
Long-Chain Acyl-Carnitines Interfere with Mitochondrial ATP Production Leading to Cardiac Dysfunction in Zebrafish

 ,
, 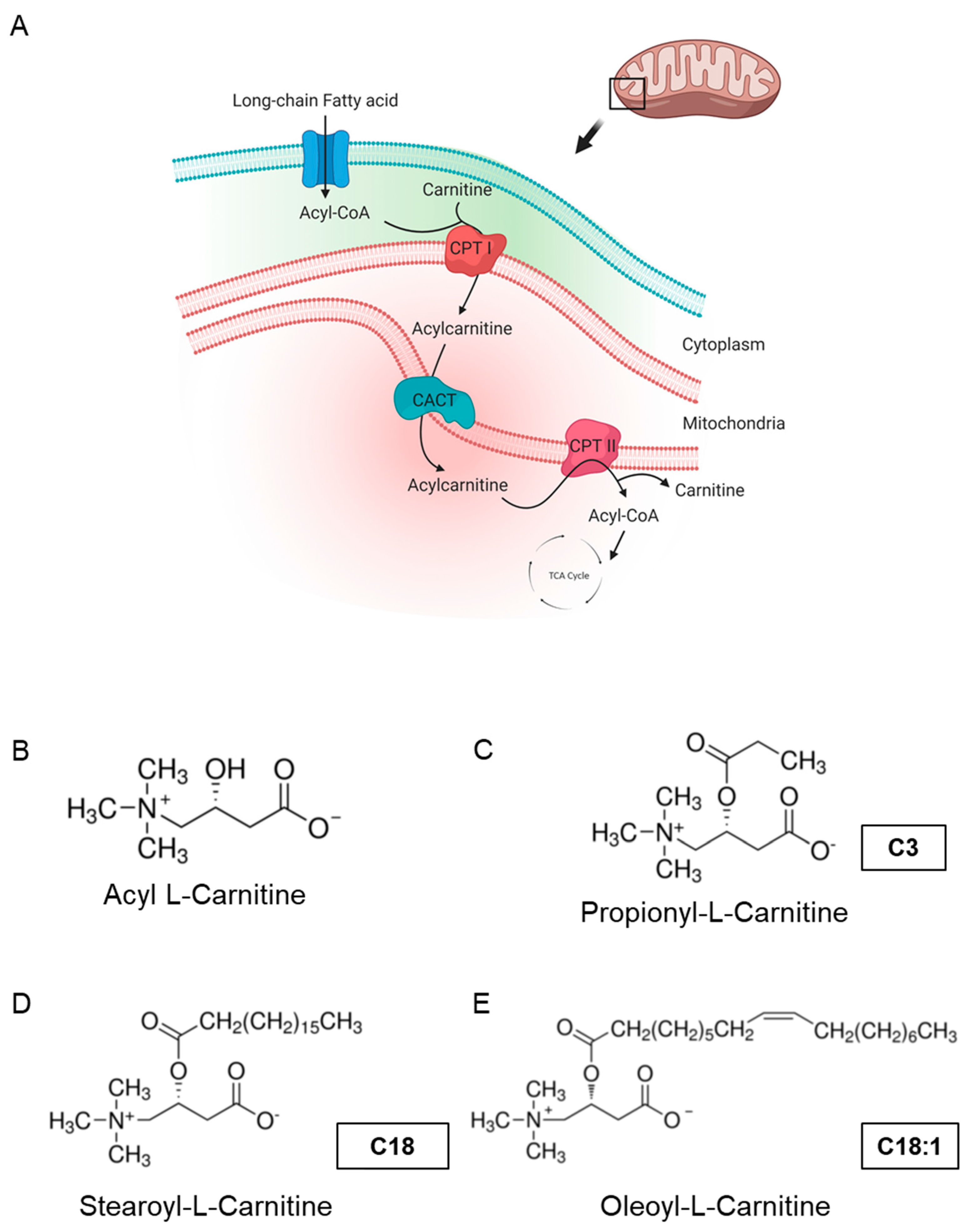{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
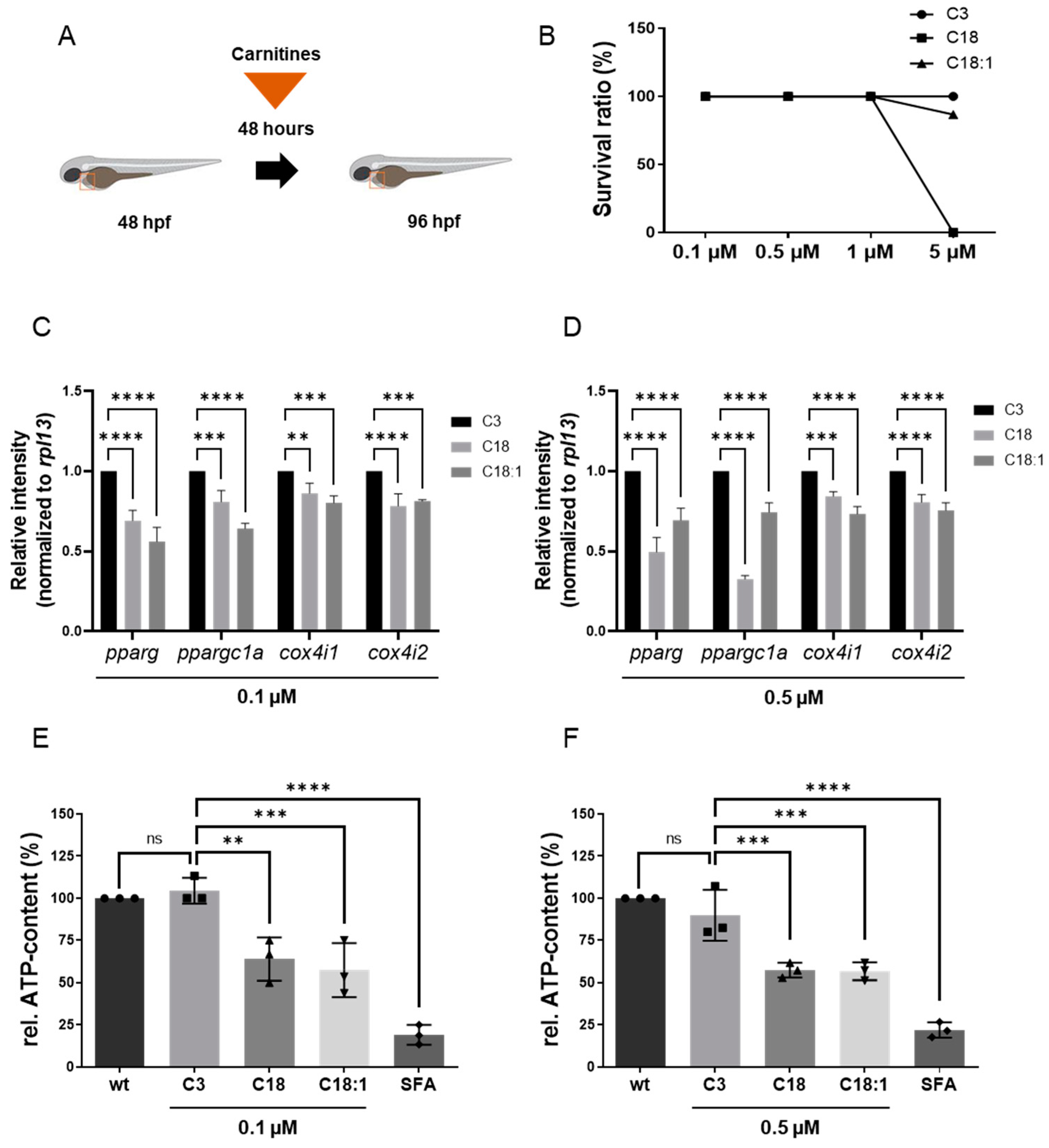
2.1. LCAC Treatment Interferes with Mitochondrial Function, Resulting in Diminished ATP Production
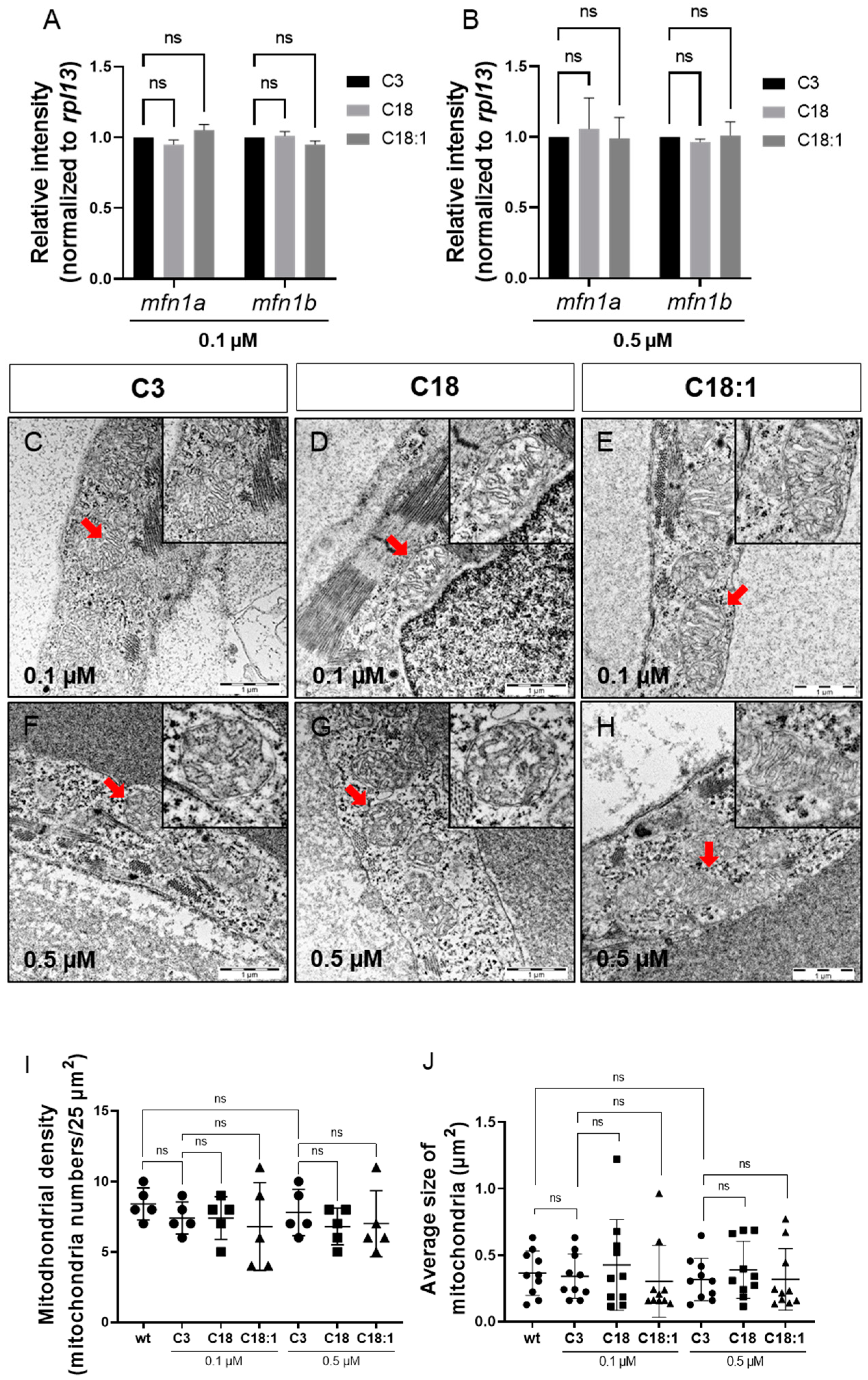
2.2. Mitochondrial Structure in Cardiomyocytes Is Not Impaired by LCAC Treatment
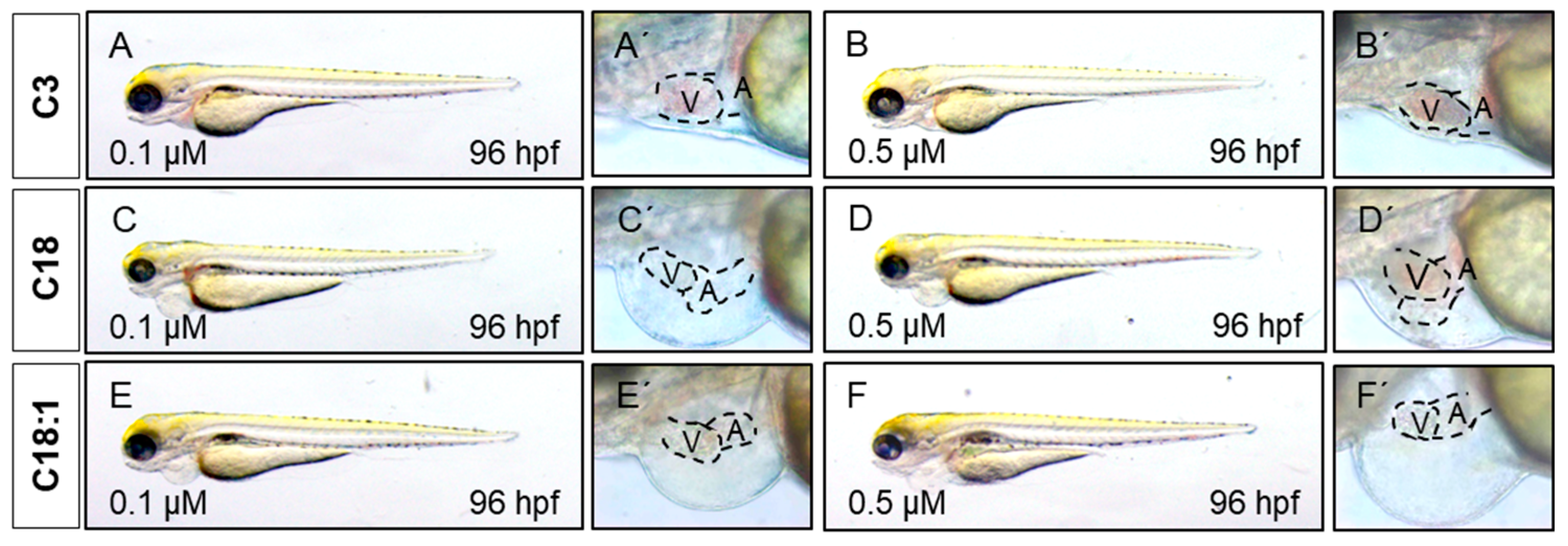
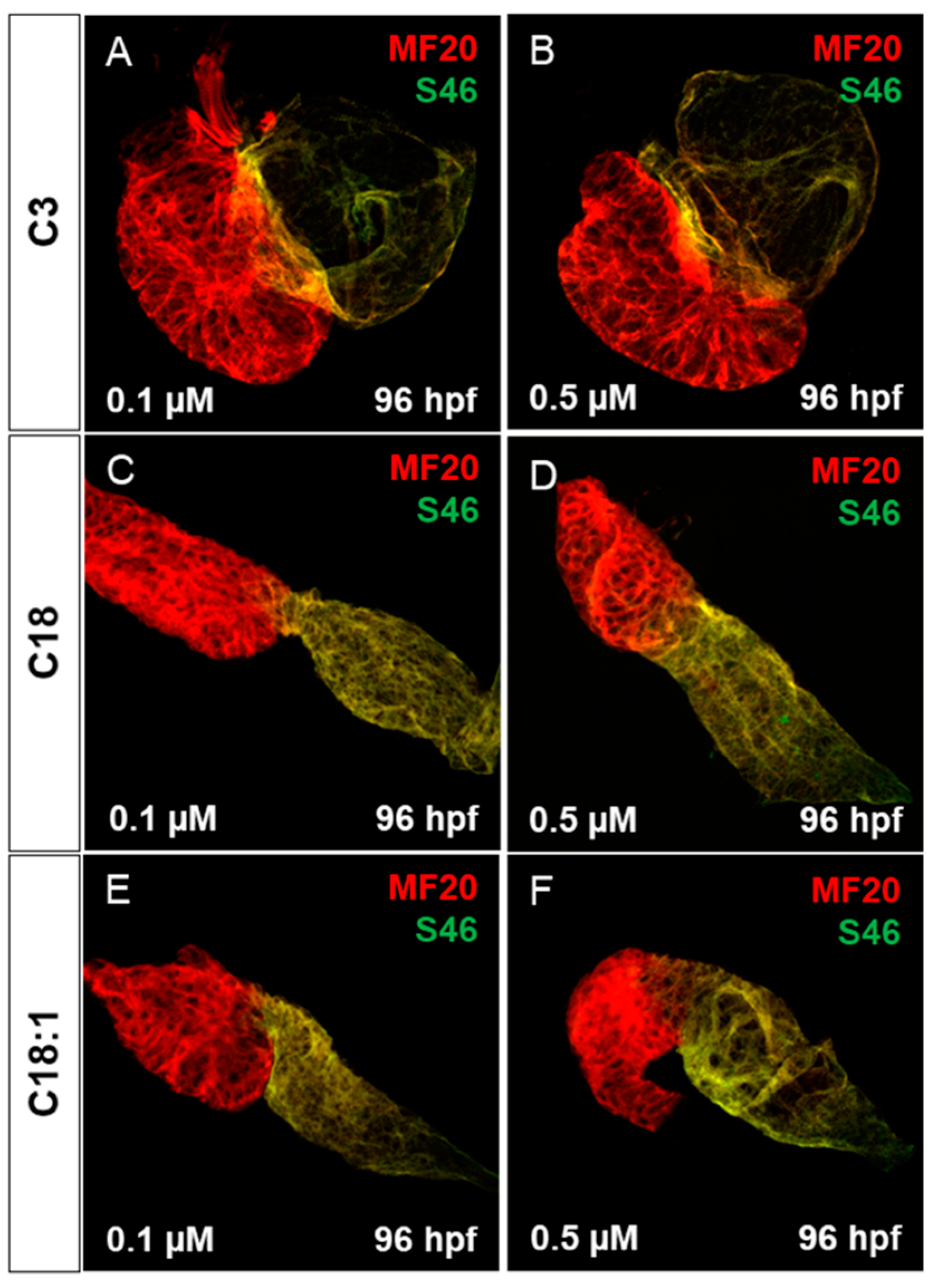
2.3. Treatment of Embryos with LCACs Leads to Cardiac Dysfunction
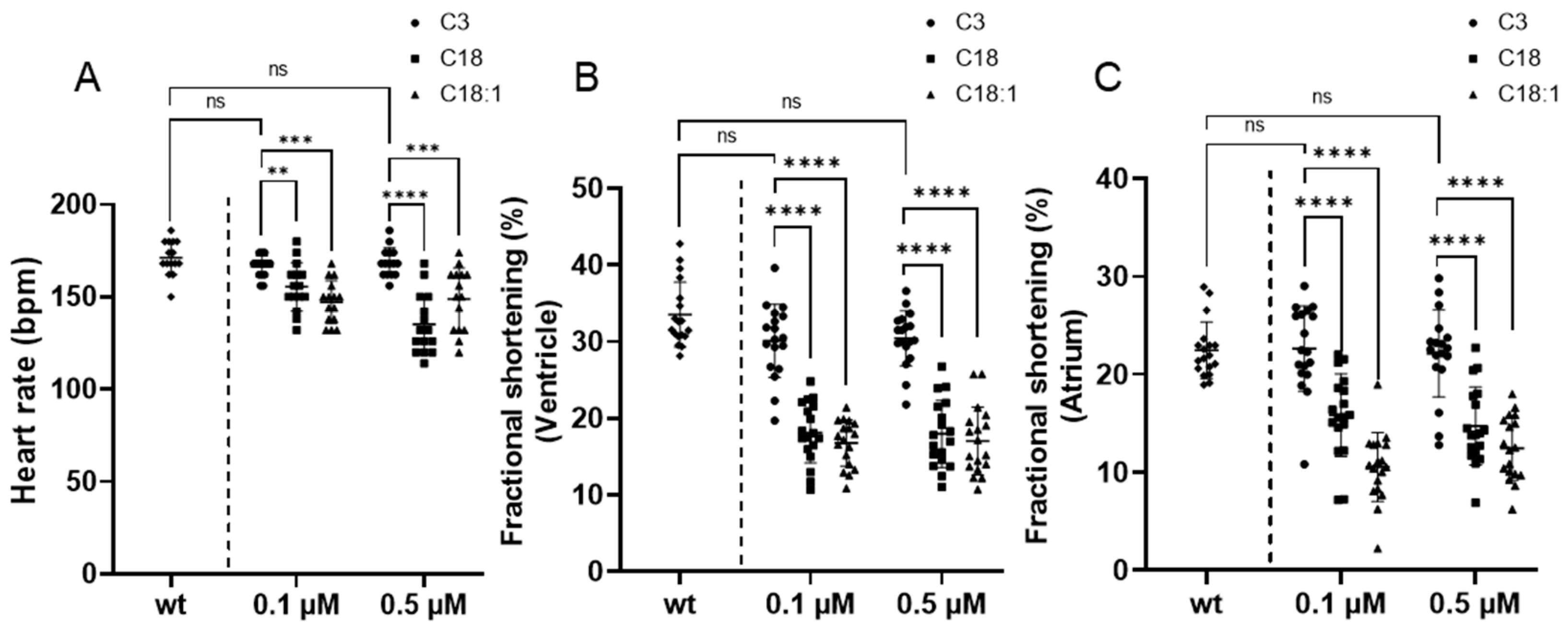
2.4. Long-Chain Acylcarnitine Treatment Impairs Cardiac Contractile Function
3. Discussion
4. Materials and Methods
4.1. Animals and Imaging
4.2. Acylcarnitine Preparation and Embryo Treatment
4.3. Fractional Shortening
4.4. MF20/S46 Immunostaining
4.5. Counting Cardiomyocytes
4.6. RNA Extraction and Quantitative Real-Time PCR
4.7. Histology
4.8. ATP Analysis
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Grevengoed, T.J.; Klett, E.L.; Coleman, R.A. Acyl-CoA metabolism and partitioning. Ann. Rev. Nutr. 2014, 34, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Knottnerus, S.J.G.; Bleeker, J.C.; Wüst, R.C.; Ferdinandusse, S.; IJlst, L.; Wijburg, F.A.; Wanders, R.J.; Visser, G.; Houtkooper, R.H. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev. Endocr. Metab. Disord. 2018, 19, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieber, L.L. Carnitine. Ann. Rev. Biochem. 