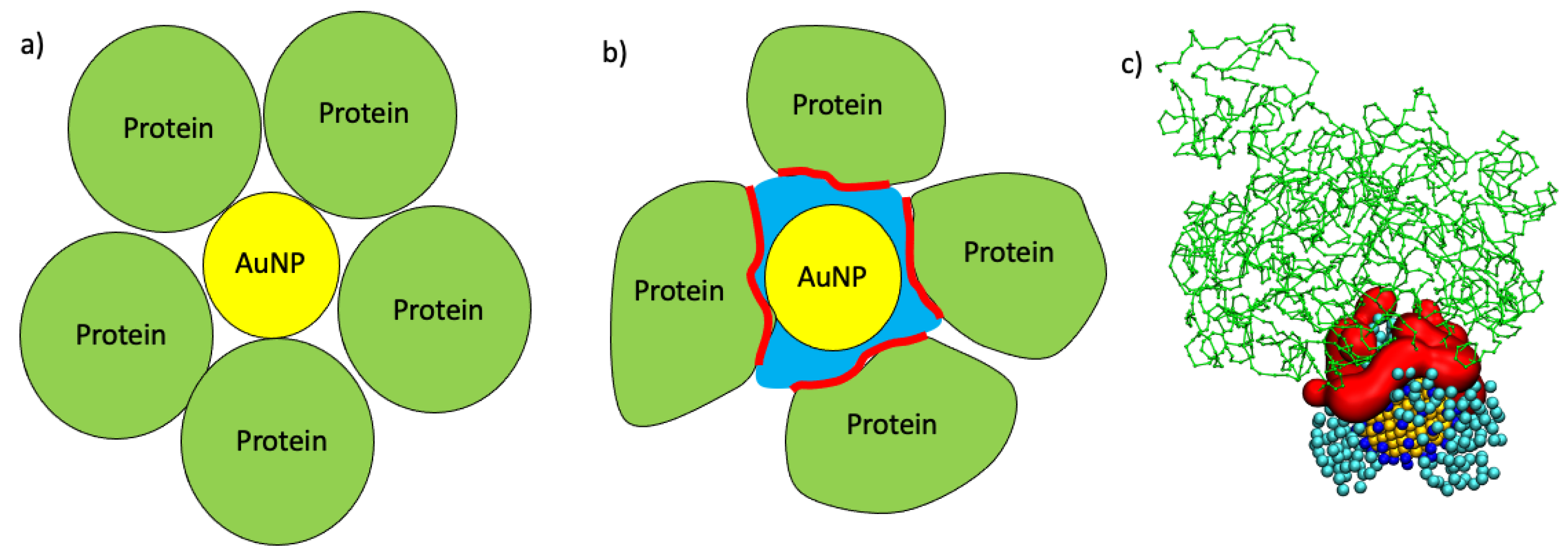


{kind=link}
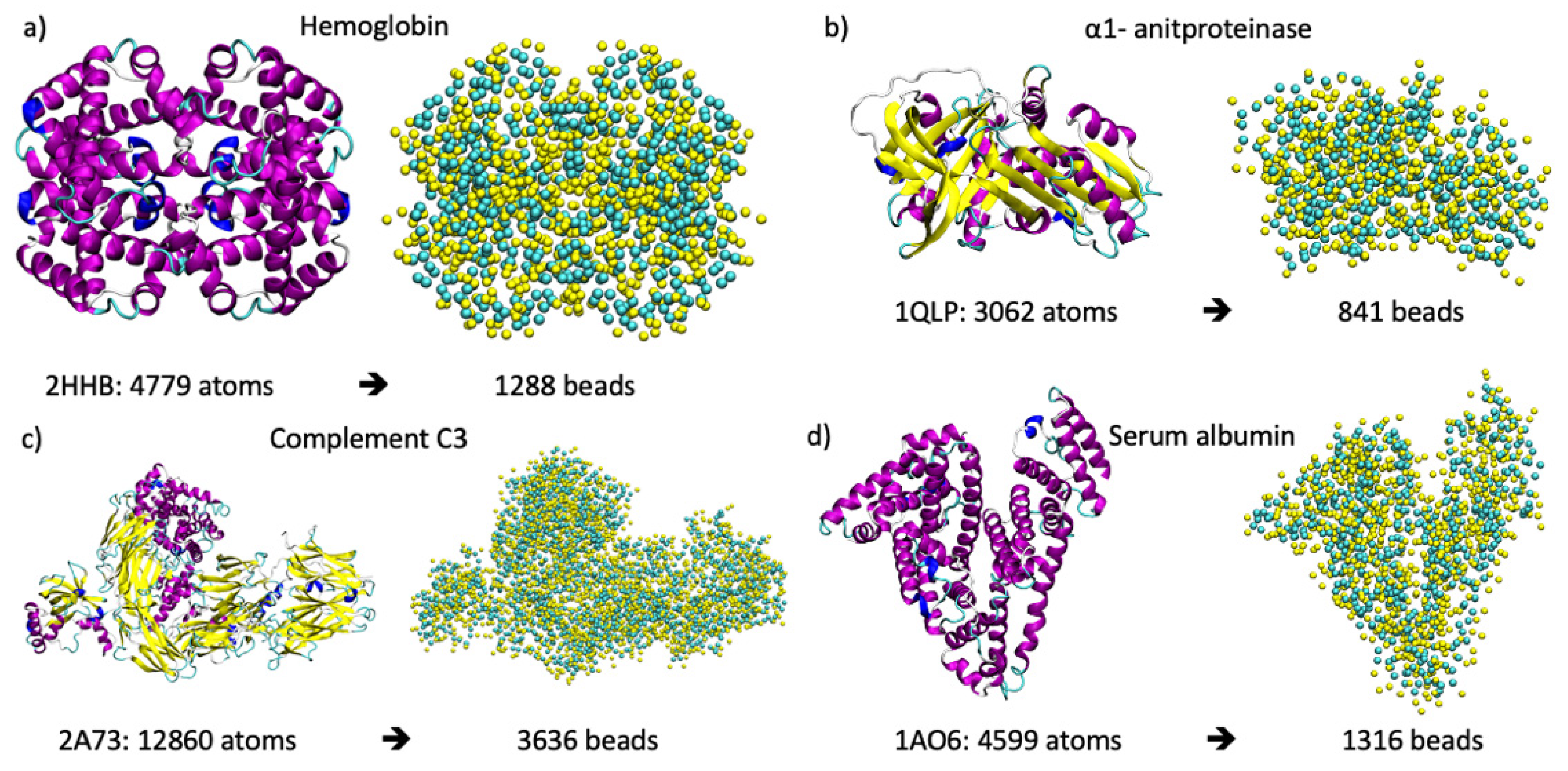
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA 1-Letter Code | AA Name | Hemoglobin | Serum Albumin | α1-Antiproteinase | Complement C3 |
|---|---|---|---|---|---|
| A | Ala | 13% | 11% | 7% | 7% |
| C | Cys | 2% | 13% | 0% | 3% |
| D | Asp | 2% | 2% | 4% | 2% |
| E | Glu | 0% | 5% | 9% | 1% |
| F | Phe | 10% | 4% | 11% | 6% |
| G | Gly | 2% | 0% | 0% | 5% |
| H | His | 3% | 5% | 0% | 2% |
| I | Ile | 0% | 4% | 9% | 4% |
| K | Lys | 8% | 4% | 4% | 5% |
| L | Leu | 3% | 16% | 20% | 10% |
| M | Met | 3% | 2% | 4% | 4% |
| N | Asn | 3% | 4% | 4% | 2% |
| O | Pyl | 0% | 0% | 0% | 0% |
| P | Pro | 10% | 5% | 2% | 7% |
| Q | Gln | 7% | 4% | 0% | 7% |
| R | Arg | 3% | 2% | 0% | 5% |
| S | Ser | 7% | 2% | 9% | 9% |
| T | Thr | 5% | 2% | 4% | 6% |
| U | Sec | 0% | 0% | 0% | 0% |
| V | Val | 15% | 9% | 7% | 11% |
| W | Trp | 2% | 0% | 2% | 2% |
| Y | Tyr | 2% | 7% | 4% | 4% |
| Hydrophobic | 48% | 53% | 64% | 46% | |
| Charged | 14% | 12% | 17% | 14% | |
| Polar | 22% | 11% | 17% | 23% | |
| Others | 17% | 24% | 2% | 17% | |
| PMF (kcal/mol) | −41 ± 11 | −36 ± 12 | −36 ± 8 | −47 ± 15 | |
| Hemoglobin | Serum Albumin | α1-Antiproteinase | Complement C3 | |||||
|---|---|---|---|---|---|---|---|---|
| Gyration Radius (nm) | before | after | before | after | before | after | before | after |
| 2.06 ± 0.02 | 2.06 ± 0.02 | 2.18 ± 0.03 | 2.23 ± 0.09 | 1.89 ± 0.01 | 1.90 ± 0.05 | 3.32 ± 0.02 | 3.29 ± 0.02 | |
