
Non-Rodent Genetic Animal Models for Studying Tauopathy: Review of Drosophila, Zebrafish, and C. elegans Models




Abstract
1. Introduction
2. Underlying Mechanisms of Tauopathy
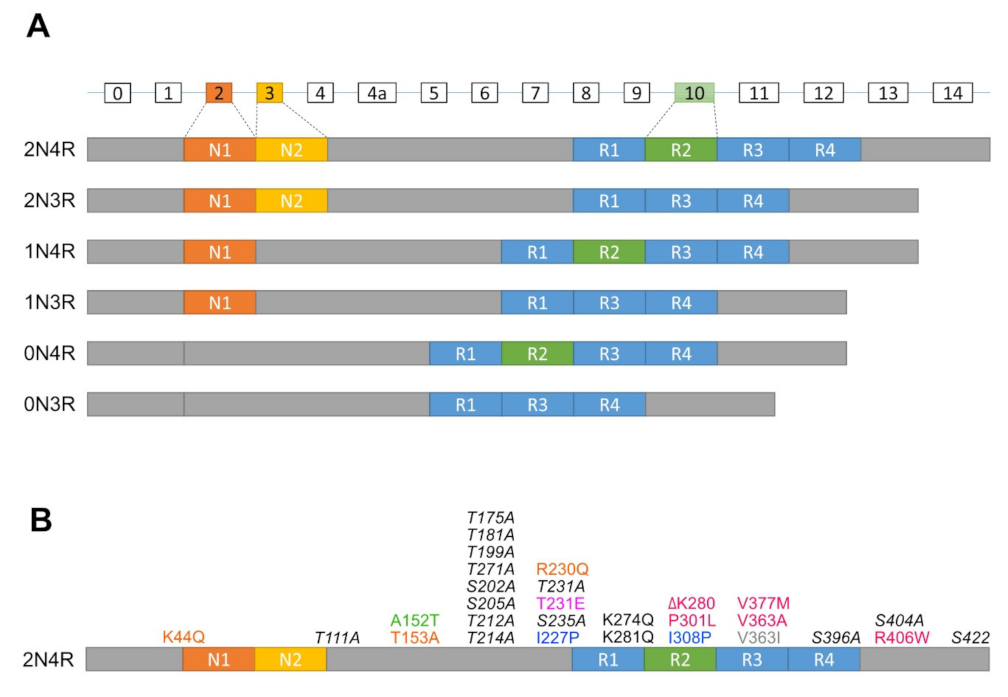
2.1. Normal Function of Tau
2.2. Phosphorylation of Tau
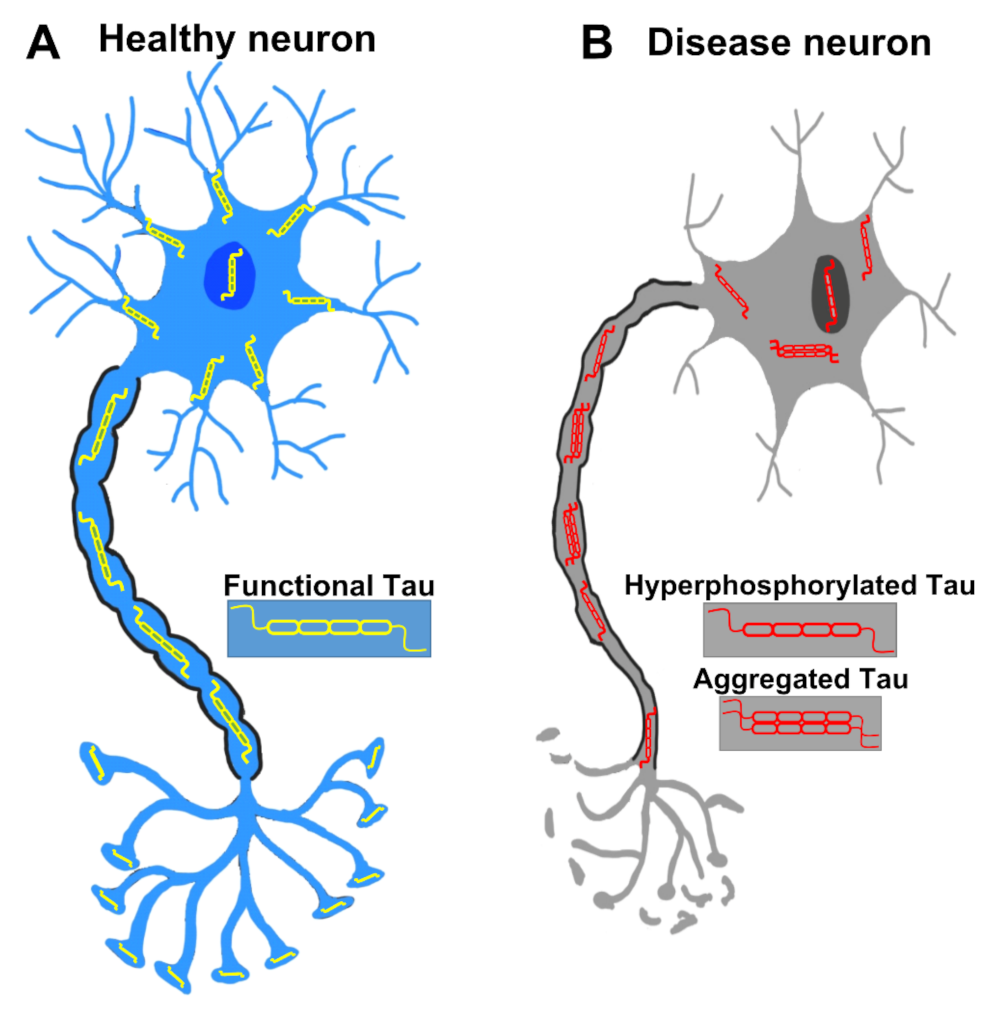
2.3. Aggregation of Tau
2.4. Toxicity of Tau
2.5. Clearance of Tau
2.5.1. Tau Is Degraded by the Proteasome
2.5.2. Tau Is Degraded by Autophagy
3. Drosophila Tauopathy Model
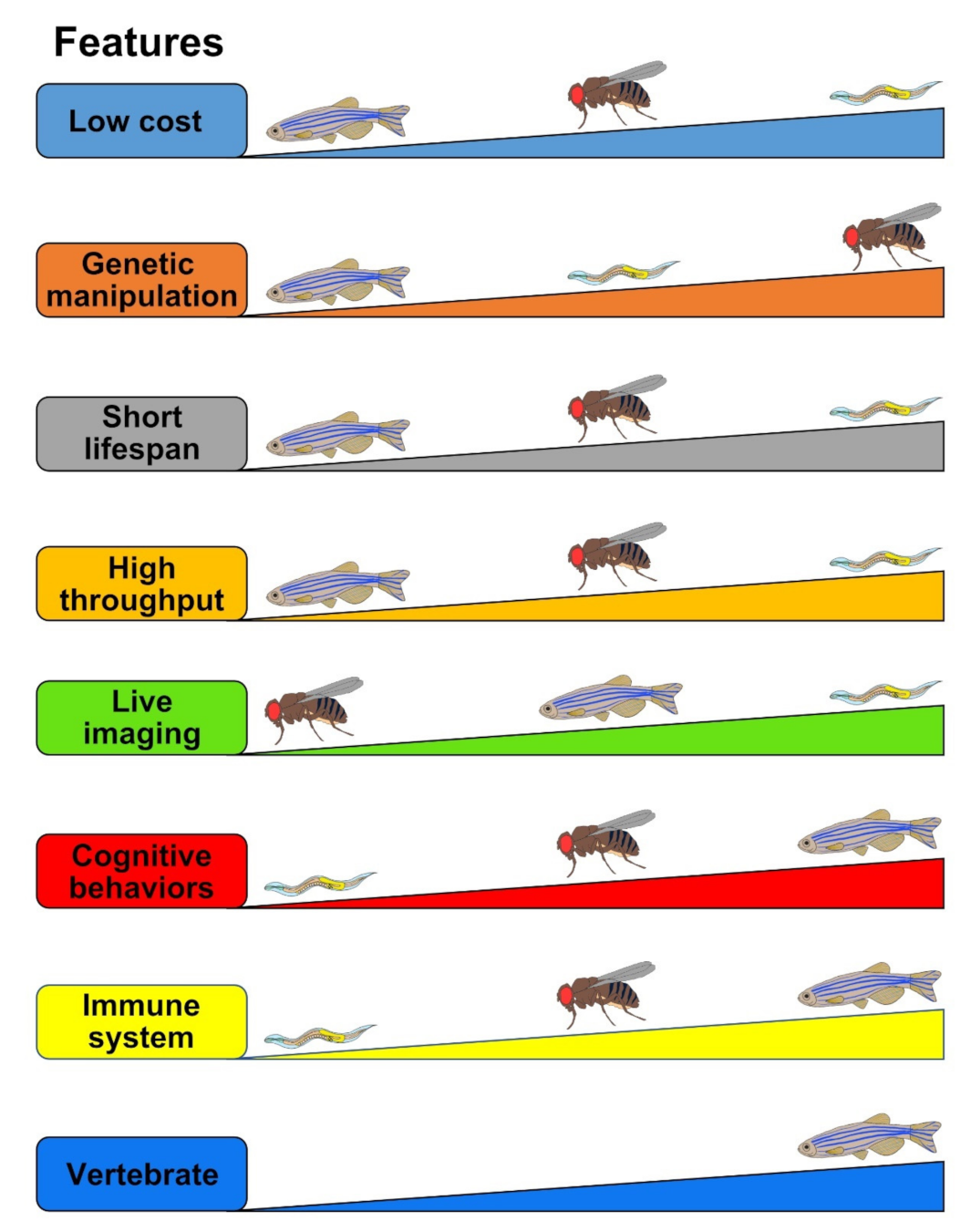
3.1. Advantages
3.2. Current Drosophila Tauopathy Models
3.3. Representative Assays
3.3.1. REP
3.3.2. Neuronal Cell Death and Neurodegeneration
3.3.3. Learning and Memory Assays
3.3.4. Lifespan
3.3.5. NMJ and Locomotion
3.3.6. Transposon Mobility
3.4. Limitations
3.5. The Utility of the Drosophila Tauopathy Models and Their Translational Applications
4. Zebrafish Tauopathy Model
4.1. Advantages
4.2. Current Zebrafish Tauopathy Models
4.3. Representative Assays
4.3.1. Neuronal Cell Death
4.3.2. Axonopathies
4.3.3. Locomotion
4.3.4. Ubiquitin-Proteasome System (UPS) and Autophagy-Lysosomal Pathway (ALP)
4.4. Limitations
4.5. The Utility of the Zebrafish Tauopathy Models and Their Translational Applications
5. C. elegans Tauopathy Models
5.1. Advantages
5.2. Current C. elegans Tauopathy Models
5.3. Representative Assays
5.3.1. Lifespan
5.3.2. Neuronal Death
5.3.3. Axonal Defects
5.3.4. Behavior Phenotypes
5.4. Limitations
5.5. The Utility of the C. elegans Tauopathy Models and Their Translational Applications
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chi, H.; Sang, T.-K.; Chang, H.-Y. Tauopathy. In Cognitive Disorders; IntechOpen: London, UK, 2018. [Google Scholar]
- Spillantini, M.G.; Goedert, M.; Crowther, R.A.; Murrell, J.R.; Farlow, M.R.; Ghetti, B. Familial multiple system tauopathy with presenile dementia: A disease with abundant neuronal and glial tau filaments. Proc. Natl. Acad. Sci. USA 1997, 94, 4113–4118. [Google Scholar] [CrossRef]
- Gotz, J.; Halliday, G.; Nisbet, R.M. Molecular Pathogenesis of the Tauopathies. Annu Rev. Pathol 2019, 14, 239–261. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.-Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef]
- Brion, J.P.; Couck, A.; Passareiro, E.; Flament-Durand, J. Neurofibrillary tangles of Alzheimer’s disease: AN immunohistochemical study. J. Submicrosc. Cytol. 1985, 17, 89–96. [Google Scholar]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.-C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Wischik, C.; Crowther, R.; Walker, J.; Klug, A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. USA 1988, 85, 4051–4055. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.; Novak, M.; Edwards, P.; Klug, A.; Tichelaar, W.; Crowther, R. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.; Novak, M.; Thøgersen, H.; Edwards, P.; Runswick, M.; Jakes, R.; Walker, J.; Milstein, C.; Roth, M.; Klug, A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4506–4510. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Colin, M.; Buée, L. Invited review: Animal models of tauopathies and their implications for research/translation into the clinic. Neuropathol. Appl. Neurobiol. 2015, 41, 59–80. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.; Potier, M.; Ulrich, J.; Crowther, R. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [CrossRef]
- D’Souza, I.; Schellenberg, G.D. Regulation of tau isoform expression and dementia. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1739, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.; Goda, Y. The actin cytoskeleton: Integrating form and function at the synapse. Annu. Rev. Neurosci. 2005, 28, 25–55. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.-C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111, 3167–3177. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef]
- Brandt, R.; Léger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef]
- Pooler, A.M.; Usardi, A.; Evans, C.J.; Philpott, K.L.; Noble, W.; Hanger, D.P. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol. Aging 2012, 33, 431.e27–431.e38. [Google Scholar] [CrossRef]
- Oliveira, J.; Costa, M.; de Almeida, M.S.C.; da Cruz e Silva, O.A.; Henriques, A.G. Protein phosphorylation is a key mechanism in Alzheimer’s disease. J. Alzheimer Dis. 2017, 58, 953–978. [Google Scholar] [CrossRef]
- Kanemaru, K.; Takio, K.; Miura, R.; Titani, K.; Ihara, Y. Fetal-type phosphorylation of the τ in paired helical filaments. J. Neurochem. 1992, 58, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.P.; Peng, C.X.; Wei, W.; Tian, Q.; Liu, Y.H.; Yao, X.Q.; Zhang, Y.; Cao, F.Y.; Wang, Q.; Wang, J.Z. Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus 2010, 20, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.-H.; Chang, J.-R.; Zhang, J.; Zhang, B.-H.; Li, Y.-L.; Teng, X.; Zhu, Y.; Du, J.; Tang, C.-S.; Qi, Y.-F. Activating transcription factor 4 is involved in endoplasmic reticulum stress-mediated apoptosis contributing to vascular calcification. Apoptosis 2013, 18, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Köpke, E.; Tung, Y.-C.; Shaikh, S.; Alonso, A.d.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Götz, J. Tau-based therapies in neurodegeneration: Opportunities and challenges. Nat. Rev. Drug Discov. 2017, 16, 863–883. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.-H.; Zhang, Y.; Hong, X.-Y.; Zhang, J.-F.; Wang, J.-Z.; Liu, G.-P. Role of microtubule-associated protein tau phosphorylation in Alzheimer’s disease. J. Huazhong Univ. Sci. Technol. [Med. Sci.] 2017, 37, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Rosseels, J.; Van den Brande, J.; Violet, M.; Jacobs, D.; Grognet, P.; Lopez, J.; Huvent, I.; Caldara, M.; Swinnen, E.; Papegaey, A. Tau monoclonal antibody generation based on humanized yeast models: Impact on Tau oligomerization and diagnostics. J. Biol. Chem. 2015, 290, 4059–4074. [Google Scholar] [CrossRef]
- Vega, I.E.; Cui, L.; Propst, J.A.; Hutton, M.L.; Lee, G.; Yen, S.-H. Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates. Mol. Brain Res. 2005, 138, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927. [Google Scholar] [CrossRef]
- Sontag, E.; Hladik, C.; Montgomery, L.; Luangpirom, A.; Mudrak, I.; Ogris, E.; White, C.L., III. Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J. Neuropathol. Exp. Neurol. 2004, 63, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, B.; Grundke-Iqbal, I.; Iqbal, K. I PP2A 1 affects Tau phosphorylation via association with the catalytic subunit of protein phosphatase 2A. J. Biol. Chem. 2008, 283, 10513–10521. [Google Scholar] [CrossRef]