1988, 57, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.H.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Crosslin, D.R.; Haynes, C.; Dungan, J.; Newby, L.K.; Hauser, E.R.; Ginsburg, G.S.; et al. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent car-diovascular events. Circ. Cardiovasc. Genet. 2010, 3, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Kalim, S.; Clish, C.B.; Wenger, J.; Elmariah, S.; Yeh, R.W.; Deferio, J.J.; Pierce, K.; Deik, A.; Gerszten, R.E.; Thadhani, R.; et al. A Plasma Long-Chain Acylcarnitine Predicts Cardiovascular Mortality in Incident Dialysis Patients. J. Am. Hear. Assoc. 2013, 2, e000542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizza, S.; Copetti, M.; Rossi, C.; Cianfarani, M.; Zucchelli, M.; Luzi, A.; Pecchioli, C.; Porzio, O.; Di Cola, G.; Urbani, A.; et al. Metabolomics signature improves the prediction of cardiovascular events in elderly subjects. Atheroscler 2014, 232, 260–264. [Google Scholar] [CrossRef]
- Zordoky, B.; Sung, M.M.; Ezekowitz, J.; Mandal, R.; Han, B.; Bjorndahl, T.C.; Bouatra, S.; Anderson, T.; Oudit, G.Y.; Wishart, D.S.; et al. Metabolomic Fingerprint of Heart Failure with Preserved Ejection Fraction. PLOS ONE 2015, 10, e0124844. [Google Scholar] [CrossRef]
- Krause, J.; Loeser, A.; Boernigen, D.; Schnabel, R.; Blankenberg, S.; Eschenhagen, T.; Stenzig, J.; Zeller, T. 433Metabolomics in translational medicine - A link between acylcarnitines and atrial fibrillation. Cardiovasc. Res. 2018, 114, S104. [Google Scholar] [CrossRef] [Green Version]
