
Immunometabolic Modulatory Role of Naltrexone in BV-2 Microglia Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
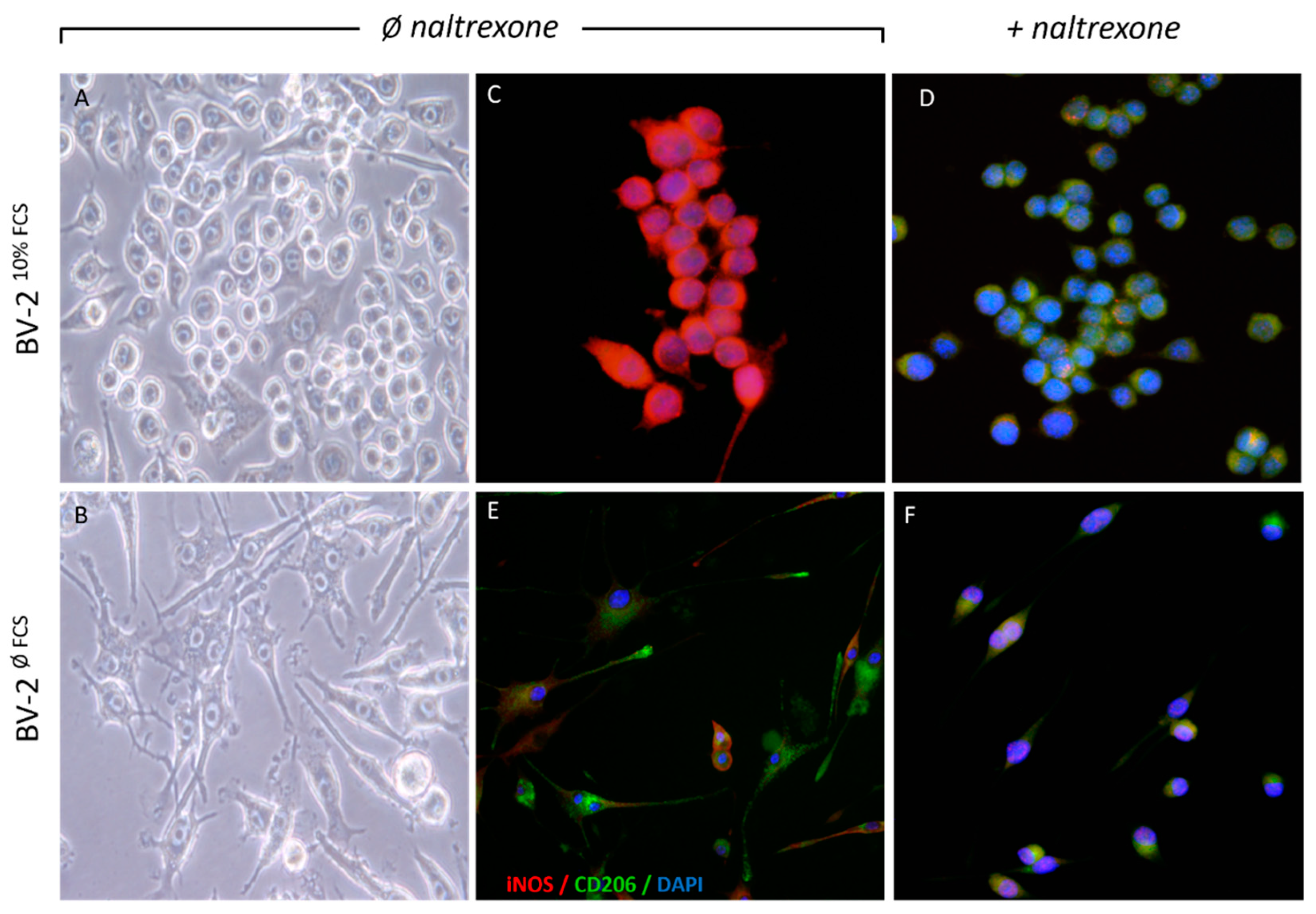
2.1. Naltrexone Induces an Anti-Inflammatory M2 Polarization State in BV-2 Cells
2.2. Naltrexone Induces an Immunometabolically-Suppressive Phenotype in BV-2 Microglia Cells
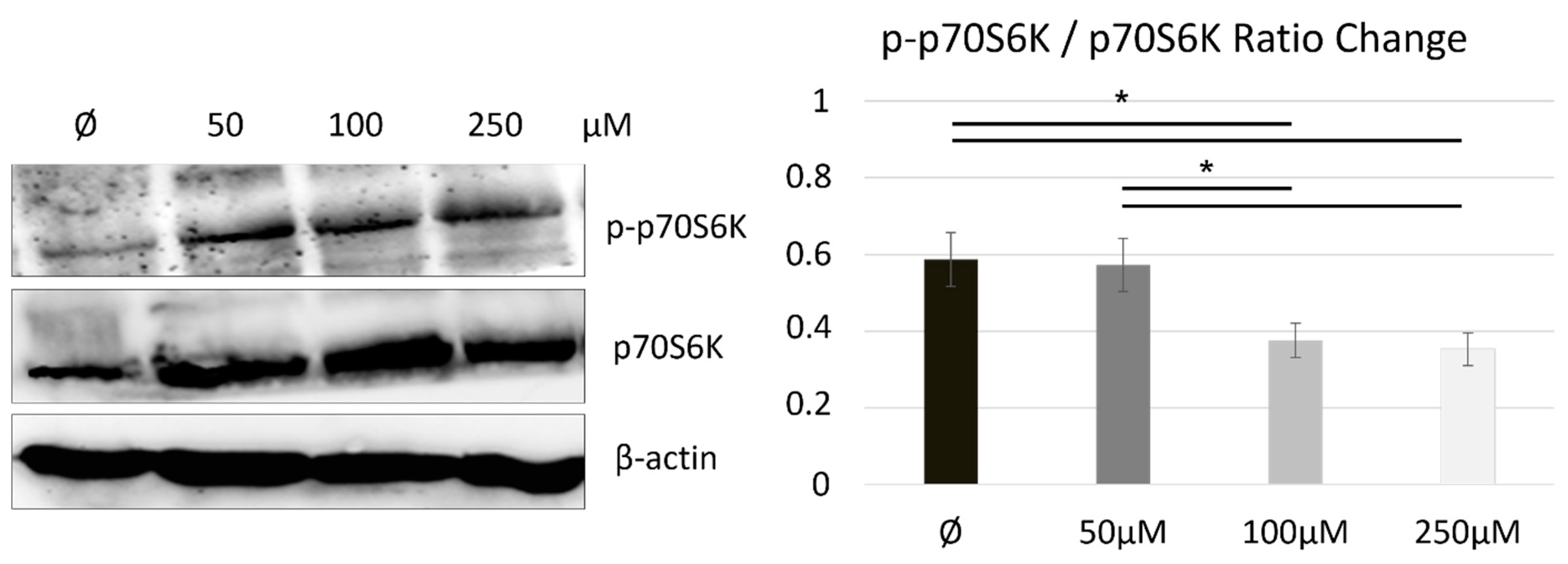
2.3. Naltrexone Calibrates a Metabolic State in BV-2 Cells by Modulating the mTOR/S6K Pathway
3. Discussion
3.1. What Is the Importance of Studying Naltrexone as an Anti-Inflammatory and Immunomodulatory Drug?
3.2. How Is Naltrexone Imposed into Microglia Immunometabolism—Possibly by Modulating the Phenotypic Features of Activated Microglia Related to Their Metabolic Switch?