- Von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.-M.; Mandelkow, E. Assembly of τ protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Khlistunova, I.; Biernat, J.; Wang, Y.; Pickhardt, M.; von Bergen, M.; Gazova, Z.; Mandelkow, E.; Mandelkow, E.-M. Inducible expression of Tau repeat domain in cell models of tauopathy: Aggregation is toxic to cells but can be reversed by inhibitor drugs. J. Biol. Chem. 2006, 281, 1205–1214. [Google Scholar] [CrossRef]
- Wille, H.; Drewes, G.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J. Cell Biol. 1992, 118, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.; Hasegawa, M.; Smith, M.; Crowther, R. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef]
- Fichou, Y.; Oberholtzer, Z.R.; Ngo, H.; Cheng, C.-Y.; Keller, T.J.; Eschmann, N.A.; Han, S. Tau-cofactor complexes as building blocks of tau fibrils. Front. Neurosci. 2019, 13, 1339. [Google Scholar] [CrossRef]
- Giustiniani, J.; Chambraud, B.; Sardin, E.; Dounane, O.; Guillemeau, K.; Nakatani, H.; Paquet, D.; Kamah, A.; Landrieu, I.; Lippens, G. Immunophilin FKBP52 induces Tau-P301L filamentous assembly in vitro and modulates its activity in a model of tauopathy. Proc. Natl. Acad. Sci. USA 2014, 111, 4584–4589. [Google Scholar] [CrossRef] [PubMed]
- Criado-Marrero, M.; Gebru, N.T.; Gould, L.A.; Blazier, D.M.; Vidal-Aguiar, Y.; Smith, T.M.; Abdelmaboud, S.S.; Shelton, L.B.; Wang, X.; Dahrendorff, J. FKBP52 overexpression accelerates hippocampal-dependent memory impairments in a tau transgenic mouse model. NPJ Aging Mech. Dis. 2021, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Braak, F.; Braak, H.; Mandelkow, E.-M. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994, 87, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.d.C.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Biernat, J.; Von Bergen, M.; Mandelkow, E.; Mandelkow, E.-M. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 1999, 38, 3549–3558. [Google Scholar] [CrossRef] [PubMed]
- Tepper, K.; Biernat, J.; Kumar, S.; Wegmann, S.; Timm, T.; Hübschmann, S.; Redecke, L.; Mandelkow, E.-M.; Müller, D.J.; Mandelkow, E. Oligomer formation of tau protein hyperphosphorylated in cells. J. Biol. Chem. 2014, 289, 34389–34407. [Google Scholar] [CrossRef] [PubMed]
- Zilka, N.; Filipcik, P.; Koson, P.; Fialova, L.; Skrabana, R.; Zilkova, M.; Rolkova, G.; Kontsekova, E.; Novak, M. Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006, 580, 3582–3588. [Google Scholar] [CrossRef]
- de Calignon, A.; Fox, L.M.; Pitstick, R.; Carlson, G.A.; Bacskai, B.J.; Spires-Jones, T.L.; Hyman, B.T. Caspase activation precedes and leads to tangles. Nature 2010, 464, 1201–1204. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, M.; Liu, X.; Kang, S.S.; Kwon, I.-S.; Duong, D.M.; Seyfried, N.T.; Hu, W.T.; Liu, Z.; Wang, J.-Z. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat. Med. 2014, 20, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Biernat, J.; Pickhardt, M.; Mandelkow, E.; Mandelkow, E.-M. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc. Natl. Acad. Sci. USA 2007, 104, 10252–10257. [Google Scholar] [CrossRef] [PubMed]
- Fatouros, C.; Pir, G.J.; Biernat, J.; Koushika, S.P.; Mandelkow, E.; Mandelkow, E.-M.; Schmidt, E.; Baumeister, R. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum. Mol. Genet. 2012, 21, 3587–3603. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Peeraer, E.; Bottelbergs, A.; Van Kolen, K.; Stancu, I.-C.; Vasconcelos, B.; Mahieu, M.; Duytschaever, H.; Ver Donck, L.; Torremans, A.; Sluydts, E. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol. Dis. 2015, 73, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Morsch, R.; Simon, W.; Coleman, P.D. Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 1999, 58, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; De Calignon, A.; Matsui, T.; Zehr, C.; Pitstick, R.; Wu, H.-Y.; Osetek, J.D.; Jones, P.B.; Bacskai, B.J.; Feany, M.B. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J. Neurosci. 2008, 28, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Sydow, A.; Van der Jeugd, A.; Zheng, F.; Ahmed, T.; Balschun, D.; Petrova, O.; Drexler, D.; Zhou, L.; Rune, G.; Mandelkow, E. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J. Neurosci. 2011, 31, 2511–2525. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeugd, A.; Hochgräfe, K.; Ahmed, T.; Decker, J.M.; Sydow, A.; Hofmann, A.; Wu, D.; Messing, L.; Balschun, D.; D’Hooge, R. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012, 123, 787–805. [Google Scholar] [CrossRef]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular tau oligomers as intermediates of tau filaments. Biochemistry 2007, 46, 3856–3861. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012, 26, 1946–1959. [Google Scholar] [CrossRef]
- Flach, K.; Hilbrich, I.; Schiffmann, A.; Gärtner, U.; Krüger, M.; Leonhardt, M.; Waschipky, H.; Wick, L.; Arendt, T.; Holzer, M. Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem. 2012, 287, 43223–43233. [Google Scholar] [CrossRef]
- Tian, H.; Davidowitz, E.; Lopez, P.; Emadi, S.; Moe, J.; Sierks, M. Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int. J. Cell Biol. 2013, 2013, 260787. [Google Scholar] [CrossRef] [PubMed]
- Gerson, J.E.; Farmer, K.M.; Henson, N.; Castillo-Carranza, D.L.; Murillo, M.C.; Sengupta, U.; Barrett, A.; Kayed, R. Tau oligomers mediate α-synuclein toxicity and can be targeted by immunotherapy. Mol. Neurodegener. 2018, 13, 1–14. [Google Scholar] [CrossRef]
- Alonso, A.d.C.; Li, B.; Grundke-Iqbal, I.; Iqbal, K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc. Natl. Acad. Sci. USA 2006, 103, 8864–8869. [Google Scholar] [CrossRef] [PubMed]
- LaPointe, N.E.; Morfini, G.; Pigino, G.; Gaisina, I.N.; Kozikowski, A.P.; Binder, L.I.; Brady, S.T. The amino terminus of tau inhibits kinesin-dependent axonal transport: Implications for filament toxicity. J. Neurosci. Res. 2009, 87, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Petrucelli, L.; Dawson, T. Mechanism of neurodegenerative disease: Role of the ubiquitin proteasome system. Ann. Med. 2004, 36, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Schwartz, D.; Gygi, S.P.; Kosik, K.S. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 2004, 279, 4869–4876. [Google Scholar] [CrossRef] [PubMed]