- Verdonschot, J.A.; Wang, P.; Van Bilsen, M.; Hazebroek, M.R.; Merken, J.J.; Vanhoutte, E.K.; Henkens, M.T.; Wijngaard, A.V.D.; Glatz, J.F.; Krapels, I.P.; et al. Metabolic Profiling Associates with Disease Severity in Nonischemic Dilated Cardiomyopathy. J. Card. Fail. 2020, 26, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Martin, D.; De Lonlay, P.; Villain, E.; Jouvet, P.; Rabier, D.; Brivet, M.; Saudubray, J.-M. Arrhythmias and Conduction Defects as Presenting Symptoms of Fatty Acid Oxidation Disorders in Children. Circulation 1999, 100, 2248–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacRae, C.A.; Peterson, R.T. Zebrafish as tools for drug discovery. Nat. Rev. Drug Discov. 2015, 14, 721–731. [Google Scholar] [CrossRef]
- Pott, A.; Rottbauer, W.; Just, S. Streamlining drug discovery assays for cardiovascular disease using zebrafish. Expert Opin. Drug Discov. 2020, 15, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paone, C.; Diofano, F.; Park, D.-D.; Rottbauer, W.; Just, S. Genetics of Cardiovascular Disease: Fishing for Causality. Front. Cardiovasc. Med. 2018, 5, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asnani, A.; Peterson, R.T. The zebrafish as a tool to identify novel therapies for human cardiovascular disease. Dis. Model. Mech. 2014, 7, 763–767. [Google Scholar] [CrossRef] [Green Version]