3.3. How Is mTOR Modulated upon Metabolic State and Polarization in the Naltrexone-Induced M2 Microglia Cell Phenotype?
3.4. Future Prospects
4. Materials and Methods
4.1. Cells
4.2. In Vitro Cell Viability Assay
4.3. Reagents and Antibodies
4.4. Immunofluorescence
4.5. Western Blot Analysis
4.6. Real-Time Metabolism Assay
4.7. Flow Cytometry
4.8. Statistical Analysis
5. Conclusions
- suppression of cell activation toward rest displaying an iNOSlowCD206high quiescence phenotype
- associated proteomic changes in mTORC1 pathway activity, analysed by p-p70S6K and p70S6K ratios
- metabolic shift from highly glycolytic to hypo-energetic cellular state, and thereby maintaining the immunometabolic balance and shape that mostly matches the protective cell microglial phenotype.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LDN | Low-dose naltrexone |
| LPS | Lipopolysaccharide |
| IFN-γ | Interferon gamma |
| mTOR | Mammalian target of rapamycin |
| OXPHOS | oxidative phosphorylation |
| DMEM | Dulbecco’s modified eagle medium |
| FCS | Fetal calf serum |
| iNOS | Inducible nitric oxide synthase |
| CD206 | Cluster of differentiation 206 |
| OCR | Oxygen consumption rate |
| ECAR | Extracellular acidification rate |
| p70S6K | Ribosomal protein S6 kinase beta-1 |
| p-p70S6K | Phosphorylated ribosomal protein S6 kinase beta-1 |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| TLR-4 | Toll-like receptor 4 |
| COVID-19 | Coronavirus disease of 2019. |
| ACE2 | Angiotensin-converting enzyme 2 |
References
- Resnick, R.B.; Volavka, J.; Freedman, A.M.; Thomas, M. Studies of EN 1639A (naltrexone): A new narcotic antagonist. Am. J. Psychiatry 1974, 131, 646–650. [Google Scholar] [CrossRef]
- Younger, J.; Parkitny, L.; McLain, D. The use of low-dose naltrexone (LDN) as a novel anti-inflammatory treatment for chronic pain. Clin. Rheumatol. 2014, 33, 451–459. [Google Scholar] [CrossRef]
- Sudakin, D. Naltrexone: Not Just for Opioids Anymore. J. Med. Toxicol. 2016, 12, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; You, Y.; Griffin, N.; Feng, J.; Shan, F. Low-dose naltrexone (LDN): A promising treatment in immune-related diseases and cancer therapy. Int. Immunopharmacol. 2018, 61, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.M.; Scott, K.A.; Dennis, J.L.; Kaminska, E.; Levett, A.J.; Dalgleish, A.G. Naltrexone at low doses upregulates a unique gene expression not seen with normal doses: Implications for its use in cancer therapy. Int. J. Oncol. 2016, 49, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Bihari, B. Bernard Bihari, MD: Low-dose naltrexone for normalizing immune system function. Altern. Ther. Health Med. 2013, 19, 56–65. [Google Scholar]
- Toljan, K.; Vrooman, B. Low-Dose Naltrexone (LDN)—Review of Therapeutic Utilization. Med. Sci. 2018, 6, 82. [Google Scholar] [CrossRef]
- Domnina, Y.M.; Suslov, V.V.; Kedik, S.A.; Volkova, P.O. Approaches to the development of a low-dose naltrexone preparation in the form of a nasal spray (Review). Drug Dev. Regist. 2021, 10, 37–47. [Google Scholar] [CrossRef]
- Selfridge, B.R.; Wang, X.; Zhang, Y.; Yin, H.; Grace, P.M.; Watkins, L.R.; Jacobson, A.E.; Rice, K.C. Structure-Activity Relationships of (+)-Naltrexone-Inspired Toll-like Receptor 4 (TLR4) Antagonists. J. Med. Chem. 2015, 58, 5038–5052. [Google Scholar] [CrossRef]
- Lehnardt, S. Innate immunity and neuroinflammation in the CNS: The role of microglia in toll-like receptor-mediated neuronal injury. Glia 2010, 58, 253–263. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Peng, Y.; Hutchinson, M.R.; Rice, K.C.; Yin, H.; Watkins, L.R. Pharmacological characterization of the opioid inactive isomers (+)-naltrexone and (+)-naloxone as antagonists of toll-like receptor 4. Br. J. Pharmacol. 2016, 173, 856–869. [Google Scholar] [CrossRef]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Dheen, S.T.; Kaur, C.; Ling, E.-A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef]
- Mittelbronn, M. The M1/M2 immune polarization concept in microglia: A fair transfer? Neuroimmunol. Neuroinflamm. 2014, 1, 6. [Google Scholar] [CrossRef]