- Cripps, D.; Thomas, S.N.; Jeng, Y.; Yang, F.; Davies, P.; Yang, A.J. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J. Biol. Chem. 2006, 281, 10825–10838. [Google Scholar] [CrossRef] [PubMed]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Botzen, D.; Engels, M.; Voss, P.; Kaiser, B.; Jung, T.; Grimm, S.; Ermak, G.; Davies, K.J. Tau protein degradation is catalyzed by the ATP/ubiquitin-independent 20S proteasome under normal cell conditions. Arch. Biochem. Biophys. 2010, 500, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Bednarski, E.; Lynch, G. Cytosolic proteolysis of τ by cathepsin D in hippocampus following suppression of cathepsins B and L. J. Neurochem. 1996, 67, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Khurana, V.; Elson-Schwab, I.; Fulga, T.A.; Sharp, K.A.; Loewen, C.A.; Mulkearns, E.; Tyynela, J.; Scherzer, C.R.; Feany, M.B. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 2010, 6, e1001026. [Google Scholar] [CrossRef] [PubMed]
- Bendiske, J.; Bahr, B.A. Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis—an approach for slowing Alzheimer disease? J. Neuropathol. Exp. Neurol. 2003, 62, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Rispoli, J.; Kaphzan, H.; Klann, E.; Chen, E.I.; Kim, J.; Komatsu, M.; Abeliovich, A. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol. Neurodegener 2012, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- St Johnston, D. The art and design of genetic screens: Drosophila melanogaster. Nat. Rev. Genet. 2002, 3, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Tickoo, S.; Russell, S. Drosophila melanogaster as a model system for drug discovery and pathway screening. Curr. Opin. Pharmacol. 2002, 2, 555–560. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef]
- Gistelinck, M.; Lambert, J.C.; Callaerts, P.; Dermaut, B.; Dourlen, P. Drosophila models of tauopathies: What have we learned? Int. J. Alzheimer Dis. 2012, 2012, 970980. [Google Scholar] [CrossRef]
- Sivanantharajah, L.; Mudher, A.; Shepherd, D. An evaluation of Drosophila as a model system for studying tauopathies such as Alzheimer’s disease. J. Neurosci. Methods 2019, 319, 77–88. [Google Scholar] [CrossRef]
- Subramanian, M.; Hyeon, S.J.; Das, T.; Suh, Y.S.; Kim, Y.K.; Lee, J.S.; Song, E.J.; Ryu, H.; Yu, K. UBE4B, a microRNA-9 target gene, promotes autophagy-mediated Tau degradation. Nat. Commun. 2021, 12, 3291. [Google Scholar] [CrossRef]
- Heidary, G.; Fortini, M.E. Identification and characterization of the Drosophila tau homolog. Mech. Dev. 2001, 108, 171–178. [Google Scholar] [CrossRef]
- Traven, A.; Jelicic, B.; Sopta, M. Yeast Gal4: A transcriptional paradigm revisited. EMBO Rep. 2006, 7, 496–499. [Google Scholar] [CrossRef]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Karsten, S.L.; Sang, T.K.; Gehman, L.T.; Chatterjee, S.; Liu, J.; Lawless, G.M.; Sengupta, S.; Berry, R.W.; Pomakian, J.; Oh, H.S.; et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron 2006, 51, 549–560. [Google Scholar] [CrossRef]
- Chatterjee, S.; Sang, T.K.; Lawless, G.M.; Jackson, G.R. Dissociation of tau toxicity and phosphorylation: Role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum. Mol. Genet. 2009, 18, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Kosmidis, S.; Grammenoudi, S.; Papanikolopoulou, K.; Skoulakis, E.M. Differential effects of Tau on the integrity and function of neurons essential for learning in Drosophila. J. Neurosci. 2010, 30, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, I.; Yang, Y.; Lu, B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 2004, 116, 671–682. [Google Scholar] [CrossRef]
- Steinhilb, M.L.; Dias-Santagata, D.; Fulga, T.A.; Felch, D.L.; Feany, M.B. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol. Biol. Cell 2007, 18, 5060–5068. [Google Scholar] [CrossRef] [PubMed]
- Iijima-Ando, K.; Zhao, L.; Gatt, A.; Shenton, C.; Iijima, K. A DNA damage-activated checkpoint kinase phosphorylates tau and enhances tau-induced neurodegeneration. Hum. Mol. Genet. 2010, 19, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Talmat-Amar, Y.; Arribat, Y.; Redt-Clouet, C.; Feuillette, S.; Bouge, A.L.; Lecourtois, M.; Parmentier, M.L. Important neuronal toxicity of microtubule-bound Tau in vivo in Drosophila. Hum. Mol. Genet. 2011, 20, 3738–3745. [Google Scholar] [CrossRef] [PubMed]
- Khurana, V.; Lu, Y.; Steinhilb, M.L.; Oldham, S.; Shulman, J.M.; Feany, M.B. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr. Biol. 2006, 16, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Reinecke, J.B.; DeVos, S.L.; McGrath, J.P.; Shepard, A.M.; Goncharoff, D.K.; Tait, D.N.; Fleming, S.R.; Vincent, M.P.; Steinhilb, M.L. Implicating calpain in tau-mediated toxicity in vivo. PLoS ONE 2011, 6, e23865. [Google Scholar] [CrossRef] [PubMed]
- Beharry, C.; Alaniz, M.E.; Alonso Adel, C. Expression of Alzheimer-like pathological human tau induces a behavioral motor and olfactory learning deficit in Drosophila melanogaster. J. Alzheimer Dis. 2013, 37, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Tyrer, M.; Shepherd, D. Tau and tau reporters disrupt central projections of sensory neurons in Drosophila. J. Comp. Neurol 2000, 428, 630–640. [Google Scholar] [CrossRef]
- Mudher, A.; Shepherd, D.; Newman, T.A.; Mildren, P.; Jukes, J.P.; Squire, A.; Mears, A.; Drummond, J.A.; Berg, S.; MacKay, D.; et al. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry 2004, 9, 522–530. [Google Scholar] [CrossRef]
- Cowan, C.M.; Bossing, T.; Page, A.; Shepherd, D.; Mudher, A. Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 2010, 120, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, H.; Benton, R.; Shulman, J.M.; St Johnston, D. The role of PAR-1 in regulating the polarised microtubule cytoskeleton in the Drosophila follicular epithelium. Development 2003, 130, 3965–3975. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; Feany, M.B. Genetic modifiers of tauopathy in Drosophila. Genetics 2003, 165, 1233–1242. [Google Scholar] [CrossRef]
- Dourlen, P. Identification of Tau Toxicity Modifiers in the Drosophila Eye. Methods Mol. Biol. 2017, 1523, 375–389. [Google Scholar] [CrossRef]
- Bolkan, B.J.; Kretzschmar, D. Loss of Tau results in defects in photoreceptor development and progressive neuronal degeneration in Drosophila. Dev. Neurobiol. 2014, 74, 1210–1225. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Maruko-Otake, A.; Ohtake, Y.; Hayashishita, M.; Sekiya, M.; Iijima, K.M. Stabilization of Microtubule-Unbound Tau via Tau Phosphorylation at Ser262/356 by Par-1/MARK Contributes to Augmentation of AD-Related Phosphorylation and Abeta42-Induced Tau Toxicity. PLoS Genet. 2016, 12, e1005917. [Google Scholar] [CrossRef]
- Dermaut, B.; Norga, K.K.; Kania, A.; Verstreken, P.; Pan, H.; Zhou, Y.; Callaerts, P.; Bellen, H.J. Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J. Cell Biol. 2005, 170, 127–139. [Google Scholar] [CrossRef]