- Milan, D.J.; Peterson, T.A.; Ruskin, J.N.; Peterson, R.T.; Macrae, C.A. Drugs That Induce Repolarization Abnormalities Cause Bradycardia in Zebrafish. Circulation 2003, 107, 1355–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, C.G.; Milan, D.J.; Grande, E.J.; Rottbauer, W.; A Macrae, C.; Fishman, M.C. High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat. Chem. Biol. 2005, 1, 263–264. [Google Scholar] [CrossRef]
- Chi, N.C.; Shaw, R.; Jungblut, B.; Huisken, J.; Ferrer, T.; Arnaout, R.; Scott, I.; Beis, D.; Xiao, T.; Baier, H.; et al. Genetic and Physiologic Dissection of the Vertebrate Cardiac Conduction System. PLoS Biol. 2008, 6, e109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, M.; Berger, I.M.; Just, S.; Rottbauer, W. Loss of dihydrolipoyl succinyltransferase (DLST) leads to reduced resting heart rate in the zebrafish. Basic Res. Cardiol. 2015, 110, 14. [Google Scholar] [CrossRef] [Green Version]
- Pott, A.; Bock, S.; Berger, I.M.; Frese, K.; Dahme, T.; Keßler, M.; Rinné, S.; Decher, N.; Just, S.; Rottbauer, W.; et al. Mutation of the Na+/K+-ATPase Atp1a1a.1 causes QT interval prolongation and bradycardia in zebrafish. J. Mol. Cell. Cardiol. 2018, 120, 42–52. [Google Scholar] [CrossRef]
- Berezhnov, A.V.; Fedotova, E.I.; Nenov, M.N.; Kasymov, V.A.; Pimenov, O.Y.; Dynnik, V.V. Dissecting cellular mechanisms of long-chain acylcarnitines-driven cardiotoxicity: Disturbance of calcium homeostasis, activation of Ca(2+)-dependent phospholipases, and mitochondrial energetics collapse. Int. J. Mol. Sci. 2020, 21, 7461. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.; Fu, Y.; Ma, J.; Li, X.; Lu, C.; Zhang, R. Functional alterations and transcriptomic changes during zebrafish cardiac aging. Biogerontology 2020, 21, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef]