| Protein Size | Hemoglobin | Serum Albumin | α1-Antiproteinase | Complement C3 |
|---|---|---|---|---|
| Mass (kDa) | 64.74 | 133.14 | 44.38 | 185.69 |
| Rg (nm) | 1.47 | 2.64 | 2.16 | 4.59 |
| Radius 1 (nm) | 2.70 | 3.10 | 1.95 | 4.40 |
| Radius 2 (nm) | 2.80 | 3.65 | 3.00 | 6.65 |
| Nmax | ||||
| Wang et al. [25] | 7 | 2 | 3 | 1 |
| 7 | 2 | 3 | 1 | |
| Calzolai et al. [10] | 13 | 12 | 17 | 10 |
| 13 | 11 | 12 | 8 | |
| Dell’Orco et al. [26] | 12 | 11 | 16 | 8 |
| 12 | 10 | 11 | 7 | |
| This work | 5 | 5 | 6 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavanti, F.; Menziani, M.C. Computational Insight on the Interaction of Common Blood Proteins with Gold Nanoparticles. Int. J. Mol. Sci. 2021, 22, 8722. https://doi.org/10.3390/ijms22168722
Tavanti F, Menziani MC. Computational Insight on the Interaction of Common Blood Proteins with Gold Nanoparticles. International Journal of Molecular Sciences. 2021; 22(16):8722. https://doi.org/10.3390/ijms22168722
Chicago/Turabian StyleTavanti, Francesco, and Maria Cristina Menziani. 2021. "Computational Insight on the Interaction of Common Blood Proteins with Gold Nanoparticles" International Journal of Molecular Sciences 22, no. 16: 8722. https://doi.org/10.3390/ijms22168722
APA StyleTavanti, F., & Menziani, M. C. (2021). Computational Insight on the Interaction of Common Blood Proteins with Gold Nanoparticles. International Journal of Molecular Sciences, 22(16), 8722. https://doi.org/10.3390/ijms22168722