
Enriched Alternative Splicing in Islets of Diabetes-Susceptible Mice

 , ,
, ,
Abstract
:1. Introduction
2. Results
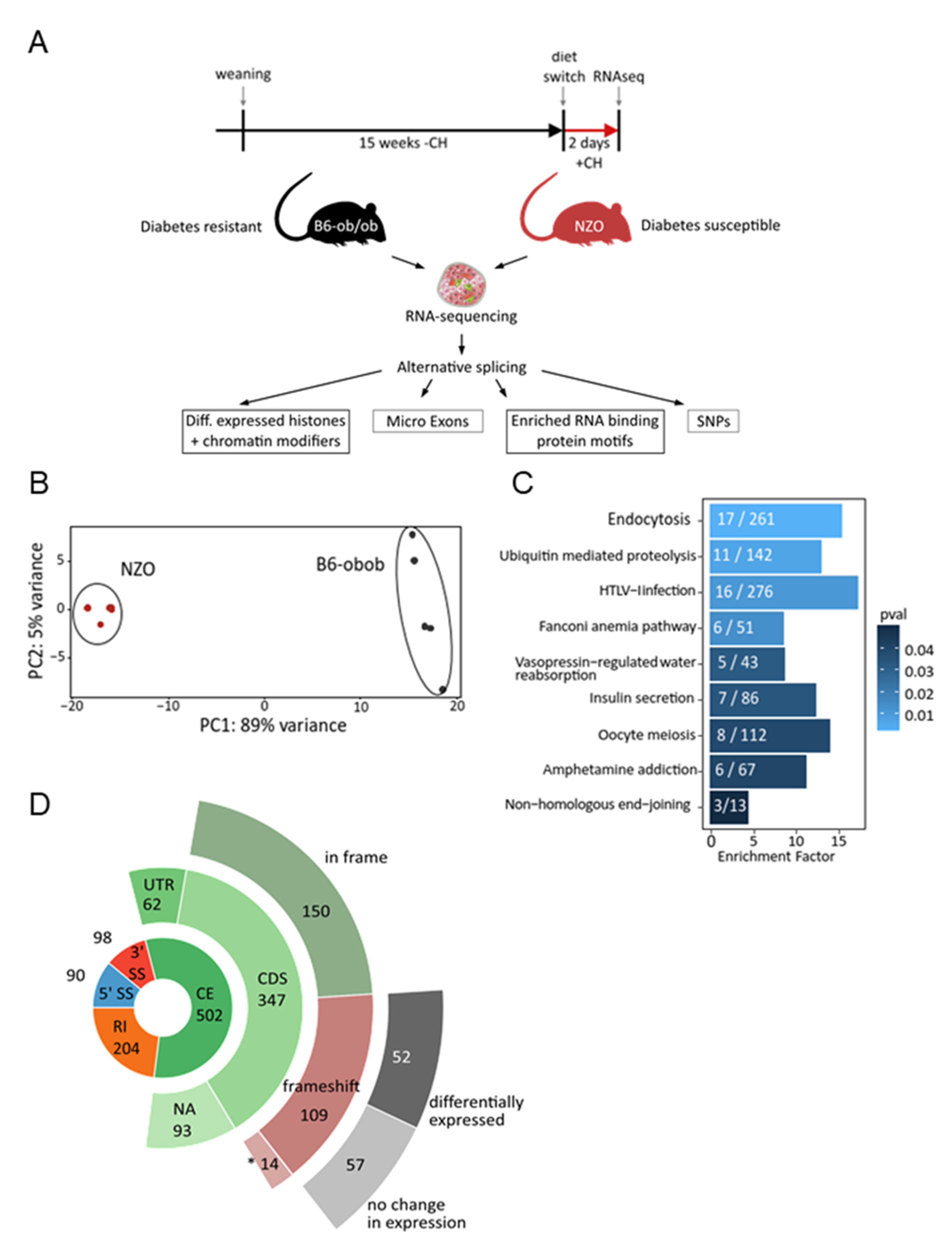
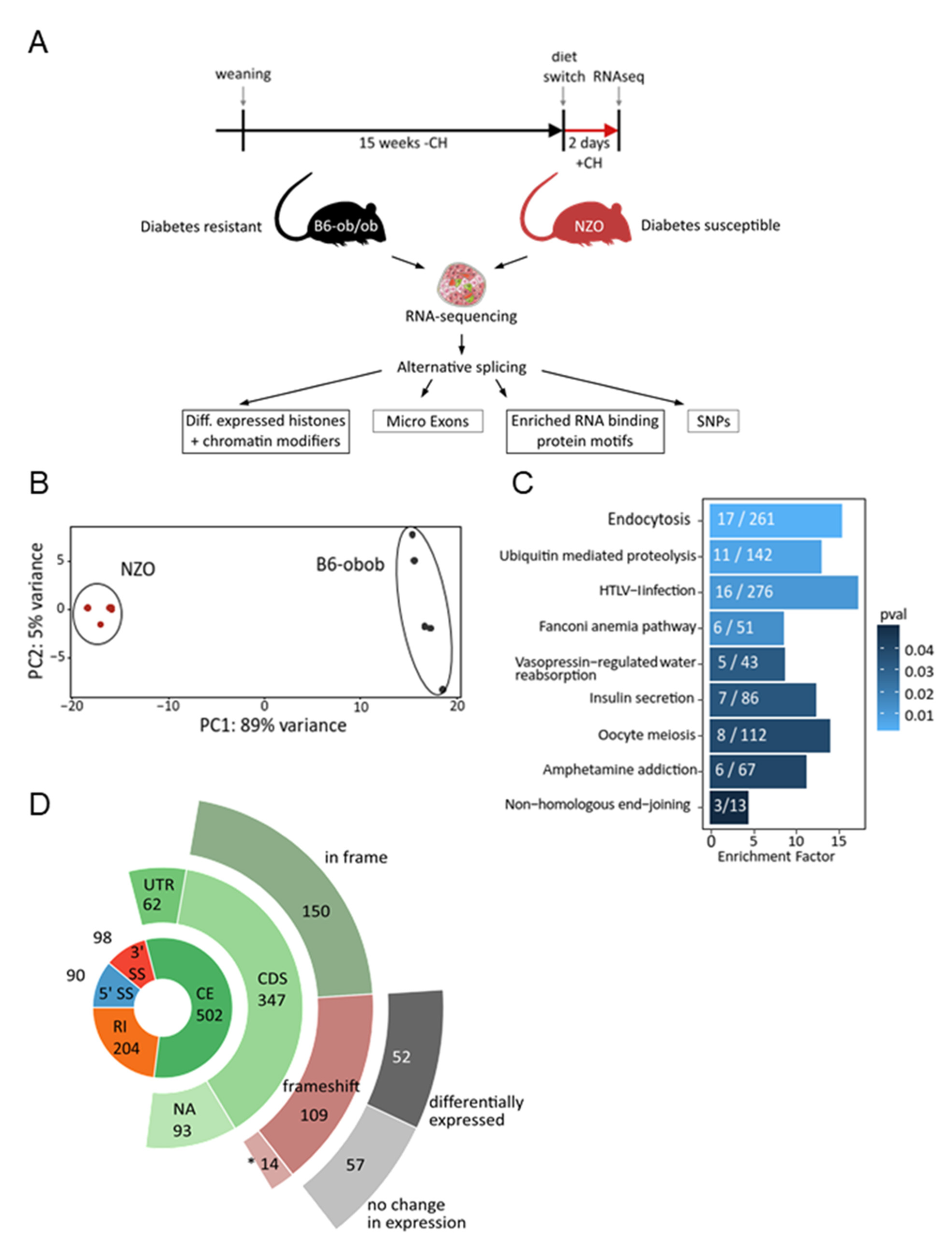
2.1. Differential Alternative Splicing in Islets of Diabetes-Prone and -Resistant Mice
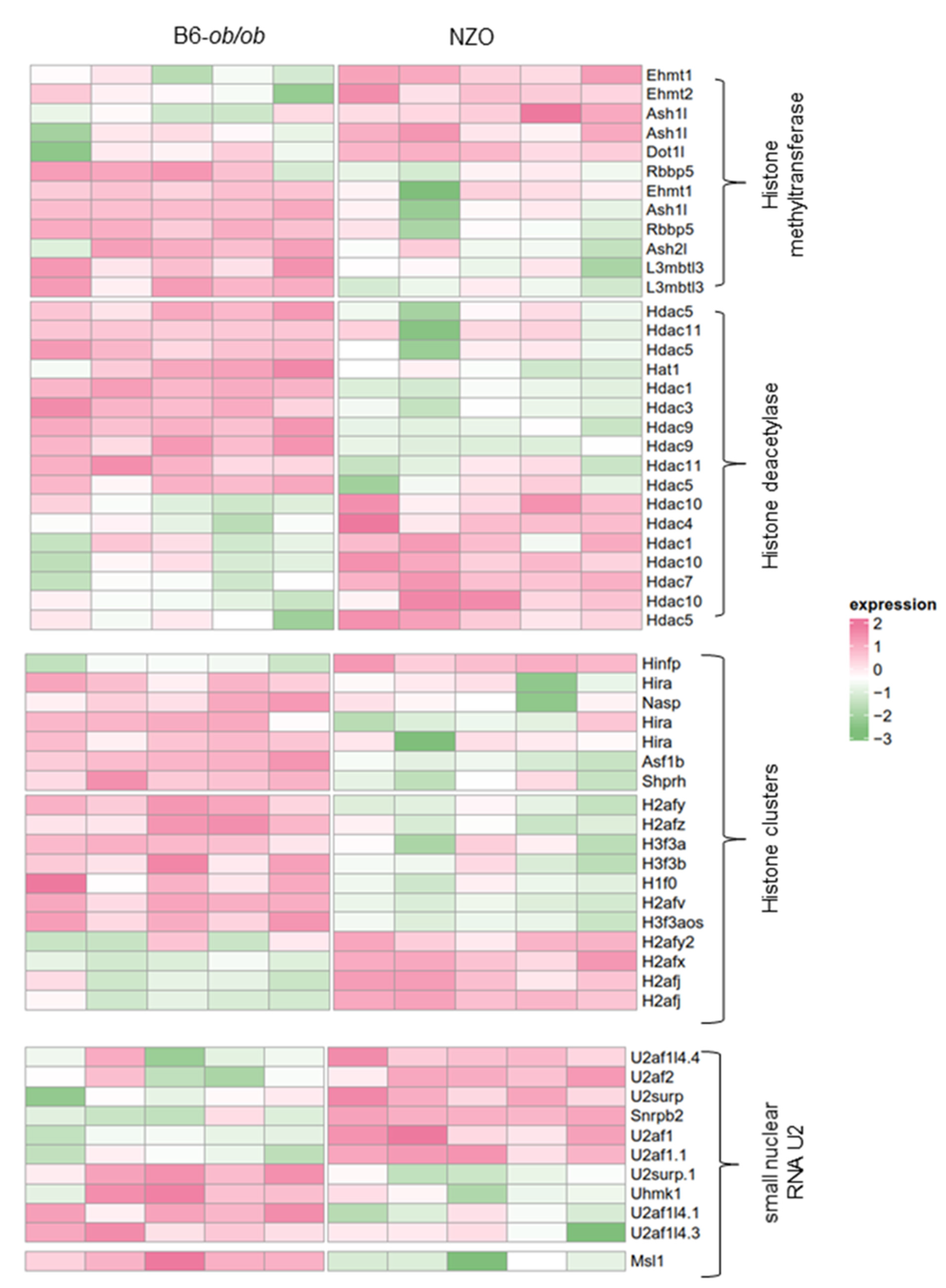
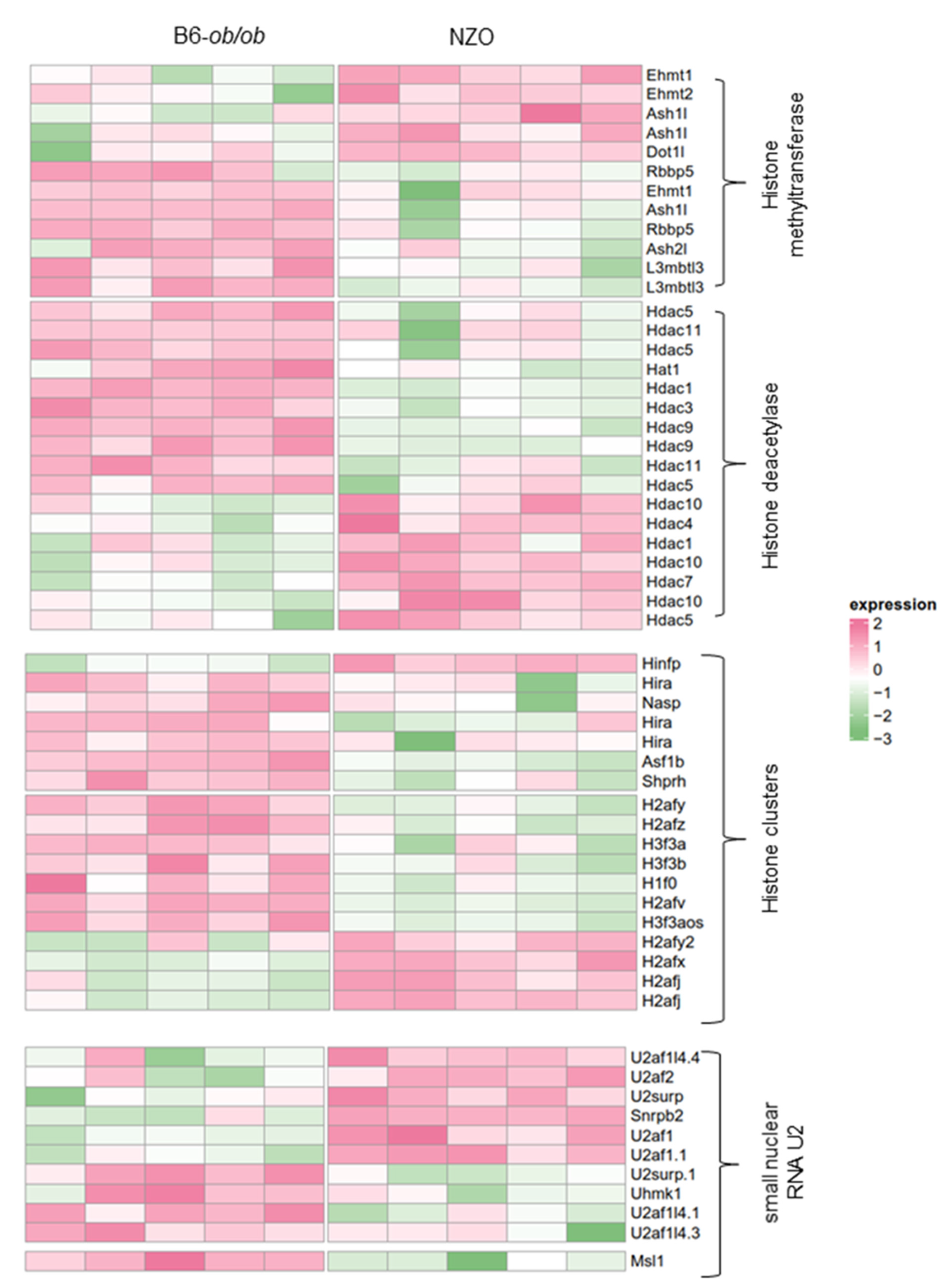
2.2. Altered Expression of Histones and Chromatin Modifiers
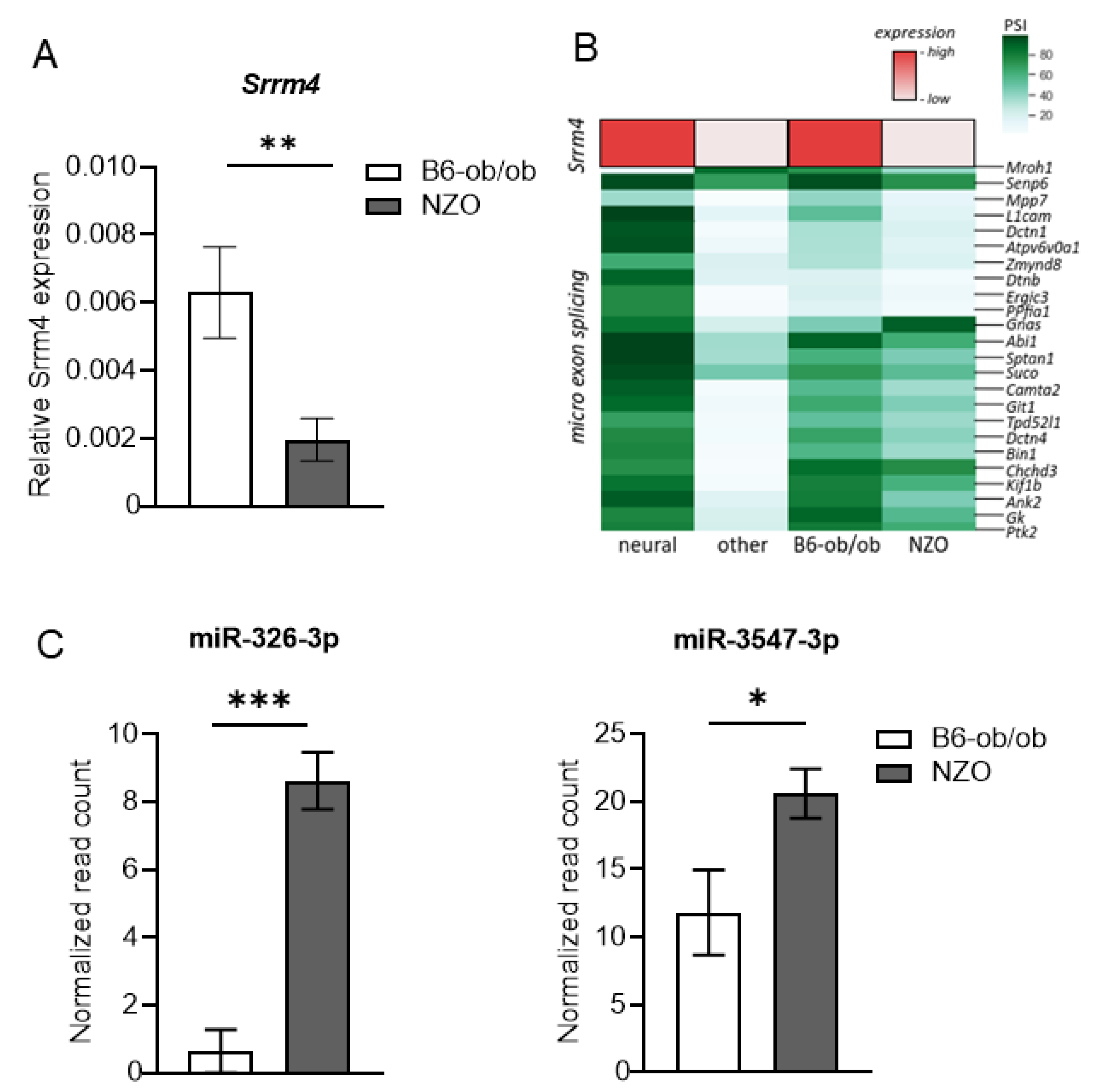
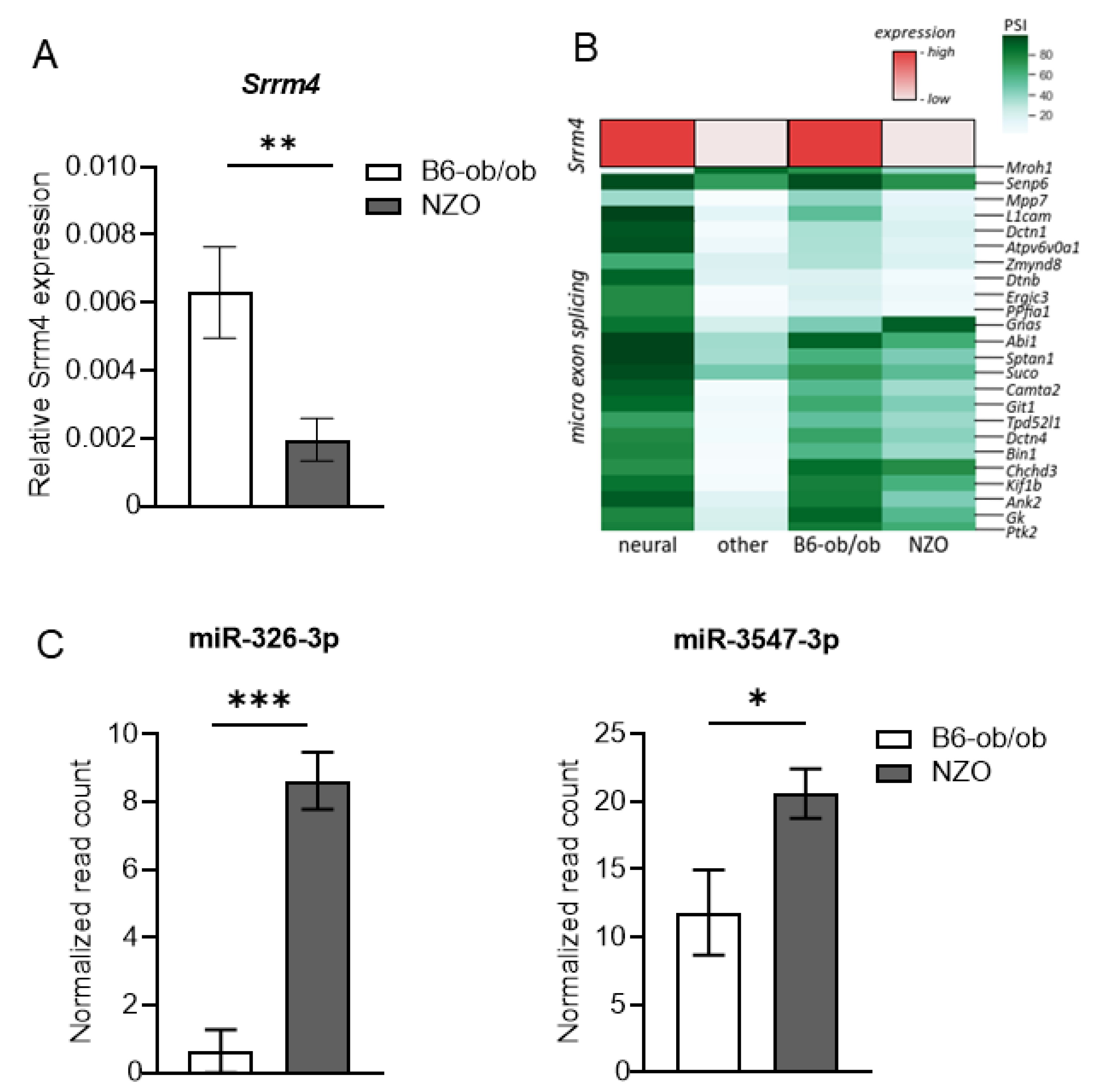
2.3. Differences in Splicing of Microexons in NZO and B6-ob/ob Islets
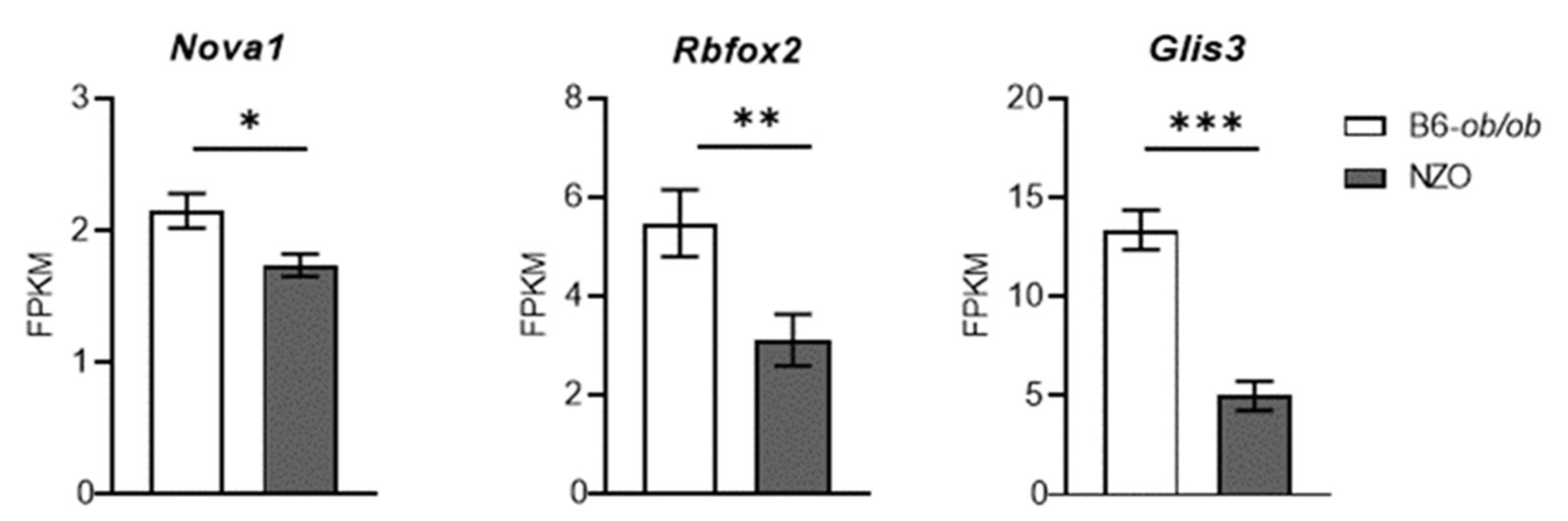
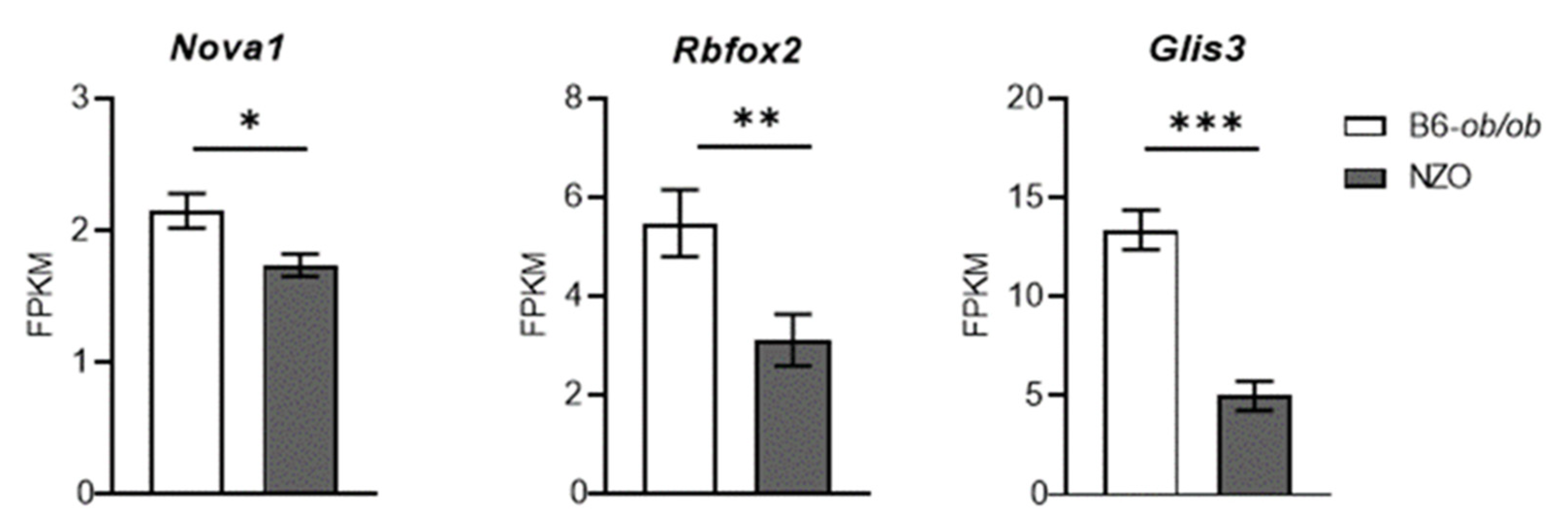
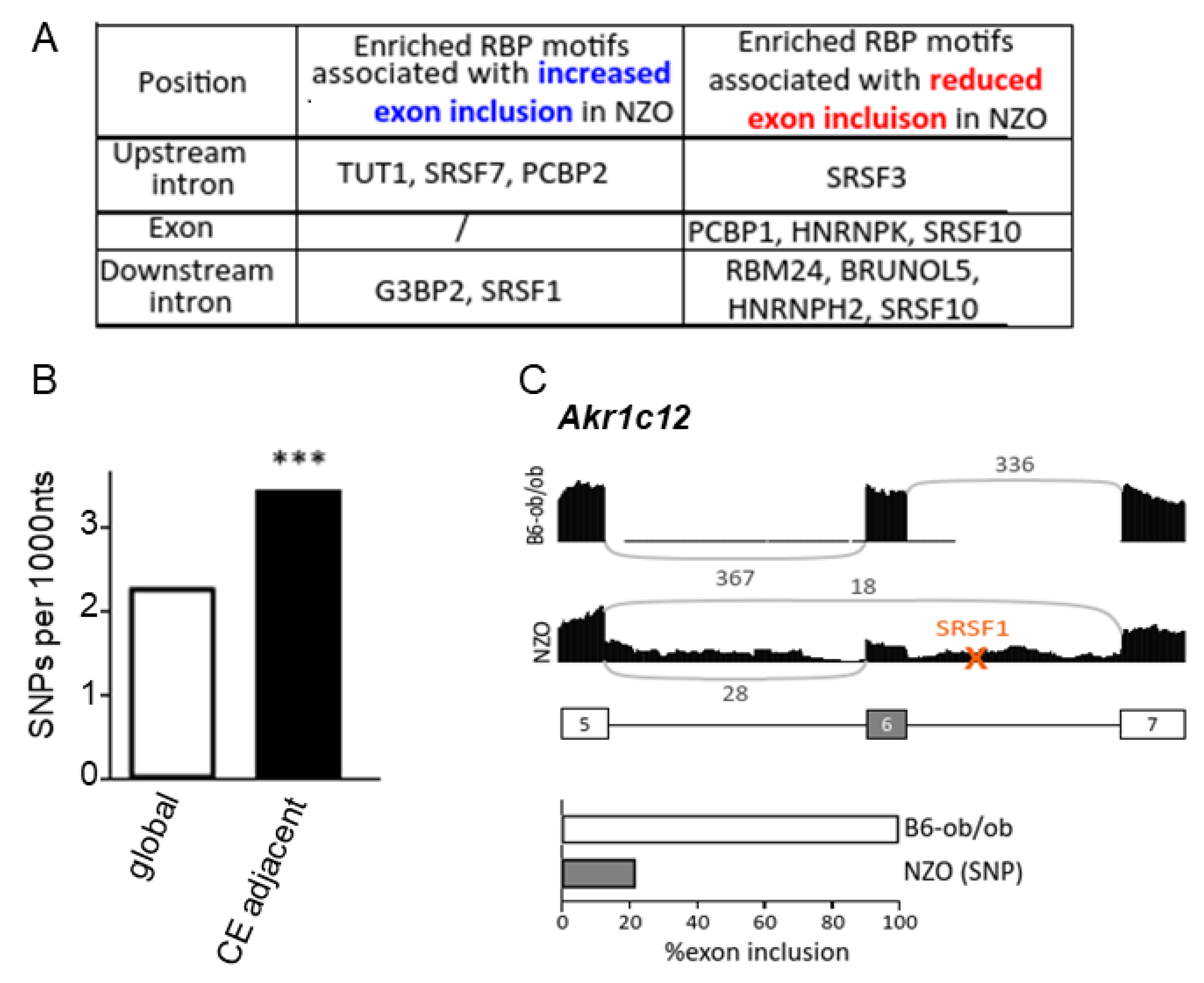
2.4. Role of Neuron-Specific Splicing Factors and Enrichment of Affected RNA Binding Protein Motifs Splicing Relevant
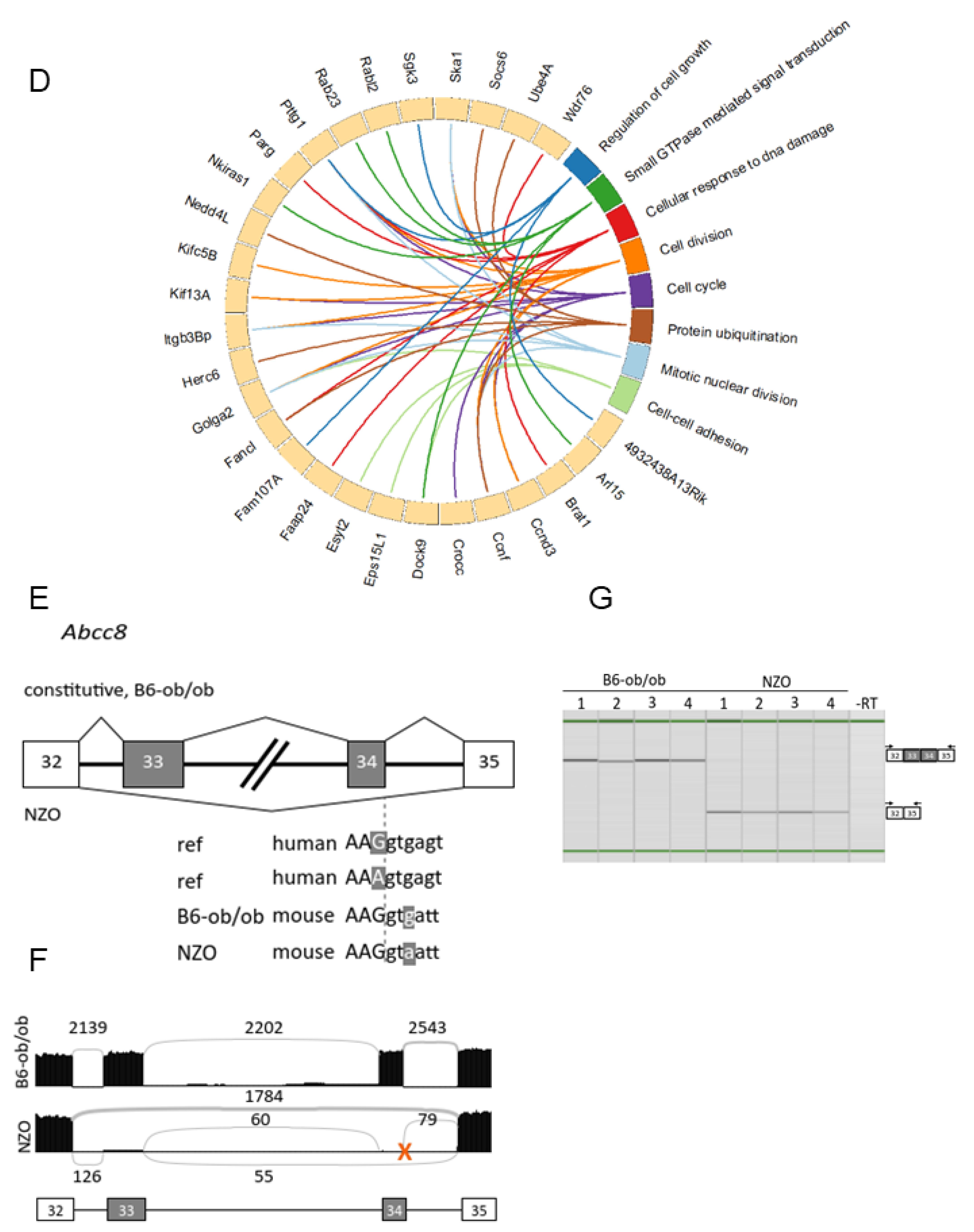
2.5. Polymorphisms in Splice Sites in Diabetes-Prone Mice
3. Discussion
4. Material and Methods
4.1. Animals and Study Design
4.2. RNA Extraction and Sequencing
4.3. Splice Sensitive RT-PCR and qRT-PCR
4.4. Bioinformatic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NZO | New Zealand obese |
| RBP | RNA binding protein |
| SNP | single nucleotide polymorphism |
| ss | splice site |
| PSI | percent spliced in |
| nts | nucleotides |
| SR | serine (S) arginine (R) repeat rich proteins |
| hnRNP | heterogeneous nuclear ribonucleoprotein |
References
- Kluth, O.; Matzke, D.; Kamitz, A.; Jähnert, M.; Vogel, H.; Scherneck, S.; Schulze, M.; Staiger, H.; Machicao, F.; Häring, H.U.; et al. Identification of Four Mouse Diabetes Candidate Genes Altering β-Cell Proliferation. PLoS Genet. 2015, 11, e1005506. [Google Scholar] [CrossRef]
- Kluth, O.; Stadion, M.; Gottmann, P.; Aga, H.; Jähnert, M.; Scherneck, S.; Vogel, H.; Krus, U.; Seelig, A.; Ling, C.; et al. Decreased Expression of Cilia Genes in Pancreatic Islets as a Risk Factor for Type 2 Diabetes in Mice and Humans. Cell Rep. 2019, 26, 3027–3036.e3. [Google Scholar] [CrossRef] [Green Version]