- Quintas-Neves, M.; Teylan, M.A.; Besser, L.; Soares-Fernandes, J.; Mock, C.N.; Kukull, W.A.; Crary, J.F.; Oliveira, T.G. Magnetic resonance imaging brain atrophy assessment in primary age-related tauopathy (PART). Acta Neuropathol. Commun. 2019, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Heisenberg, M.; Borst, A.; Wagner, S.; Byers, D. Drosophila mushroom body mutants are deficient in olfactory learning. J. Neurogenet. 1985, 2, 1–30. [Google Scholar] [CrossRef] [PubMed]
- de Belle, J.S.; Heisenberg, M. Associative odor learning in Drosophila abolished by chemical ablation of mushroom bodies. Science 1994, 263, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Dubnau, J.; Grady, L.; Kitamoto, T.; Tully, T. Disruption of neurotransmission in Drosophila mushroom body blocks retrieval but not acquisition of memory. Nature 2001, 411, 476–480. [Google Scholar] [CrossRef]
- McGuire, S.E.; Le, P.T.; Davis, R.L. The role of Drosophila mushroom body signaling in olfactory memory. Science 2001, 293, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.; Davis, R.L. Molecular biology and anatomy of Drosophila olfactory associative learning. Bioessays 2001, 23, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Mershin, A.; Pavlopoulos, E.; Fitch, O.; Braden, B.C.; Nanopoulos, D.V.; Skoulakis, E.M. Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn. Mem. 2004, 11, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.O.; Ruan, K.; Zhai, R.G. NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy. Hum. Mol. Genet. 2012, 21, 237–250. [Google Scholar] [CrossRef]
- Kim, M.; Subramanian, M.; Cho, Y.H.; Kim, G.H.; Lee, E.; Park, J.J. Short-term exposure to dim light at night disrupts rhythmic behaviors and causes neurodegeneration in fly models of tauopathy and Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2018, 495, 1722–1729. [Google Scholar] [CrossRef]
- Keshishian, H.; Broadie, K.; Chiba, A.; Bate, M. The drosophila neuromuscular junction: A model system for studying synaptic development and function. Annu. Rev. Neurosci. 1996, 19, 545–575. [Google Scholar] [CrossRef]
- Menon, K.P.; Carrillo, R.A.; Zinn, K. Development and plasticity of the Drosophila larval neuromuscular junction. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 647–670. [Google Scholar] [CrossRef]
- Chee, F.C.; Mudher, A.; Cuttle, M.F.; Newman, T.A.; MacKay, D.; Lovestone, S.; Shepherd, D. Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol. Dis. 2005, 20, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Mhatre, S.D.; Satyasi, V.; Killen, M.; Paddock, B.E.; Moir, R.D.; Saunders, A.J.; Marenda, D.R. Synaptic abnormalities in a Drosophila model of Alzheimer’s disease. Dis. Models Mech. 2014, 7, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.I.; Suh, Y.S.; Chung, Y.J.; Yu, K.; Park, C.B. Shedding Light on Alzheimer’s beta-Amyloidosis: Photosensitized Methylene Blue Inhibits Self-Assembly of beta-Amyloid Peptides and Disintegrates Their Aggregates. Sci. Rep. 2017, 7, 7523. [Google Scholar] [CrossRef] [PubMed]
- Levin, H.L.; Moran, J.V. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 2011, 12, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef]
- Evrony, G.D.; Cai, X.; Lee, E.; Hills, L.B.; Elhosary, P.C.; Lehmann, H.S.; Parker, J.J.; Atabay, K.D.; Gilmore, E.C.; Poduri, A.; et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 2012, 151, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Upton, K.R.; Gerhardt, D.J.; Jesuadian, J.S.; Richardson, S.R.; Sanchez-Luque, F.J.; Bodea, G.O.; Ewing, A.D.; Salvador-Palomeque, C.; van der Knaap, M.S.; Brennan, P.M.; et al. Ubiquitous L1 mosaicism in hippocampal neurons. Cell 2015, 161, 228–239. [Google Scholar] [CrossRef]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Perrat, P.N.; DasGupta, S.; Wang, J.; Theurkauf, W.; Weng, Z.; Rosbash, M.; Waddell, S. Transposition-driven genomic heterogeneity in the Drosophila brain. Science 2013, 340, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Moschetti, R.; Palazzo, A.; Lorusso, P.; Viggiano, L.; Marsano, R.M. “What You Need, Baby, I Got It”: Transposable Elements as Suppliers of Cis-Operating Sequences in Drosophila. Biology 2020, 9, 25. [Google Scholar] [CrossRef]
- Guo, C.; Jeong, H.H.; Hsieh, Y.C.; Klein, H.U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Samimi, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci. 2018, 21, 1038–1048. [Google Scholar] [CrossRef]
- Peng, H.; Chung, P.; Long, F.; Qu, L.; Jenett, A.; Seeds, A.M.; Myers, E.W.; Simpson, J.H. BrainAligner: 3D registration atlases of Drosophila brains. Nat. Methods 2011, 8, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M. Neuroanatomy: Connectome connects fly and mammalian brain networks. Curr. Biol. 2015, 25, R416–R418. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Li, H.; Li, Y.; Liu, L.; Liu, Q.; Lu, H.; Pan, Y.; Wu, Z.; Zhang, K.; Zhu, Y. Vision, memory, and cognition in Drosophila. Learn. Theory Behav. 2017, 483–503. [Google Scholar]
- Hindle, S.J.; Bainton, R.J. Barrier mechanisms in the Drosophila blood-brain barrier. Front. Neurosci. 2014, 8, 414. [Google Scholar] [CrossRef] [PubMed]
- Salminen, T.S.; Vale, P.F. Drosophila as a model system to investigate the effects of mitochondrial variation on innate immunity. Front. Immunol. 2020, 11, 521. [Google Scholar] [CrossRef]
- Pandey, M.K.; DeGrado, T.R. Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapy and Imaging. Theranostics 2016, 6, 571–593. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Sun, C.; Zheng, M.; Liu, S.; Shi, R. Amentoflavone suppresses amyloid beta1-42 neurotoxicity in Alzheimer’s disease through the inhibition of pyroptosis. Life Sci. 2019, 239, 117043. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Song, C.; Du, Y.; Gaur, U.; Yang, M. Pharmacological Treatment of Alzheimer’s Disease: Insights from Drosophila melanogaster. Int. J. Mol. Sci. 2020, 21, 4621. [Google Scholar] [CrossRef]
- Colodner, K.J.; Feany, M.B. Glial fibrillary tangles and JAK/STAT-mediated glial and neuronal cell death in a Drosophila model of glial tauopathy. J. Neurosci. 2010, 30, 16102–16113. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; de Haro, M.; Hao, S.; Park, J.; Rousseaux, M.W.; Al-Ramahi, I.; Jafar-Nejad, P.; Vilanova-Velez, L.; See, L.; De Maio, A.; et al. Reduction of Nuak1 Decreases Tau and Reverses Phenotypes in a Tauopathy Mouse Model. Neuron 2016, 92, 407–418. [Google Scholar] [CrossRef]
