
miR-302 Attenuates Mutant Huntingtin-Induced Cytotoxicity through Restoration of Autophagy and Insulin Sensitivity

 , , , ,
, , , ,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
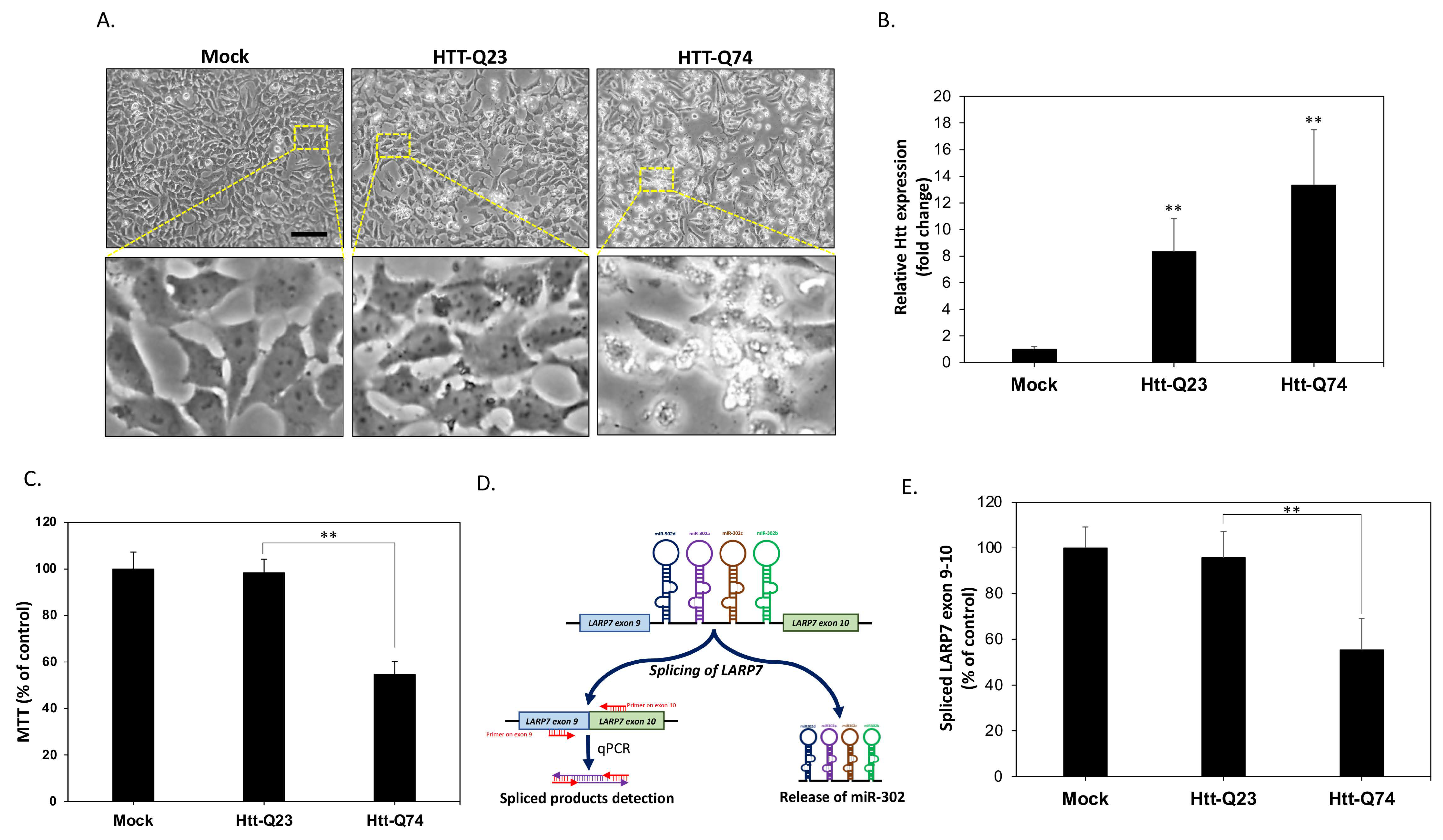
2.1. mHtt-Q74 Significantly Reduces miR-302 Cluster Generation in Neuronal SK-N-MC Cells
2.2. Overexpression of miR-302 Cluster Reduces Apoptosis from mHtt-Induced Neurotoxicity
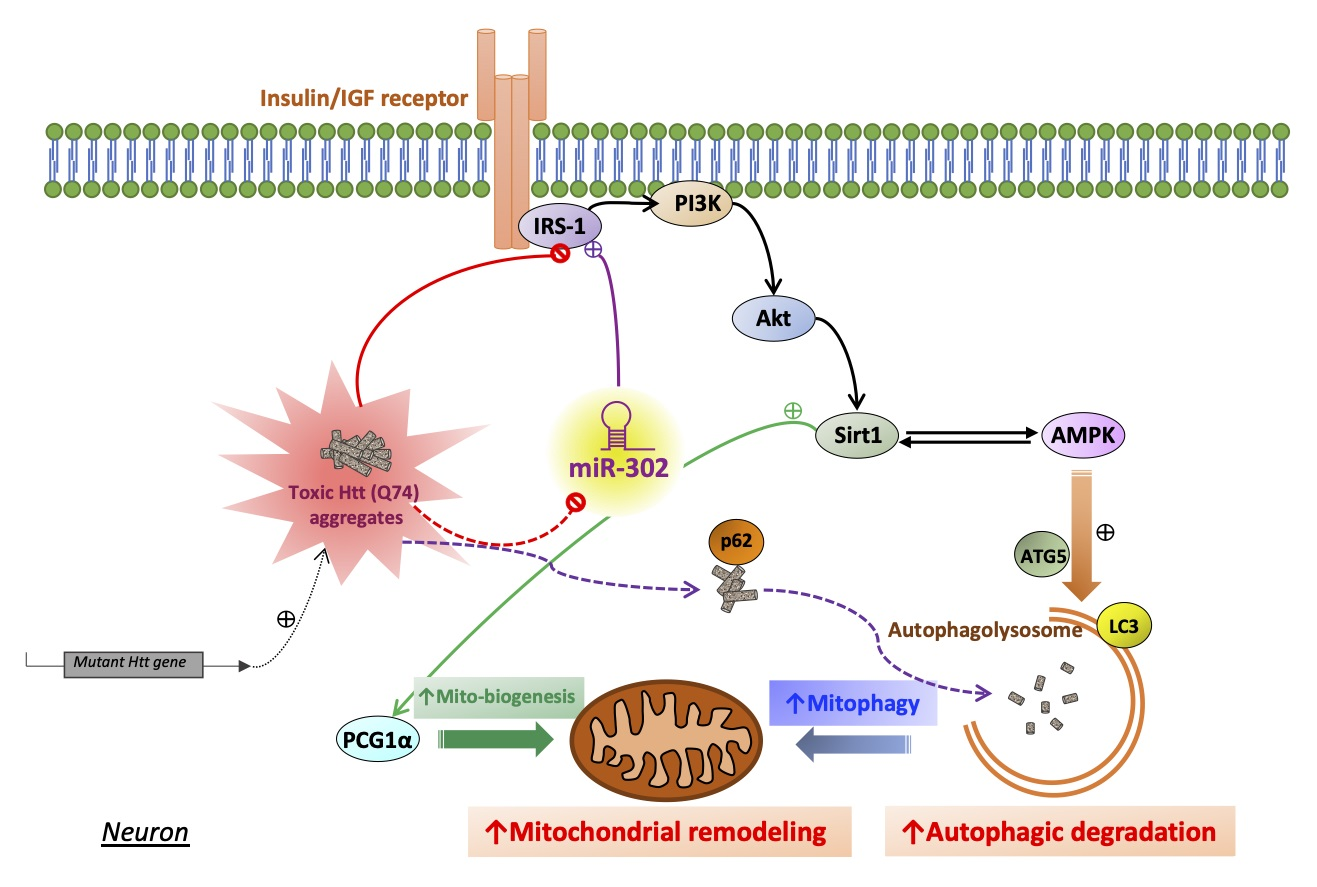
2.3. Upregulation of miR-302 Cluster Facilitates Mitochondrial Autophagy in mHtt-Overexpressing Cells
2.4. miR-302 Cluster Upregulates Sirt1/AMPK-PGC1α Pathway through the Restoration of Insulin Signaling
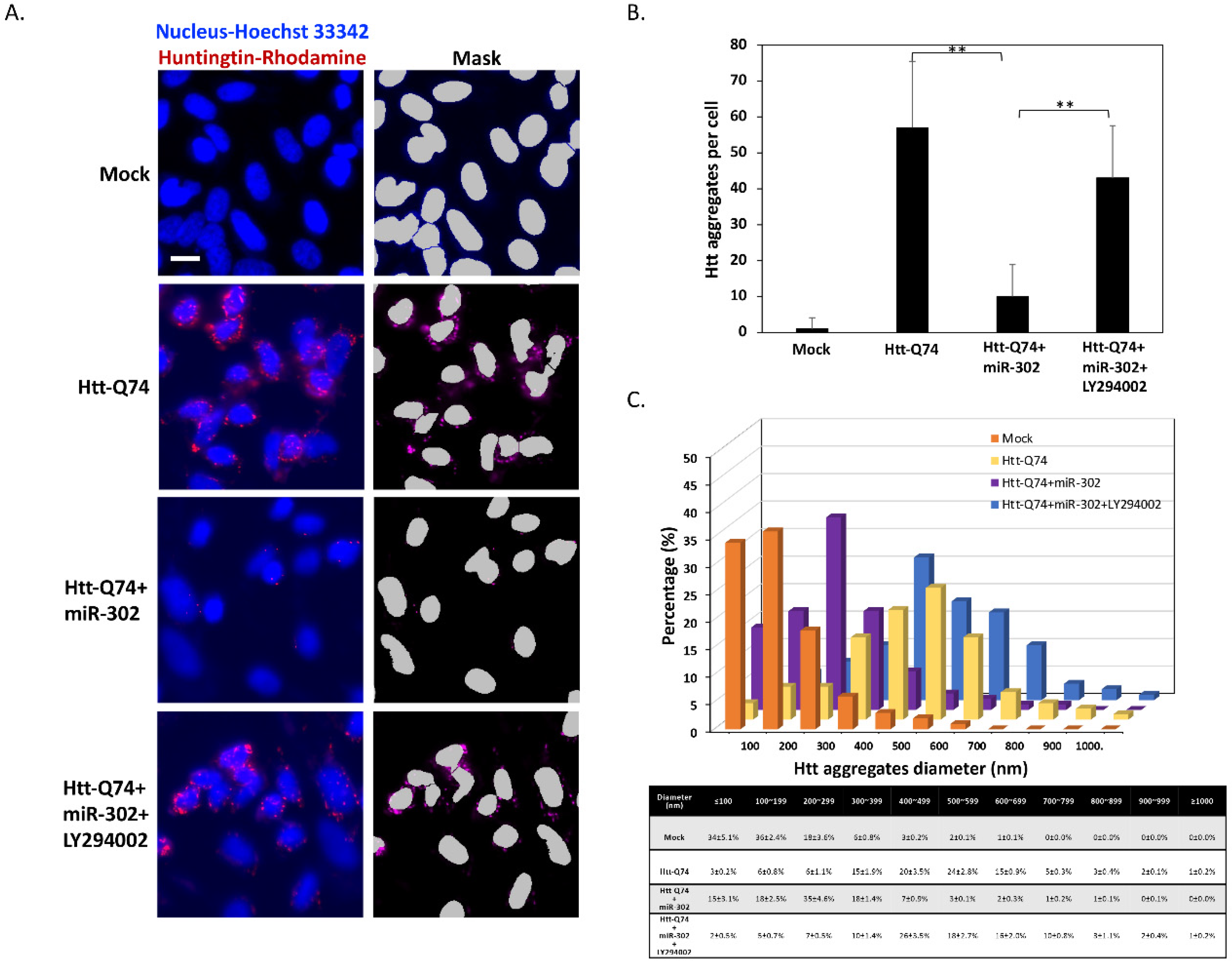
2.5. Upregulation of miR-302 Cluster Reduces the Number and Size of mHtt Aggregates in mHtt-Overexpressing Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture, Transfection and Viability Assay
4.3. mRNA Expression Analysis by Reverse-Transcription Quantitative PCR (qPCR)
4.4. Western Blot Analysis
4.5. DAPI Staining on Nuclei
4.6. TdT-Mediated dUTP-X Nick End Labeling (TUNEL) Assay
4.7. Immunocytochemistry and Acridine Orange (AO) Staining
4.8. Analysis of Mitochondrial Membrane Potential by JC-1 Staining
4.9. High-Content Fluorescence Microscopy
4.10. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soares, T.R.; Reis, S.D.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019, 49, 92–103. [Google Scholar] [CrossRef]
- Moldovean, S.N.; Chiş, V. Molecular Dynamics Simulations Applied to Structural and Dynamical Transitions of the Huntingtin Protein: A Review. ACS Chem. Neurosci. 2020, 11, 105–120. [Google Scholar] [CrossRef]