- Gässler, A.; Quiclet, C.; Kluth, O.; Gottmann, P.; Schwerbel, K.; Helms, A.; Stadion, M.; Wilhelmi, I.; Jonas, W.; Ouni, M.; et al. Overexpression of Gjb4 impairs cell proliferation and insulin secretion in primary islet cells. Mol. Metab. 2020, 41, 101042. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, E.C.; Schaal, T.D.; Hertel, K.J.; Reed, R.; Maniatis, T. Serine/arginine-rich protein-dependent suppression of exon skipping by exonic splicing enhancers. Proc. Natl. Acad. Sci. USA 2005, 102, 5002–5007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krecic, A.M.; Swanson, M.S. hnRNP complexes: Composition, structure, and function. Curr. Opin. Cell Biol. 1999, 11, 363–371. [Google Scholar] [CrossRef]
- Han, S.P.; Tang, Y.H.; Smith, R. Functional diversity of the hnRNPs: Past, present and perspectives. Biochem. J. 2010, 430, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Erkelenz, S.; Mueller, W.F.; Evans, M.S.; Busch, A.; Schöneweis, K.; Hertel, K.J.; Schaal, H. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA 2013, 19, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Soemedi, R.; Cygan, K.J.; Rhine, C.L.; Wang, J.; Bulacan, C.; Yang, J.; Bayrak-Toydemir, P.; McDonald, J.; Fairbrother, W.G. Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet. 2017, 49, 848–855. [Google Scholar] [CrossRef]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell. Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmi, I.; Kanski, R.; Neumann, A.; Herdt, O.; Hoff, F.; Jacob, R.; Preußner, M.; Heyd, F. Sec16 alternative splicing dynamically controls COPII transport efficiency. Nat. Commun. 2016, 7, 12347. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.M.; Wood, M.J. Genetic therapies for RNA mis-splicing diseases. Trends Genet. 2011, 27, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Alvelos, M.I.; Juan-Mateu, J.; Colli, M.L.; Turatsinze, J.V.; Eizirik, D.L. When one becomes many-Alternative splicing in β-cell function and failure. Diabetes Obes. Metab. 2018, 20 (Suppl. 2), 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cnop, M.; Abdulkarim, B.; Bottu, G.; Cunha, D.A.; Igoillo-Esteve, M.; Masini, M.; Turatsinze, J.V.; Griebel, T.; Villate, O.; Santin, I.; et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 2014, 63, 1978–1993. [Google Scholar] [CrossRef] [Green Version]