- Blard, O.; Feuillette, S.; Bou, J.; Chaumette, B.; Frebourg, T.; Campion, D.; Lecourtois, M. Cytoskeleton proteins are modulators of mutant tau-induced neurodegeneration in Drosophila. Hum. Mol. Genet. 2007, 16, 555–566. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ambegaokar, S.S.; Jackson, G.R. Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum. Mol. Genet. 2011, 20, 4947–4977. [Google Scholar] [CrossRef]
- Shim, K.H.; Kim, S.H.; Hur, J.; Kim, D.H.; Demirev, A.V.; Yoon, S.Y. Small-molecule drug screening identifies drug Ro 31-8220 that reduces toxic phosphorylated tau in Drosophila melanogaster. Neurobiol. Dis. 2019, 130, 104519. [Google Scholar] [CrossRef]
- Appocher, C.; Klima, R.; Feiguin, F. Functional screening in Drosophila reveals the conserved role of REEP1 in promoting stress resistance and preventing the formation of Tau aggregates. Hum. Mol. Genet. 2014, 23, 6762–6772. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; Imboywa, S.; Giagtzoglou, N.; Powers, M.P.; Hu, Y.; Devenport, D.; Chipendo, P.; Chibnik, L.B.; Diamond, A.; Perrimon, N.; et al. Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates Tau-mediated mechanisms. Hum. Mol. Genet. 2014, 23, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Zon, L.I.; Peterson, R.T. In vivo drug discovery in the zebrafish. Nat. Rev. Drug Discov. 2005, 4, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B. Patterning the brain of the zebrafish embryo. Annu. Rev. Neurosci. 1993, 16, 707–732. [Google Scholar] [CrossRef] [PubMed]
- Rupp, B.; Reichert, H. Neuroanatomy of the Zebrafish Brain: A Topological Atlas; Birkhauser: Basel, Switzerland, 1996. [Google Scholar]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K. Transposon tools and methods in zebrafish. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2005, 234, 244–254. [Google Scholar] [CrossRef]
- Halpern, M.E.; Rhee, J.; Goll, M.G.; Akitake, C.M.; Parsons, M.; Leach, S.D. Gal4/UAS transgenic tools and their application to zebrafish. Zebrafish 2008, 5, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Hans, S.; Kaslin, J.; Freudenreich, D.; Brand, M. Temporally-controlled site-specific recombination in zebrafish. PLoS ONE 2009, 4, e4640. [Google Scholar] [CrossRef]
- Schmid, B.; Haass, C. Genomic editing opens new avenues for zebrafish as a model for neurodegeneration. J. Neurochem. 2013, 127, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Poskanzer, K.E.; Freeman, M.R.; Monk, K.R. Live-imaging of astrocyte morphogenesis and function in zebrafish neural circuits. Nat. Neurosci. 2020, 23, 1297–1306. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Overall, C.M.; Power, C. Deciphering complex mechanisms in neurodegenerative diseases: The advent of systems biology. Trends Neurosci. 2009, 32, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Sabaliauskas, N.A.; Foutz, C.A.; Mest, J.R.; Budgeon, L.R.; Sidor, A.T.; Gershenson, J.A.; Joshi, S.B.; Cheng, K.C. High-throughput zebrafish histology. Methods 2006, 39, 246–254. [Google Scholar] [CrossRef]
- Green, J.; Collins, C.; Kyzar, E.J.; Pham, M.; Roth, A.; Gaikwad, S.; Cachat, J.; Stewart, A.M.; Landsman, S.; Grieco, F. Automated high-throughput neurophenotyping of zebrafish social behavior. J. Neurosci. Methods 2012, 210, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Lee, J.G.; Kim, J.H.; Kim, S.Y.; Huh, Y.H.; Kim, H.J.; Lee, K.S.; Yu, K.; Lee, J.S. Vascular defects of DYRK1A knockouts are ameliorated by modulating calcium signaling in zebrafish. Dis. Model. Mech. 2019, 12, dmm037044. [Google Scholar] [CrossRef] [PubMed]
- Patton, E.E.; Zon, L.I.; Langenau, D.M. Zebrafish disease models in drug discovery: From preclinical modelling to clinical trials. Nat. Rev. Drug Discov. 2021, 20, 611–628. [Google Scholar] [CrossRef]
- Griffin, A.; Hamling, K.R.; Knupp, K.; Hong, S.; Lee, L.P.; Baraban, S.C. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 2017, 140, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Martins, R.N.; Lardelli, M. Complex splicing and neural expression of duplicated tau genes in zebrafish embryos. J. Alzheimer Dis. 2009, 18, 305–317. [Google Scholar] [CrossRef]
- Paquet, D.; Bhat, R.; Sydow, A.; Mandelkow, E.-M.; Berg, S.; Hellberg, S.; Fälting, J.; Distel, M.; Köster, R.W.; Schmid, B. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J. Clin. Investig. 2009, 119, 1382–1395. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Lee, S.E.; Wojta, K.; Ramos, E.M.; Klein, E.; Chen, J.; Boxer, A.L.; Gorno-Tempini, M.L.; Geschwind, D.H.; Schlotawa, L. A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain 2017, 140, 1128–1146. [Google Scholar] [CrossRef]
- Tomasiewicz, H.G.; Flaherty, D.B.; Soria, J.; Wood, J.G. Transgenic zebrafish model of neurodegeneration. J. Neurosci. Res. 2002, 70, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Garver, J.A.; Hukriede, N.A.; Burton, E.A. Generation of a transgenic zebrafish model of Tauopathy using a novel promoter element derived from the zebrafish eno2 gene. Nucleic Acids Res. 2007, 35, 6501–6516. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.-K.; Yuan, R.-Y.; Lien, H.-W.; Hung, C.-C.; Hwang, P.-P.; Chen, R.P.-Y.; Chang, C.-C.; Liao, Y.-F.; Huang, C.-J. Multiple signaling factors and drugs alleviate neuronal death induced by expression of human and zebrafish tau proteins in vivo. J. Biomed. Sci. 2016, 23, 25. [Google Scholar] [CrossRef]
- Cosacak, M.I.; Bhattarai, P.; Bocova, L.; Dzewas, T.; Mashkaryan, V.; Papadimitriou, C.; Brandt, K.; Hollak, H.; Antos, C.L.; Kizil, C. Human TAU P301L overexpression results in TAU hyperphosphorylation without neurofibrillary tangles in adult zebrafish brain. Sci. Rep. 2017, 7, 12959. [Google Scholar] [CrossRef]
- Alyenbaawi, H.; Kanyo, R.; Locskai, L.F.; Kamali-Jamil, R.; DuVal, M.G.; Bai, Q.; Wille, H.; Burton, E.A.; Allison, W.T. Seizures are a druggable mechanistic link between TBI and subsequent tauopathy. eLife 2021, 10, e58744. [Google Scholar] [CrossRef] [PubMed]
- Curado, S.; Anderson, R.M.; Jungblut, B.; Mumm, J.; Schroeter, E.; Stainier, D.Y. Conditional targeted cell ablation in zebrafish: A new tool for regeneration studies. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 1025–1035. [Google Scholar] [CrossRef]
- van Ham, T.J.; Mapes, J.; Kokel, D.; Peterson, R.T. Live imaging of apoptotic cells in zebrafish. FASEB J. 2010, 24, 4336–4342. [Google Scholar] [PubMed]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Babin, P.J.; Goizet, C.; Raldúa, D. Zebrafish models of human motor neuron diseases: Advantages and limitations. Prog. Neurobiol. 2014, 118, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Zelenchuk, T.A.; Brusés, J.L. In vivo labeling of zebrafish motor neurons using an mnx1 enhancer and Gal4/UAS. Genesis 2011, 49, 546–554. [Google Scholar] [CrossRef]