- Pinho, B.R.; Santos, M.M.; Fonseca-Silva, A.; Valentao, P.; Andrade, P.B.; Oliveira, J.M. How mitochondrial dysfunction affects zebrafish development and cardiovascular function: An in vivo model for testing mitochondria-targeted drugs. Br. J. Pharmacol. 2013, 169, 1072–1090. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.; Leone, T.C.; Keller, M.P.; Martin, O.J.; Broman, A.T.; Nigro, J.; Kapoor, K.; Koves, T.R.; Stevens, R.; Ilkayeva, O.R.; et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: A multisystems approach. Circ. Heart Fail 2014, 7, 1022–1031. [Google Scholar] [CrossRef] [Green Version]
- McGarrah, R.W.; Crown, S.B.; Zhang, G.F.; Shah, S.H.; Newgard, C.B. Cardiovascular metabolomics. Circ. Res. 2018, 122, 1238–1258. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Kelly, J.P.; McGarrah, R.W.; Hellkamp, A.S.; Fiuzat, M.; Testani, J.M.; Wang, T.S.; Verma, A.; Samsky, M.D.; Donahue, M.P.; et al. Prognostic implications of long-chain acylcarnitines in heart failure and reversibility with mechanical circulatory support. J. Am. Coll. Cardiol. 2016, 67, 291–299. [Google Scholar] [CrossRef]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 2008, 80, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taegtmeyer, H.; Lubrano, G. Rethinking cardiac metabolism: Metabolic cycles to refuel and rebuild the failing heart. F1000Prime Rep. 2014, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Gramer, G.; Haege, G.; Fang-Hoffmann, J.; Schwab, K.O.; Tacke, U.; Trefz, F.K.; Mengel, E.; Wendel, U.; Leichsenring, M.; et