- Juan-Mateu, J.; Villate, O.; Eizirik, D.L. Mechanisms in Endocrinology: Alternative splicing: The new frontier in diabetes research. Eur. J. Endocrinol. 2016, 174, R225–R238. [Google Scholar] [CrossRef] [Green Version]
- Luco, R.F.; Pan, Q.; Tominaga, K.; Blencowe, B.J.; Pereira-Smith, O.M.; Misteli, T. Regulation of alternative splicing by histone modifications. Science 2010, 327, 996–1000. [Google Scholar] [CrossRef] [Green Version]
- Kolathur, K.K. Role of promoters in regulating alternative splicing. Gene 2021, 782, 145523. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.L.; Luo, G.; Wise, J.A.; Lou, H. Regulation of alternative splicing by local histone modifications: Potential roles for RNA-guided mechanisms. Nucleic Acids Res. 2014, 42, 701–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, F.Q.; Johnson, T.L. Acetylation by the transcriptional coactivator Gcn5 plays a novel role in co-transcriptional spliceosome assembly. PLoS Genet. 2009, 5, e1000682. [Google Scholar] [CrossRef] [Green Version]
- Gunderson, F.Q.; Merkhofer, E.C.; Johnson, T.L. Dynamic histone acetylation is critical for cotranscriptional spliceosome assembly and spliceosomal rearrangements. Proc. Natl. Acad. Sci. USA 2011, 108, 2004–2009. [Google Scholar] [CrossRef] [Green Version]
- Hnilicova, J.; Hozeifi, S.; Duskova, E.; Icha, J.; Tomankova, T.; Stanek, D. Histone deacetylase activity modulates alternative splicing. PLoS ONE 2011, 6, e16727. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.I.; Sanchez-Pulido, L.; Haerty, W.; Ponting, C.P. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015, 25, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesnel-Vallières, M.; Irimia, M.; Cordes, S.P.; Blencowe, B.J. Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development. Genes Dev. 2015, 29, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atouf, F.; Czernichow, P.; Scharfmann, R. Expression of neuronal traits in pancreatic beta cells. Implication of neuron-restrictive silencing factor/repressor element silencing transcription factor, a neuron-restrictive silencer. J. Biol. Chem. 1997, 272, 1929–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irimia, M.; Weatheritt, R.J.; Ellis, J.D.; Parikshak, N.N.; Gonatopoulos-Pournatzis, T.; Babor, M.; Quesnel-Vallières, M.; Tapial, J.; Raj, B.; O’Hanlon, D.; et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 2014, 159, 1511–1523. [Google Scholar] [CrossRef] [Green Version]
- Tamura, Y.; Ozkan, E.D.; Bole, D.G.; Ueda, T. IPF, a vesicular uptake inhibitory protein factor, can reduce the Ca(2+)-dependent, evoked release of glutamate, GABA and serotonin. J. Neurochem. 2001, 76, 1153–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouni, M.; Gottmann, P.; Westholm, E.; Schwerbel, K.; Jahnert, M.; Stadion, M.; Rittig, K.; Vogel, H.; Schurmann, A. MiR-205 is upregulated in islets of diabetes-susceptible mice and targets the diabetes gene Tcf7l2. Acta Physiol. 2021, 232, e13693. [Google Scholar] [CrossRef] [PubMed]
- Gottmann, P.; Ouni, M.; Saussenthaler, S.; Roos, J.; Stirm, L.; Jahnert, M.; Kamitz, A.; Hallahan, N.; Jonas, W.; Fritsche, A.; et al. A computational biology approach of a genome-wide screen connected miRNAs to obesity and type 2 diabetes. Mol. Metab. 2018, 11, 145–159. [Google Scholar] [CrossRef]
- Ustianenko, D.; Weyn-Vanhentenryck, S.M.; Zhang, C. Microexons: Discovery, regulation, and function. Wiley Interdiscip. Rev. RNA 2017, 8, e1418. [Google Scholar] [CrossRef]
- Villate, O.; Turatsinze, J.V.; Mascali, L.G.; Grieco, F.A.; Nogueira, T.C.; Cunha, D.A.; Nardelli, T.R.; Sammeth, M.; Salunkhe, V.A.; Esguerra, J.L.; et al. Nova1 is a master regulator of alternative splicing in pancreatic beta cells. Nucleic Acids Res. 2014, 42, 11818–11830. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, T.C.; Paula, F.M.; Villate, O.; Colli, M.L.; Moura, R.F.; Cunha, D.A.; Marselli, L.; Marchetti, P.; Cnop, M.; Julier, C.; et al. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet. 2013, 9, e1003532. [Google Scholar] [CrossRef]