- van der Burg, J.M.; Björkqvist, M.; Brundin, P. Beyond the brain: Widespread pathology in Huntington’s disease. Lancet Neurol. 2009, 8, 765–774. [Google Scholar] [CrossRef]
- Montojo, M.T.; Aganzo, M.; González, N. Huntington’s Disease and Diabetes: Chronological Sequence of its Association. J. Huntingtons Dis. 2017, 6, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrer, L.A. Diabetes mellitus in Huntington disease. Clin. Genet. 1985, 27, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, A.C.; Gonzalez-Alegre, P.; Schultz, J.L. Diabetes Mellitus Is Associated With an Earlier Age of Onset of Huntington’s Disease. Mov. Disord. 2020. [Google Scholar] [CrossRef]
- Lalić, N.M.; Marić, J.; Svetel, M.; Jotić, A.; Stefanova, E.; Lalić, K.; Dragasević, N.; Milicić, T.; Lukić, L.; Kostić, V.S. Glucose homeostasis in Huntington disease: Abnormalities in insulin sensitivity and early-phase insulin secretion. Arch. Neurol. 2008, 65, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Lin, T.C.; Ho, H.L.; Kuo, C.Y.; Li, H.H.; Korolenko, T.A.; Chen, W.J.; Lai, T.J.; Ho, Y.J.; Lin, C.L. GLP-1 Analogue Liraglutide Attenuates Mutant Huntingtin-Induced Neurotoxicity by Restoration of Neuronal Insulin Signaling. Int. J. Mol. Sci. 2018, 19, 2505. [Google Scholar] [CrossRef] [Green Version]
- Li, H.H.; Lin, S.L.; Huang, C.N.; Lu, F.J.; Chiu, P.Y.; Huang, W.N.; Lai, T.J.; Lin, C.L. miR-302 Attenuates Amyloid-beta-Induced Neurotoxicity through Activation of Akt Signaling. J. Alzheimers Dis. JAD 2016, 50, 1083–1098. [Google Scholar] [CrossRef]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein Misfolding and ER Stress in Huntington’s Disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, H.; Martinez-Vicente, M.; Arias, E.; Kaushik, S.; Sulzer, D.; Cuervo, A.M. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J. Neurosci. 2011, 31, 18492–18505. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2021, 17, 672–689. [Google Scholar] [CrossRef]
- Cordeiro, L.M.; Machado, M.L.; da Silva, A.F.; Obetine Baptista, F.B.; da Silveira, T.L.; Soares, F.A.A.; Arantes, L.P. Rutin protects Huntington’s disease through the insulin/IGF1 (IIS) signaling pathway and autophagy activity: Study in Caenorhabditis elegans model. Food Chem. Toxicol. 2020, 141, 111323. [Google Scholar] [CrossRef]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef] [Green Version]
- Corti, O.; Blomgren, K.; Poletti, A.; Beart, P.M. Autophagy in neurodegeneration: New insights underpinning therapy for neurological diseases. J. Neurochem. 2020, 154, 354–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, A.; Cremona, M.L.; Rothman, J.E. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol. 2006, 172, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Barroso-del Jesus, A.; Lucena-Aguilar, G.; Menendez, P. The miR-302-367 cluster as a potential stemness regulator in ESCs. Cell Cycle 2009, 8, 394–398. [Google Scholar] [CrossRef]
- Ibrayeva, A.; Bay, M.; Pu, E.; Jörg, D.J.; Peng, L.; Jun, H.; Zhang, N.; Aaron, D.; Lin, C.; Resler, G.; et al. Early stem cell aging in the mature brain. Cell Stem Cell 2021, 28, 955–966.e957. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, L.; Wang, L.; Tang, F.; Hua, J. MiR-302 enhances the viability and stemness of male germline stem cells. Reprod. Domest. Anim. 2018, 53, 1580–1588. [Google Scholar] [CrossRef]