al. Efficacy and outcome of expanded newborn screening for metabolic diseases—Report of 10 years from South-West Germany. Orphanet. J. Rare Dis. 2011, 6, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef] [Green Version]
- Ingwall, J.S. Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 2009, 81, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Ingwall, J.S.; Weiss, R.G. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ. Res. 2004, 95, 135–145. [Google Scholar] [CrossRef]
- Hunter, W.G.; Kelly, J.P.; McGarrah, R.W., 3rd; Khouri, M.G.; Craig, D.; Haynes, C.; Ilkayeva, O.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.J.; et al. Metabolomic profiling identifies novel circulating biomarkers of mitochondrial dysfunction differentially elevated in heart failure with preserved versus reduced ejection fraction: Evidence for shared metabolic impairments in clinical heart failure. J. Am. Heart Assoc. 2016, 5, e003190. [Google Scholar] [CrossRef] [Green Version]
- Vissing, C.R.; Hedermann, G.; Vissing, J. Moderate-intensity aerobic exercise improves physical fitness in bethlem myopathy. Muscle Nerve 2019, 60, 183–188. [Google Scholar] [CrossRef]
- McCann, M.R.; George De la Rosa, M.V.; Rosania, G.R.; Stringer, K.A. L-carnitine and acylcarnitines: Mitochondrial biomarkers for precision medicine. Metabolites 2021, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Aitken-Buck, H.M.; Krause, J.; Zeller, T.; Jones, P.P.; Lamberts, R.R. Long-chain acylcarnitines and cardiac excitation-contraction coupling: Links to arrhythmias. Front. Physiol. 