- Papasaikas, P.; Tejedor, J.R.; Vigevani, L.; Valcarcel, J. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol. Cell 2015, 57, 7–22. [Google Scholar] [CrossRef] [Green Version]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Georgiadi, A.; Lopez-Salazar, V.; Merahbi, R.E.; Karikari, R.A.; Ma, X.; Mourao, A.; Klepac, K.; Buhler, L.; Alfaro, A.J.; Kaczmarek, I.; et al. Orphan GPR116 mediates the insulin sensitizing effects of the hepatokine FNDC4 in adipose tissue. Nat. Commun. 2021, 12, 2999. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, M.; Deng, W.; Zhong, D.; Jiang, Y.; Liao, Y.; Chen, B.; Zhang, X. ADP-ribosylation factor-like GTPase 15 enhances insulin-induced AKT phosphorylation in the IR/IRS1/AKT pathway by interacting with ASAP2 and regulating PDPK1 activity. Biochem. Biophys. Res. Commun. 2017, 486, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.Y.; Mellet, N.; Thai, L.M.; Meikle, P.J.; Biden, T.J. Short-term inhibition of autophagy benefits pancreatic beta-cells by augmenting ether lipids and peroxisomal function, and by countering depletion of n-3 polyunsaturated fatty acids after fat-feeding. Mol. Metab. 2020, 40, 101023. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Chu, W.S.; Hale, T.C.; Wang, X.; Craig, R.L.; Wang, H.; Shuldiner, A.R.; Froguel, P.; Deloukas, P.; McCarthy, M.I.; et al. Polymorphisms in the glucokinase-associated, dual-specificity phosphatase 12 (DUSP12) gene under chromosome 1q21 linkage peak are associated with type 2 diabetes. Diabetes 2006, 55, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Talerico, M.; Berget, S.M. Effect of 5’ splice site mutations on splicing of the preceding intron. Mol. Cell. Biol. 1990, 10, 6299–6305. [Google Scholar] [CrossRef] [Green Version]
- Roca, X.; Olson, A.J.; Rao, A.R.; Enerly, E.; Kristensen, V.N.; Børresen-Dale, A.L.; Andresen, B.S.; Krainer, A.R.; Sachidanandam, R. Features of 5’-splice-site efficiency derived from disease-causing mutations and comparative genomics. Genome Res. 2008, 18, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Rajan, A.S.; Aguilar-Bryan, L.; Nelson, D.A.; Yaney, G.C.; Hsu, W.H.; Kunze, D.L.; Boyd, A.E., 3rd. Ion channels and insulin secretion. Diabetes Care 1990, 13, 340–363. [Google Scholar] [CrossRef]