- Trevarrow, B.; Marks, D.L.; Kimmel, C.B. Organization of hindbrain segments in the zebrafish embryo. Neuron 1990, 4, 669–679. [Google Scholar] [CrossRef]
- Kamei, M.; Weinstein, B.M. Long-term time-lapse fluorescence imaging of developing zebrafish. Zebrafish 2005, 2, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Colwill, R.M.; Creton, R. Locomotor behaviors in zebrafish (Danio rerio) larvae. Behav. Process. 2011, 86, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Kyriakatos, A.; Mahmood, R.; Ausborn, J.; Porres, C.P.; Büschges, A.; El Manira, A. Initiation of locomotion in adult zebrafish. J. Neurosci. 2011, 31, 8422–8431. [Google Scholar] [CrossRef]
- Osmulski, P.A.; Hochstrasser, M.; Gaczynska, M. A tetrahedral transition state at the active sites of the 20S proteasome is coupled to opening of the α-ring channel. Structure 2009, 17, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Joberty, G.; Buist, A.; Vanoosthuyse, A.; Stancu, I.-C.; Vasconcelos, B.; Pierrot, N.; Faelth-Savitski, M.; Kienlen-Campard, P.; Octave, J.-N. Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta Neuropathol. 2017, 133, 731–749. [Google Scholar] [CrossRef] [PubMed]
- Njomen, E.; Osmulski, P.A.; Jones, C.L.; Gaczynska, M.; Tepe, J.J. Small molecule modulation of proteasome assembly. Biochemistry 2018, 57, 4214–4224. [Google Scholar] [CrossRef]
- McKinnon, C.; Tabrizi, S.J. The ubiquitin-proteasome system in neurodegeneration. Antioxid. Redox Signal. 2014, 21, 2302–2321. [Google Scholar] [CrossRef]
- Imamura, S.; Yabu, T.; Yamashita, M. Protective role of cell division cycle 48 (CDC48) protein against neurodegeneration via ubiquitin-proteasome system dysfunction during zebrafish development. J. Biol. Chem. 2012, 287, 23047–23056. [Google Scholar] [CrossRef]
- Mathai, B.J.; Meijer, A.H.; Simonsen, A. Studying Autophagy in Zebrafish. Cells 2017, 6, 21. [Google Scholar] [CrossRef]
- Plucinska, G.; Paquet, D.; Hruscha, A.; Godinho, L.; Haass, C.; Schmid, B.; Misgeld, T. In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J. Neurosci. 2012, 32, 16203–16212. [Google Scholar] [CrossRef] [PubMed]
- Drewes, G.; Ebneth, A.; Preuss, U.; Mandelkow, E.-M.; Mandelkow, E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 1997, 89, 297–308. [Google Scholar] [CrossRef]
- Hassan-Abdi, R.; Brenet, A.; Bennis, M.; Yanicostas, C.; Soussi-Yanicostas, N. Neurons Expressing Pathological Tau Protein Trigger Dramatic Changes in Microglial Morphology and Dynamics. Front. Neurosci. 2019, 13, 1199. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Barbereau, C.; Yehya, A.; Silhol, M.; Cubedo, N.; Verdier, J.M.; Maurice, T.; Rossel, M. Neuroprotective brain-derived neurotrophic factor signaling in the TAU-P301L tauopathy zebrafish model. Pharm. Res. 2020, 158, 104865. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda-Diaz, J.E.; Alavi Naini, S.M.; Huynh, M.B.; Ouidja, M.O.; Yanicostas, C.; Chantepie, S.; Villares, J.; Lamari, F.; Jospin, E.; van Kuppevelt, T.H.; et al. HS3ST2 expression is critical for the abnormal phosphorylation of tau in Alzheimer’s disease-related tau pathology. Brain 2015, 138, 1339–1354. [Google Scholar] [CrossRef] [PubMed]
- Moussaed, M.; Huc-Brandt, S.; Cubedo, N.; Silhol, M.; Murat, S.; Lebart, M.C.; Kovacs, G.; Verdier, J.M.; Trousse, F.; Rossel, M.; et al. Regenerating islet-derived 1alpha (REG-1alpha) protein increases tau phosphorylation in cell and animal models of tauopathies. Neurobiol. Dis. 2018, 119, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.; Buffet, M.; Fenwick, A.E.; Haigh, D.; Ife, R.J.; Saunders, M.; Slingsby, B.P.; Stacey, R.; Ward, R.W. 3-Anilino-4-arylmaleimides: Potent and selective inhibitors of glycogen synthase kinase-3 (GSK-3). Bioorganic Med. Chem. Lett. 2001, 11, 635–639. [Google Scholar] [CrossRef]
- Faria-Ramos, I.; Poças, J.; Marques, C.; Santos-Antunes, J.; Macedo, G.; Reis, C.A.; Magalhães, A. Heparan Sulfate Glycosaminoglycans:(Un) Expected Allies in Cancer Clinical Management. Biomolecules 2021, 11, 136. [Google Scholar] [CrossRef]
- Lanzi, C.; Cassinelli, G. Heparan sulfate mimetics in cancer therapy: The challenge to define structural determinants and the relevance of targets for optimal activity. Molecules 2018, 23, 2915. [Google Scholar] [CrossRef]
- Alavi Naini, S.M.; Yanicostas, C.; Hassan-Abdi, R.; Blondeel, S.; Bennis, M.; Weiss, R.J.; Tor, Y.; Esko, J.D.; Soussi-Yanicostas, N. Surfen and oxalyl surfen decrease tau hyperphosphorylation and mitigate neuron deficits in vivo in a zebrafish model of tauopathy. Transl. Neurodegener. 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- VerPlank, J.J.S.; Tyrkalska, S.D.; Fleming, A.; Rubinsztein, D.C.; Goldberg, A.L. cGMP via PKG activates 26S proteasomes and enhances degradation of proteins, including ones that cause neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 14220–14230. [Google Scholar] [CrossRef]
- Wu, B.K.; Yuan, R.Y.; Chang, Y.P.; Lien, H.W.; Chen, T.S.; Chien, H.C.; Tong, T.S.; Tsai, H.P.; Fang, C.L.; Liao, Y.F.; et al. Epicatechin isolated from Tripterygium wilfordii extract reduces tau-GFP-induced neurotoxicity in zebrafish embryo through the activation of Nrf2. Biochem. Biophys. Res. Commun. 2016, 477, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.; Carrel, J.S. Fertilization and sperm competition in the nematodeCaenorhabditis elegans. Dev. Biol. 1979, 73, 304–321. [Google Scholar] [CrossRef]
- Kimble, J.; Crittenden, S.L. Germline Proliferation and Its Control. In WormBook; The C. elegans Research Community: Pasadena, CA, USA, 2005. [Google Scholar] [CrossRef]
- Kobet, R.A.; Pan, X.; Zhang, B.; Pak, S.C.; Asch, A.S.; Lee, M.-H. Caenorhabditis elegans: A model system for anti-cancer drug discovery and therapeutic target identification. Biomol. Ther. 2014, 22, 371. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Hendricks, G.L.; Lee, K.; Mylonakis, E. An update on the use of C. elegans for preclinical drug discovery: Screening and identifying anti-infective drugs. Expert Opin. Drug Discov. 2017, 12, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.J.; Jarrell, T.A.; Brittin, C.A.; Wang, Y.; Bloniarz, A.E.; Yakovlev, M.A.; Nguyen, K.C.; Tang, L.T.-H.; Bayer, E.A.; Duerr, J.S. Whole-animal connectomes of both Caenorhabditis elegans sexes. Nature 2019, 571, 63–71. [Google Scholar] [CrossRef]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef]
- Chen, X.; Barclay, J.W.; Burgoyne, R.D.; Morgan, A. Using C. elegans to discover therapeutic compounds for ageing-associated neurodegenerative diseases. Chem. Cent. J. 2015, 9, 1–20. [Google Scholar] [CrossRef]