2020, 11, 577856. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Kim, T.; Long, Q.; Liu, J.; Wang, P.; Zhou, Y.; Ding, Y.; Prasain, J.; Wood, P.A.; Yang, Q. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation 2012, 126, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Haynie, K.R.; Vandanmagsar, B.; Wicks, S.E.; Zhang, J.; Mynatt, R.L. Inhibition of carnitine palymitoyltransferase1b induces cardiac hypertrophy and mortality in mice. Diabetes Obes. Metab. 2014, 16, 757–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Fang, X.; Dai, M.; Cao, Q.; Tan, T.; He, W.; Huang, Y.; Chu, L.; Bao, M. Cardiac-specific down-regulation of carnitine palmitoyltransferase-1b (CPT-1b) prevents cardiac remodeling in obese mice. Obesity 2016, 24, 2533–2543. [Google Scholar] [CrossRef] [Green Version]
- Makrecka-Kuka, M.; Korzh, S.; Videja, M.; Vilskersts, R.; Sevostjanovs, E.; Zharkova-Malkova, O.; Arsenyan, P.; Kuka, J.; Dambrova, M.; Liepinsh, E. Inhibition of CPT2 exacerbates cardiac dysfunction and inflammation in experimental endotoxaemia. J. Cell Mol. Med. 2020, 24, 11903–11911. [Google Scholar] [CrossRef] [PubMed]
- Meder, B.; Scholz, E.P.; Hassel, D.; Wolff, C.; Just, S.; Berger, I.M.; Patzel, E.; Karle, C.; Katus, H.A.; Rottbauer, W. Reconstitution of defective protein trafficking rescues Long-QT syndrome in zebrafish. Biochem. Biophys. Res. Commun. 