- Aguilar-Bryan, L.; Nichols, C.G.; Wechsler, S.W.; Clement, J.P.; Boyd, A.E., 3rd; González, G.; Herrera-Sosa, H.; Nguy, K.; Bryan, J.; Nelson, D.A. Cloning of the beta cell high-affinity sulfonylurea receptor: A regulator of insulin secretion. Science 1995, 268, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Edghill, E.L.; Flanagan, S.E.; Ellard, S. Permanent neonatal diabetes due to activating mutations in ABCC8 and KCNJ11. Rev. Endocr. Metab. Disord. 2010, 11, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Nestorowicz, A.; Glaser, B.; Wilson, B.A.; Shyng, S.L.; Nichols, C.G.; Stanley, C.A.; Thornton, P.S.; Permutt, M.A. Genetic heterogeneity in familial hyperinsulinism. Hum. Mol. Genet. 1998, 7, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluth, O.; Matzke, D.; Schulze, G.; Schwenk, R.W.; Joost, H.G.; Schürmann, A. Differential transcriptome analysis of diabetes-resistant and -sensitive mouse islets reveals significant overlap with human diabetes susceptibility genes. Diabetes 2014, 63, 4230–4238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwerbel, K.; Kamitz, A.; Krahmer, N.; Hallahan, N.; Jähnert, M.; Gottmann, P.; Lebek, S.; Schallschmidt, T.; Arends, D.; Schumacher, F.; et al. Immunity-related GTPase induces lipophagy to prevent excess hepatic lipid accumulation. J. Hepatol. 2020, 73, 771–782. [Google Scholar] [CrossRef]
- Bazrgar, M.; Khodabakhsh, P.; Mohagheghi, F.; Prudencio, M.; Ahmadiani, A. Brain microRNAs dysregulation: Implication for missplicing and abnormal post-translational modifications of tau protein in Alzheimer’s disease and related tauopathies. Pharmacol. Res. 2020, 155, 104729. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.Y.; Delay, C.; Girard, J.; Papon, M.A.; Planel, E.; Sergeant, N.; Buee, L.; Hebert, S.S. MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum. Mol. Genet. 