- Franco, R.; Fernández-Suárez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Stunault, M.I.; Bories, G.; Guinamard, R.R.; Ivanov, S. Metabolism plays a key role during macrophage activation. Mediat. Inflamm. 2018, 2018, 2426138. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Angiari, S. Microglia immunometabolism: From metabolic disorders to single cell metabolism. Semin. Cell Dev. Biol. 2019, 94, 129–137. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yao, X.; Taylor, N.; Bai, Y.; Lovenberg, T.; Bhattacharya, A. RNA sequencing analysis reveals quiescent microglia isolation methods from postnatal mouse brains and limitations of BV2 cells. J. Neuroinflamm. 2018, 15, 153. [Google Scholar] [CrossRef]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef]
- Wang, R.; Green, D.R. Metabolic checkpoints in activated T cells. Nat. Immunol. 2012, 13, 907–915. [Google Scholar] [CrossRef]
- Benmoussa, K.; Garaude, J.; Acín-Pérez, R. How Mitochondrial Metabolism Contributes to Macrophage Phenotype and Functions. J. Mol. Biol. 2018, 430, 3906–3921. [Google Scholar] [CrossRef]
- de Breda, C.N.S.; Davanzo, G.G.; Basso, P.J.; Saraiva Câmara, N.O.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. MTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. MTOR coordinates protein synthesis, mitochondrial activity. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef]
- Jones, R.G.; Pearce, E.J. MenTORing Immunity: mTOR Signaling in the Development and Function of Tissue-Resident Immune Cells. Immunity 2017, 46, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Gravel, S.P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. MTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.Y.; Ma, C.H.E. Bipolar/rod-shaped microglia are proliferating microglia with distinct M1/M2 phenotypes. Sci. Rep. 2014, 4, 7279. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Nigro, A.; Romano, A.; De Domenico, S.; Damato, M.; Franck, J.; Coricciati, C.; Wistorski, M.; Cardon, T.; Fournier, I.; et al. Distinct Protein Expression Networks are Activated in Microglia Cells after Stimulation with IFN-γ and IL-4. Cells 2019, 8, 580. [Google Scholar] [CrossRef]
- Srivastava, A.B.; Gold, M.S. Naltrexone: A History and Future Directions. Cerebrum 2018, 2018, cer-13-18. [Google Scholar]
- Parkitny, L.; Younger, J. Reduced pro-inflammatory cytokines after eight weeks of low-dose naltrexone for Fibromyalgia. Biomedicines 2017, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.S.; Fishman, M.A. Low-Dose Naltrexone for Chronic Pain: Update and Systemic Review. Curr. Pain Headache Rep. 2020, 24, 64. [Google Scholar] [CrossRef] [PubMed]
- Trofimovitch, D.; Baumrucker, S.J. Pharmacology Update: Low-Dose Naltrexone as a Possible Nonopioid Modality for Some Chronic, Nonmalignant Pain Syndromes. Am. J. Hosp. Palliat. Med. 2019, 36, 907–912. [Google Scholar] [CrossRef]
- Jarosz, J.; Miernik, K.; Wa̧chal, M.; Walczak, J.; Krumpl, G. Naltrexone (50 mg) plus psychotherapy in alcohol-dependent patients: A meta-analysis of randomized controlled trials. Am. J. Drug Alcohol Abuse 2013, 39, 144–160. [Google Scholar] [CrossRef]
- Oschwald, A.; Petry, P.; Kierdorf, K.; Erny, D. CNS Macrophages and Infant Infections. Front. Immunol. 2020, 11, 2123. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Borst, K.; Schwabenland, M.; Prinz, M. Microglia metabolism in health and disease. Neurochem. Int. 2019, 130, 104331. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.A. Can the emerging field of immunometabolism provide insights into neuroinflammation? Prog. Neurobiol. 2020, 184, 101719. [Google Scholar] [CrossRef] [PubMed]
- Kučić, N.; Rački, V.; Jurdana, K.; Marcelić, M.; Grabušić, K. Immunometabolic phenotype of BV-2 microglia cells upon murine cytomegalovirus infection. J. Neurovirol. 2019, 25, 496–507. [Google Scholar] [CrossRef]
- Andersen, M.N.; Andersen, N.F.; Rødgaard-Hansen, S.; Hokland, M.; Abildgaard, N.; Møller, H.J. The novel biomarker of alternative macrophage activation, soluble mannose receptor (sMR/sCD206): Implications in multiple myeloma. Leuk. Res. 2015, 39, 971–975. [Google Scholar] [CrossRef]
- Prosniak, M.; Harshyne, L.A.; Andrews, D.W.; Kenyon, L.C.; Bedelbaeva, K.; Apanasovich, T.V.; Heber-Katz, E.; Curtis, M.T.; Cotzia, P.; Hooper, D.C. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin. Cancer Res. 2013, 19, 3776–3786. [Google Scholar] [CrossRef]