2011, 408, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Rottbauer, W.; Baker, K.; Wo, Z.G.; Mohideen, M.A.; Cantiello, H.F.; Fishman, M.C. Growth and function of the embryonic heart depend upon the cardiac-specific L-type calcium channel alpha1 subunit. Dev. Cell 2001, 1, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, A.T.; Bradberry, S.M.; Vale, J.A. Sodium fluoroacetate poisoning. Toxicol. Rev. 2006, 25, 213–219. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, D.-D.; Gahr, B.M.; Krause, J.; Rottbauer, W.; Zeller, T.; Just, S. Long-Chain Acyl-Carnitines Interfere with Mitochondrial ATP Production Leading to Cardiac Dysfunction in Zebrafish. Int. J. Mol. Sci. 2021, 22, 8468. https://doi.org/10.3390/ijms22168468
Park D-D, Gahr BM, Krause J, Rottbauer W, Zeller T, Just S. Long-Chain Acyl-Carnitines Interfere with Mitochondrial ATP Production Leading to Cardiac Dysfunction in Zebrafish. International Journal of Molecular Sciences. 2021; 22(16):8468. https://doi.org/10.3390/ijms22168468
Chicago/Turabian StylePark, Deung-Dae, Bernd M. Gahr, Julia Krause, Wolfgang Rottbauer, Tanja Zeller, and Steffen Just. 2021. "Long-Chain Acyl-Carnitines Interfere with Mitochondrial ATP Production Leading to Cardiac Dysfunction in Zebrafish" International Journal of Molecular Sciences 22, no. 16: 8468. https://doi.org/10.3390/ijms22168468
APA StylePark, D.-D., Gahr, B. M., Krause, J., Rottbauer, W., Zeller, T., & Just, S. (2021). Long-Chain Acyl-Carnitines Interfere with Mitochondrial ATP Production Leading to Cardiac Dysfunction in Zebrafish. International Journal of Molecular Sciences, 22(16), 8468. https://doi.org/10.3390/ijms22168468