2011, 20, 4016–4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.E.; Park, C.; Kim, K.E.; Kim, K.K. Histone and RNA-binding protein interaction creates crosstalk network for regulation of alternative splicing. Biochem. Biophys. Res. Commun. 2018, 499, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.H.; Gonzalez, C.; Tailor, N.; Hamedani, M.K.; Leygue, E.; Davie, J.R. Dynamic Histone Acetylation of H3K4me3 Nucleosome Regulates MCL1 Pre-mRNA Splicing. J. Cell Physiol. 2016, 231, 2196–2204. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Type | Psi_NZO | Psi_obob | DeltaPsi | Probability | Category | Log2FC | p-Value | Expression Change | #snps Intronic Upstream | #snps 3 Prime ss Adjacent | #snps Exonic | #snps 5 Prime ss Adjacent | #snps Intronic Downstream | #snps Total | #CG | Phenotypes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abcc8 | CE | 0.039 | 0.999 | –0.959 | 1 | strong exclusion | –0.14 | 6.35 × 10–3 | not significant | 0 | 0 | 0 | 1 | 0 | 1 | 6 | Impaired glucose tolerance |
| Tcf7l2 | CE | 0.079 | 0.629 | –0.551 | 0.98 | strong exclusion | –0.71 | 1.52 × 10–5 | significant | 1 | 0 | 0 | 0 | 1 | 2 | 3 | Incomplete penetrance (HOM), decrease total body fat |
| Pbx4 | CE | 0.237 | 0.647 | –0.410 | 0.95 | moderate exclusion | –1.29 | 7.82 × 10–4 | significant | 1 | 0 | 0 | 0 | 5 | 6 | 3 | Not tested |
| Nudt6 | CE | 0.104 | 0.488 | –0.385 | 0.952 | moderate exclusion | 0.13 | 3.01 × 10–1 | not significant | 11 | 2 | 0 | 1 | 5 | 19 | 0 | Not tested |
| Tpcn2 | CE | 0.562 | 0.863 | –0.301 | 0.909 | moderate exclusion | –1.26 | 5.51 × 10–11 | significant | 1 | 0 | 0 | 0 | 7 | 8 | 3 | Increased circulating triglyceride level |
| Zfp945 | CE | 0.143 | 0.424 | –0.281 | 0.919 | moderate exclusion | –0.66 | 1.45 × 10–5 | significant | 0 | 0 | 0 | 0 | 2 | 2 | 3 | Not tested |
| Myt1l | CE | 0.746 | 0.984 | –0.238 | 0.995 | moderate exclusion | –0.05 | 5.68 × 10–1 | not significant | 6 | 3 | 3 | 1 | 14 | 27 | 0 | Decreased circulating glycerol level |
| Aktip | CE | 0.217 | 0.061 | 0.155 | 0.995 | moderate inclusion | –0.02 | 7.07 × 10–1 | not significant | 1 | 0 | 1 | 0 | 4 | 6 | 0 | Decreased subcutaneous adipose tissue amount, decreased total body fat amount, decreased body weight |