- Kim, W.; Underwood, R.S.; Greenwald, I.; Shaye, D.D. OrthoList 2: A new comparative genomic analysis of human and Caenorhabditis elegans genes. Genetics 2018, 210, 445–461. [Google Scholar] [CrossRef]
- The C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 1998, 282, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund, M.; Hobert, O.; Miller, D.M., 3rd; Sestan, N. The CeNGEN project: The complete gene expression map of an entire nervous system. Neuron 2018, 99, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, L.A.; Knight, A.L.; Caldwell, G.A.; Caldwell, K.A. Generation of stable transgenic C. elegans using microinjection. J. Vis. Exp. 2008. [Google Scholar] [CrossRef] [PubMed]
- Schweinsberg, P.J.; Grant, B.D. C. elegans gene transformation by microparticle bombardment. In WormBook; The C. elegans Research Community: Pasadena, CA, USA, 2018. [Google Scholar] [CrossRef]
- McDermott, J.B.; Aamodt, S.; Aamodt, E. ptl-1, a Caenorhabditis elegans gene whose products are homologous to the τ microtubule-associated proteins. Biochemistry 1996, 35, 9415–9423. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.C.; Zhang, B.; Leverenz, J.B.; Thomas, J.H.; Trojanowski, J.Q.; Schellenberg, G.D. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. USA 2003, 100, 9980–9985. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, T.; Ding, Z.; Gengyo-Ando, K.; Oue, M.; Yamaguchi, H.; Mitani, S.; Ihara, Y. Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol. Dis. 2005, 20, 372–383. [Google Scholar] [CrossRef]
- Brandt, R.; Gergou, A.; Wacker, I.; Fath, T.; Hutter, H. A Caenorhabditis elegans model of tau hyperphosphorylation: Induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol. Aging 2009, 30, 22–33. [Google Scholar] [CrossRef]
- Pir, G.J.; Choudhary, B.; Mandelkow, E.; Mandelkow, E.-M. Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol. Neurodegener. 2016, 11, 1–21. [Google Scholar] [CrossRef]
- Morelli, F.; Romeo, M.; Barzago, M.M.; Bolis, M.; Mattioni, D.; Rossi, G.; Tagliavini, F.; Bastone, A.; Salmona, M.; Diomede, L. V363I and V363A mutated tau affect aggregation and neuronal dysfunction differently in C. elegans. Neurobiol. Dis. 2018, 117, 226–234. [Google Scholar] [CrossRef]
- Guha, S.; Fischer, S.; Johnson, G.V.; Nehrke, K. Tauopathy-associated tau modifications selectively impact neurodegeneration and mitophagy in a novel C. elegans single-copy transgenic model. Mol. Neurodegener. 2020, 15, 65. [Google Scholar] [CrossRef]
- Park, H.-E.H.; Jung, Y.; Lee, S.-J.V. Survival assays using Caenorhabditis elegans. Mol. Cells 2017, 40, 90. [Google Scholar] [CrossRef]
- Stiernagle, T. Maintenance of C. elegans. C. Elegans 1999, 2, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Amrit, F.R.G.; Ratnappan, R.; Keith, S.A.; Ghazi, A. The C. elegans lifespan assay toolkit. Methods 2014, 68, 465–475. [Google Scholar] [CrossRef]
- Lettre, G.; Hengartner, M.O. Developmental apoptosis in C. elegans: A complex CEDnario. Nat. Rev. Mol. Cell Biol. 2006, 7, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Bargmann, C.I. Chemosensation in C. elegans. In WormBook; The C. elegans Research Community: Pasadena, CA, USA, 2006. [Google Scholar] [CrossRef]
- Laughlin, S.T.; Bertozzi, C.R. In vivo imaging of Caenorhabditis elegans glycans. ACS Chem. Biol. 2009, 4, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Kerk, S.Y.; Kratsios, P.; Hart, M.; Mourao, R.; Hobert, O. Diversification of C. elegans motor neuron identity via selective effector gene repression. Neuron 2017, 93, 80–98. [Google Scholar] [CrossRef]
- Van Krugten, J.; TARIS, K.K.; Peterman, E.J. Imaging adult C. elegans live using light-sheet microscopy. J. Microsc. 2021, 281, 214–223. [Google Scholar] [CrossRef]
- Atakan, H.B.; Alkanat, T.; Cornaglia, M.; Trouillon, R.; Gijs, M.A. Automated phenotyping of Caenorhabditis elegans embryos with a high-throughput-screening microfluidic platform. Microsyst. Nanoeng. 2020, 6, 24. [Google Scholar] [CrossRef]
- Nawa, M.; Matsuoka, M. The method of the body bending assay using Caenorhabditis elegans. Bio-Protocol 2012, 2, e253. [Google Scholar] [CrossRef]
- Buckingham, S.D.; Sattelle, D.B. Strategies for automated analysis of C. elegans locomotion. Invertebr. Neurosci. 2008, 8, 121. [Google Scholar] [CrossRef]
- Zhen, M.; Samuel, A.D. C. elegans locomotion: Small circuits, complex functions. Curr. Opin. Neurobiol. 2015, 33, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Celniker, S.E.; Dillon, L.A.; Gerstein, M.B.; Gunsalus, K.C.; Henikoff, S.; Karpen, G.H.; Kellis, M.; Lai, E.C.; Lieb, J.D.; MacAlpine, D.M. Unlocking the secrets of the genome. Nature 2009, 459, 927–930. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Timmons, L.; Tabara, H.; Mello, C.C.; Fire, A.Z. Inducible systemic RNA silencing in Caenorhabditis elegans. Mol. Biol. Cell 2003, 14, 2972–2983. [Google Scholar] [CrossRef]
- Asikainen, S.; Vartiainen, S.; Lakso, M.; Nass, R.; Wong, G. Selective sensitivity of Caenorhabditis elegans neurons to RNA interference. Neuroreport 2005, 16, 1995–1999. [Google Scholar] [CrossRef]
- Corsi, A.K.; Wightman, B.; Chalfie, M. A transparent window into biology: A primer on Caenorhabditis elegans. Genetics 2015, 200, 387–407. [Google Scholar] [CrossRef]
- Katz, M.; Shaham, S. Learning and Memory: Mind over Matter in C. elegans. Curr. Biol. 2019, 29, R365–R367. [Google Scholar] [CrossRef] [PubMed]
- Rankin, C.H. But can they learn? My accidental discovery of learning and memory in C. elegans. J. Neurogenet. 2020, 34, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Schlipalius, D.I.; Valmas, N.; Tuck, A.G.; Jagadeesan, R.; Ma, L.; Kaur, R.; Goldinger, A.; Anderson, C.; Kuang, J.; Zuryn, S. A core metabolic enzyme mediates resistance to phosphine gas. Science 2012, 338, 807–810. [Google Scholar] [CrossRef]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/RNS generation. J. Biomed. Sci. 2017, 24, 1–10. [Google Scholar] [CrossRef]
- Ahmad, W. Dihydrolipoamide dehydrogenase suppression induces human tau phosphorylation by increasing whole body glucose levels in a C. elegans model of Alzheimer’s Disease. Exp. Brain Res. 2018, 236, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Dickson, D.; Robinson, C.; Ross, O.; Dächsel, J.; Lincoln, S.; Cobb, S.; Rajput, M.; Farrer, M. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 2006, 67, 1506–1508. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Sultana, R.; Ferree, A.; Smith, K.; Barone, E.; Perluigi, M.; Coccia, R.; Pierce, W.; Cai, J.; Mancuso, C.; et al. Redox proteomics analyses of the influence of co-expression of wild-type or mutated LRRK2 and Tau on C. elegans protein expression and oxidative modification: Relevance to Parkinson disease. Antioxid Redox Signal. 2012, 17, 1490–1506. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, B.; Mandelkow, E.; Mandelkow, E.M.; Pir, G.J. Glutamatergic nervous system degeneration in a C. elegans Tau(A152T) tauopathy model involves pathways of excitotoxicity and Ca(2+) dysregulation. Neurobiol. Dis. 2018, 117, 189–202. [Google Scholar] [CrossRef]