- Mas-Bargues, C.; Sanz-Ros, J.; Román-Domínguez, A.; Gimeno-Mallench, L.; Inglés, M.; Viña, J.; Borrás, C. Extracellular Vesicles from Healthy Cells Improves Cell Function and Stemness in Premature Senescent Stem Cells by miR-302b and HIF-1α Activation. Biomolecules 2020, 10, 957. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.Y.; Fang, W.; Lin, S.L. The miR-302-Mediated Induction of Pluripotent Stem Cells (iPSC): Multiple Synergistic Reprogramming Mechanisms. Methods Mol. Biol. 2018, 1733, 283–304. [Google Scholar] [CrossRef]
- Kandasamy, M.; Aigner, L. Reactive Neuroblastosis in Huntington’s Disease: A Putative Therapeutic Target for Striatal Regeneration in the Adult Brain. Front. Cell. Neurosci. 2018, 12, 37. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [Green Version]
- Candeias, E.; Sebastião, I.; Cardoso, S.; Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I.; Duarte, A.I. Brain GLP-1/IGF-1 Signaling and Autophagy Mediate Exendin-4 Protection Against Apoptosis in Type 2 Diabetic Rats. Mol. Neurobiol. 2018, 55, 4030–4050. [Google Scholar] [CrossRef]
- Wamaitha, S.E.; Grybel, K.J.; Alanis-Lobato, G.; Gerri, C.; Ogushi, S.; McCarthy, A.; Mahadevaiah, S.K.; Healy, L.; Lea, R.A.; Molina-Arcas, M.; et al. IGF1-mediated human embryonic stem cell self-renewal recapitulates the embryonic niche. Nat. Commun. 