| Fbrsl1 | RI | 0.295 | 0.114 | 0.180 | 0.982 | moderate inclusion | 0.13 | 1.69 × 10–1 | not significant | 1 | 0 | 1 | 0 | 2 | 4 | 15 | Preweaning lethality, incomplete penetrance (HOM) |
| Nrxn1 | CE | 0.908 | 0.568 | 0.340 | 0.975 | moderate inclusion | –2.29 | 3.09 × 10–61 | significant | 1 | 0 | 0 | 0 | 0 | 1 | 1 | Decreased fasting circulating glucose level, impaired glucose tolerance, decreased circulating HDL cholesterol level, decreased body fat amount, decreased circulating cholesterol level |
| Nrxn1 | CE | 0.917 | 0.569 | 0.348 | 0.979 | moderate inclusion | –2.29 | 3.09 × 10–61 | significant | 1 | 0 | 0 | 0 | 1 | 2 | 0 | Decreased fasting circulating glucose level, impaired glucose tolerance, decreased circulating HDL cholesterol level, decreased body fat amount, decreased circulating cholesterol level |
| Arl15 | CE | 0.613 | 0.103 | 0.510 | 0.995 | strong inclusion | –0.17 | 3.31 × 10–2 | not significant | 5 | 1 | 0 | 0 | 4 | 10 | 1 | Preweaning lethality, complete penetrance (HOM) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilhelmi, I.; Neumann, A.; Jähnert, M.; Ouni, M.; Schürmann, A. Enriched Alternative Splicing in Islets of Diabetes-Susceptible Mice. Int. J. Mol. Sci. 2021, 22, 8597. https://doi.org/10.3390/ijms22168597
Wilhelmi I, Neumann A, Jähnert M, Ouni M, Schürmann A. Enriched Alternative Splicing in Islets of Diabetes-Susceptible Mice. International Journal of Molecular Sciences. 2021; 22(16):8597. https://doi.org/10.3390/ijms22168597
Chicago/Turabian StyleWilhelmi, Ilka, Alexander Neumann, Markus Jähnert, Meriem Ouni, and Annette Schürmann. 2021. "Enriched Alternative Splicing in Islets of Diabetes-Susceptible Mice" International Journal of Molecular Sciences 22, no. 16: 8597. https://doi.org/10.3390/ijms22168597
APA StyleWilhelmi, I., Neumann, A., Jähnert, M., Ouni, M., & Schürmann, A. (2021). Enriched Alternative Splicing in Islets of Diabetes-Susceptible Mice. International Journal of Molecular Sciences, 22(16), 8597. https://doi.org/10.3390/ijms22168597