- Salehi, B.; Stojanović-Radić, Z.; Matejić, J.; Sharifi-Rad, M.; Kumar, N.V.A.; Martins, N.; Sharifi-Rad, J. The therapeutic potential of curcumin: A review of clinical trials. Eur. J. Med. Chem. 2019, 163, 527–545. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Pandav, R.; Dodge, H.; Johnston, J.; Belle, S.; DeKosky, S.; Ganguli, M. Incidence of Alzheimer’s disease in a rural community in India: The Indo–US study. Neurology 2001, 57, 985–989. [Google Scholar] [CrossRef]
- Miyasaka, T.; Xie, C.; Yoshimura, S.; Shinzaki, Y.; Yoshina, S.; Kage-Nakadai, E.; Mitani, S.; Ihara, Y. Curcumin improves tau-induced neuronal dysfunction of nematodes. Neurobiol. Aging 2016, 39, 69–81. [Google Scholar] [CrossRef]
- Miyasaka, T.; Shinzaki, Y.; Yoshimura, S.; Yoshina, S.; Kage-Nakadai, E.; Mitani, S.; Ihara, Y. Imbalanced Expression of Tau and Tubulin Induces Neuronal Dysfunction in C. elegans Models of Tauopathy. Front. Neurosci. 2018, 12, 415. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Burgess, J.K.; Chen, J.H.; Thomas, J.H.; Schellenberg, G.D. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum. Mol. Genet. 2006, 15, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.C.; Schellenberg, G.D. SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum. Mol. Genet. 2007, 16, 1959–1971. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, C.R.; Schellenberg, G.D.; Kraemer, B.C. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum. Mol. Genet. 2009, 18, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, C.R.; Greenup, L.; Leverenz, J.B.; Kraemer, B.C. MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum. Mol. Genet. 2011, 20, 1989–1999. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Tau Isoform | Constructs (UAS) | GAL4 Driver | Phenotypes | References |
|---|---|---|---|---|
| 2N4R | hTauWT | GMR-GAL4, Ealv-GAL4 | REP, ND | [95] |
| hTauP301L | GMR-GAL4 | REP, ND | [96] | |
| hTauWT, hTauS2A hTauS11A | GMR-GAL4 | REP, ND | [97] | |
| hTauWT:FLAG hTauSTA:FLAG | Elav-GAL4, OK107-GAL4 | ND, LMD | [98] | |
| 0N4R | hTauWT hTauV337M hTauR406W | Elav-GAL4 OK6-GAL4 | RL, ND TL, MT | [89] |
| hTauR406W/S2A hTauR406W/S202A | GMR-GAL4 | REP, ND | [99] | |
| hTauT111A/T153A hTauT175A/T181A hTauT199A/T217A hTauS202A/S205A hTauT212A hTauS214A hTauT231A/S235A hTauAP5 hTauS422A hTauS396A/S404A | GMR-GAL4 | REP, pTL | [100] | |
| hTauS262A | GMR-GAL4 | REP, ND | [101] | |
| TauAP | Elav-GAL4, OK6GAL4 | MT defects | [102] | |
| hTauE14 | GMR-GAL4, | REP, pTL, ND, | [103] | |
| hTauK44Q/R230Q hTau44–230 | GMR-GAL4 | REP, calpain activity | [104] | |
| hTau1–421 | Elav-GAL4 | RL, ND, pTL | [83] | |
| PH-Tau | Elav-GAL4, MB-GAL4 | climbing, MB defects, LMD | [105] | |
| 0N3R | hTauWT | C161-GAL4 | sensory neurons defects | [106] |
| hTauWT | D42-GAL4 | locomotion, axon, NMJ defects | [107] | |
| hTauWT | Elav-GAL4, D42-GAL4 | pTL, MT stability defects | [108] | |
| dTau | tauEP3203 | MT stability defects | [109] | |
| tauEP3597 | MT stability defects | [109] |
| Tau Isoform (Mutation) | Promoter | Biochemical Phenotype | Biological and/or Behavioral Phenotype | Rescue | References |
|---|---|---|---|---|---|
| hTau 2N4R WT | GATA-2 |
| disrupted cytoskeletal filaments | N/A | [171] |
| hTau 0N4R WT | eno2 | Tau accumulation | N/A | N/A | [172] |
| hTau 2N4R (P301L) | (UAS/GAL4) HuC |
|
| GSK3β inhibitor reduced Tau hyperphosphorylation | [169] |
| truncated hTau | HuC | AT8+ | neuronal cell death | overexpression of Bcl2-L1, Nrf2, and GDNF rescued neuronal cell death | [173] |
| hTau 2N4R (P301L) | HuC | AT8+ | N/A | N/A | [174] |
| hTau 2N4R (A152T) | (UAS/GAL4) HuC |
|
| activated autophagy rescued all phenotypes | [170] |
| hTau 2N4R WT | eno2 | N/A | TBI induces
| dynamin inhibitors/anti-convulsant drugs rescued TBI-induced Tau inclusions/cell death | [175] |
| Tau Isoform (Mutation) | Promoter | Biochemical Phenotypes | Biological Phenotypes | Behavioral Phenotypes | Rescue | References |
|---|---|---|---|---|---|---|
| hTau 1N4R (WT, P301L, V337M) | aex-3 (pan neuronal) | 12E8, AT8, pT205, AT270, pT181, CP13, PHF-1, pS422+ |
|
| N/A | [219] |
| hTau 0N3R(WT), 0N4R(WT, P301L, R406W) | mec-7 (touch neuron) | PHF-1, AT8+ |
| age-dependent touch response defect | HSP70 expression improved touch response | [220] |
| hTau 0N3R(WT, PHP) | F25B3.3 (pan-neuronal) | Tau5, AT180, PHF-1, AT8, TG3+ | defective motor axons by PHP-Tau | progressive age-dependent Unc | N/A | [221] |
| aex-3 for 1N4R (V337M); Rab-3 for ∆K280 (pan-neuronal) |
|
| early-onset paralysis (1N4R(V337M) + ∆K280) | TAI reduced Tau expression/rescued the locomotion | [59] |
| hTau 2N4R (WT, A152T) | Snb-1 (pan-neuronal) |
| A152T showed:
| early-onset paralysis (A152T) | TAI did not rescue paralysis | [222] |
| hTau 2N4R (V363I, V363A) | aex-3 (pan-neuronal) |
|
|
| N/A | [223] |
| hTau 0N4R (T231E, K274/281Q) | mec-7 (touch neuron) | N/A |
| reduced touch sensation | N/A | [224] |
| Strength | Limitation | |
|---|---|---|
| Mouse (not covered in this review) |
|
|
| Drosophila |
|
|
| Zebrafish |
|
|
| C. elegans |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giong, H.-K.; Subramanian, M.; Yu, K.; Lee, J.-S. Non-Rodent Genetic Animal Models for Studying Tauopathy: Review of Drosophila, Zebrafish, and C. elegans Models. Int. J. Mol. Sci. 2021, 22, 8465. https://doi.org/10.3390/ijms22168465
Giong H-K, Subramanian M, Yu K, Lee J-S. Non-Rodent Genetic Animal Models for Studying Tauopathy: Review of Drosophila, Zebrafish, and C. elegans Models. International Journal of Molecular Sciences. 2021; 22(16):8465. https://doi.org/10.3390/ijms22168465
Chicago/Turabian StyleGiong, Hoi-Khoanh, Manivannan Subramanian, Kweon Yu, and Jeong-Soo Lee. 2021. "Non-Rodent Genetic Animal Models for Studying Tauopathy: Review of Drosophila, Zebrafish, and C. elegans Models" International Journal of Molecular Sciences 22, no. 16: 8465. https://doi.org/10.3390/ijms22168465
APA StyleGiong, H.-K., Subramanian, M., Yu, K., & Lee, J.-S. (2021). Non-Rodent Genetic Animal Models for Studying Tauopathy: Review of Drosophila, Zebrafish, and C. elegans Models. International Journal of Molecular Sciences, 22(16), 8465. https://doi.org/10.3390/ijms22168465