2020, 11, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.L.; Chang, D.C.; Ying, S.Y.; Leu, D.; Wu, D.T. MicroRNA miR-302 inhibits the tumorigenecity of human pluripotent stem cells by coordinate suppression of the CDK2 and CDK4/6 cell cycle pathways. Cancer Res. 2010, 70, 9473–9482. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-C.; Tsou, S.-H.; Chen, W.-J.; Ho, Y.-J.; Hung, H.-C.; Liu, G.-Y.; Singh, S.K.; Li, H.-H.; Lin, C.-L. miR-302 Attenuates Mutant Huntingtin-Induced Cytotoxicity through Restoration of Autophagy and Insulin Sensitivity. Int. J. Mol. Sci. 2021, 22, 8424. https://doi.org/10.3390/ijms22168424
Chang C-C, Tsou S-H, Chen W-J, Ho Y-J, Hung H-C, Liu G-Y, Singh SK, Li H-H, Lin C-L. miR-302 Attenuates Mutant Huntingtin-Induced Cytotoxicity through Restoration of Autophagy and Insulin Sensitivity. International Journal of Molecular Sciences. 2021; 22(16):8424. https://doi.org/10.3390/ijms22168424
Chicago/Turabian StyleChang, Ching-Chi, Sing-Hua Tsou, Wei-Jen Chen, Ying-Jui Ho, Hui-Chih Hung, Guang-Yaw Liu, Sandeep Kumar Singh, Hsin-Hua Li, and Chih-Li Lin. 2021. "miR-302 Attenuates Mutant Huntingtin-Induced Cytotoxicity through Restoration of Autophagy and Insulin Sensitivity" International Journal of Molecular Sciences 22, no. 16: 8424. https://doi.org/10.3390/ijms22168424
APA StyleChang, C.-C., Tsou, S.-H., Chen, W.-J., Ho, Y.-J., Hung, H.-C., Liu, G.-Y., Singh, S. K., Li, H.-H., & Lin, C.-L. (2021). miR-302 Attenuates Mutant Huntingtin-Induced Cytotoxicity through Restoration of Autophagy and Insulin Sensitivity. International Journal of Molecular Sciences, 22(16), 8424. https://doi.org/10.3390/ijms22168424