- Kazuo, T.; Suzuki, Y.; Yoshimura, K.; Yasui, H.; Karayama, M.; Hozumi, H.; Furuhashi, K.; Enomoto, N.; Fujisawa, T.; Nakamura, Y.; et al. Macrophage Mannose Receptor CD206 Predicts Prognosis in Community-acquired Pneumonia. Sci. Rep. 2019, 9, 18750. [Google Scholar] [CrossRef]
- Zhou, T.; Huang, Z.; Sun, X.; Zhu, X.; Zhou, L.; Li, M.; Cheng, B.; Liu, X.; He, C. Microglia polarization with M1/M2 phenotype changes in rd1 mouse model of retinal degeneration. Front. Neuroanat. 2017, 11, 77. [Google Scholar] [CrossRef]
- Choubey, A.; Dehury, B.; Kumar, S.; Medhi, B.; Mondal, P. Naltrexone a potential therapeutic candidate for COVID-19. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 2834. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, W.; Wang, J.; Yan, J.; Shi, Y.; Zhang, C.; Ge, W.; Wu, J.; Du, P.; Chen, Y. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-κB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine 2018, 35, 345. [Google Scholar] [CrossRef]
- Ye, X.; Zhu, M.; Che, X.; Wang, H.; Liang, X.-J.; Wu, C.; Xue, X.; Yang, J. Lipopolysaccharide induces neuroinflammation in microglia by activating the MTOR pathway and downregulating Vps34 to inhibit autophagosome formation. J. Neuroinflamm. 2020, 17, 18. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef]
- Afridi, R.; Lee, W.-H.; Suk, K. Microglia Gone Awry: Linking Immunometabolism to Neurodegeneration. Front. Cell. Neurosci. 2020, 14, 246. [Google Scholar] [CrossRef] [PubMed]
- Devanney, N.A.; Stewart, A.N.; Gensel, J.C. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp. Neurol. 2020, 329, 113310. [Google Scholar] [CrossRef] [PubMed]
- Grace, P.M.; Shimizu, K.; Strand, K.A.; Rice, K.C.; Deng, G.; Watkins, L.R.; Herson, P.S. (+)-Naltrexone is neuroprotective and promotes alternative activation in the mouse hippocampus after cardiac arrest/cardiopulmonary resuscitation. Brain. Behav. Immun. 2015, 48, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Bordon, Y. Old drug, new trick. Nat. Rev. Immunol. 2018, 18, 295. [Google Scholar] [CrossRef]
- Henn, A.; Lund, S.; Hedtjärn, M.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 2009, 26, 83–94. [Google Scholar] [CrossRef]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of murine microglial cells by a v-raf / v-myc carrying retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]
- Racki, V.; Marcelic, M.; Stimac, I.; Petric, D.; Kucic, N. Effects of haloperidol, risperidone and aripiprazole on the immunometabolic properties of BV-2 microglial cells. Int. J. Mol. Sci. 2021, 22, 4399. [Google Scholar] [CrossRef] [PubMed]
- Cadena-Herrera, D.; Esparza-De Lara, J.E.; Ramírez-Ibañez, N.D.; López-Morales, C.A.; Pérez, N.O.; Flores-Ortiz, L.F.; Medina-Rivero, E. Validation of three viable-cell counting methods: Manual, semi-automated, and automated. Biotechnol. Rep. 2015, 7, 9–16. [Google Scholar] [CrossRef]
- Hartmann, B. P70S6 kinase phosphorylation for pharmacodynamic monitoring. Clin. Chim. Acta 2012, 413, 1387–1390. [Google Scholar] [CrossRef]
- Bass, J.J.; Wilkinson, D.J.; Rankin, D.; Phillips, B.E.; Szewczyk, N.J.; Smith, K.; Atherton, P.J. An overview of technical considerations for Western blotting applications to physiological research. Scand. J. Med. Sci. Sports 2017, 27, 4–25. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kučić, N.; Rački, V.; Šverko, R.; Vidović, T.; Grahovac, I.; Mršić-Pelčić, J. Immunometabolic Modulatory Role of Naltrexone in BV-2 Microglia Cells. Int. J. Mol. Sci. 2021, 22, 8429. https://doi.org/10.3390/ijms22168429
Kučić N, Rački V, Šverko R, Vidović T, Grahovac I, Mršić-Pelčić J. Immunometabolic Modulatory Role of Naltrexone in BV-2 Microglia Cells. International Journal of Molecular Sciences. 2021; 22(16):8429. https://doi.org/10.3390/ijms22168429
Chicago/Turabian StyleKučić, Natalia, Valentino Rački, Roberta Šverko, Toni Vidović, Irena Grahovac, and Jasenka Mršić-Pelčić. 2021. "Immunometabolic Modulatory Role of Naltrexone in BV-2 Microglia Cells" International Journal of Molecular Sciences 22, no. 16: 8429. https://doi.org/10.3390/ijms22168429
APA StyleKučić, N., Rački, V., Šverko, R., Vidović, T., Grahovac, I., & Mršić-Pelčić, J. (2021). Immunometabolic Modulatory Role of Naltrexone in BV-2 Microglia Cells. International Journal of Molecular Sciences, 22(16), 8429. https://doi.org/10.3390/ijms22168429