
Red Light Irradiation In Vivo Upregulates DJ-1 in the Retinal Ganglion Cell Layer and Protects against Axotomy-Related Dendritic Pruning

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
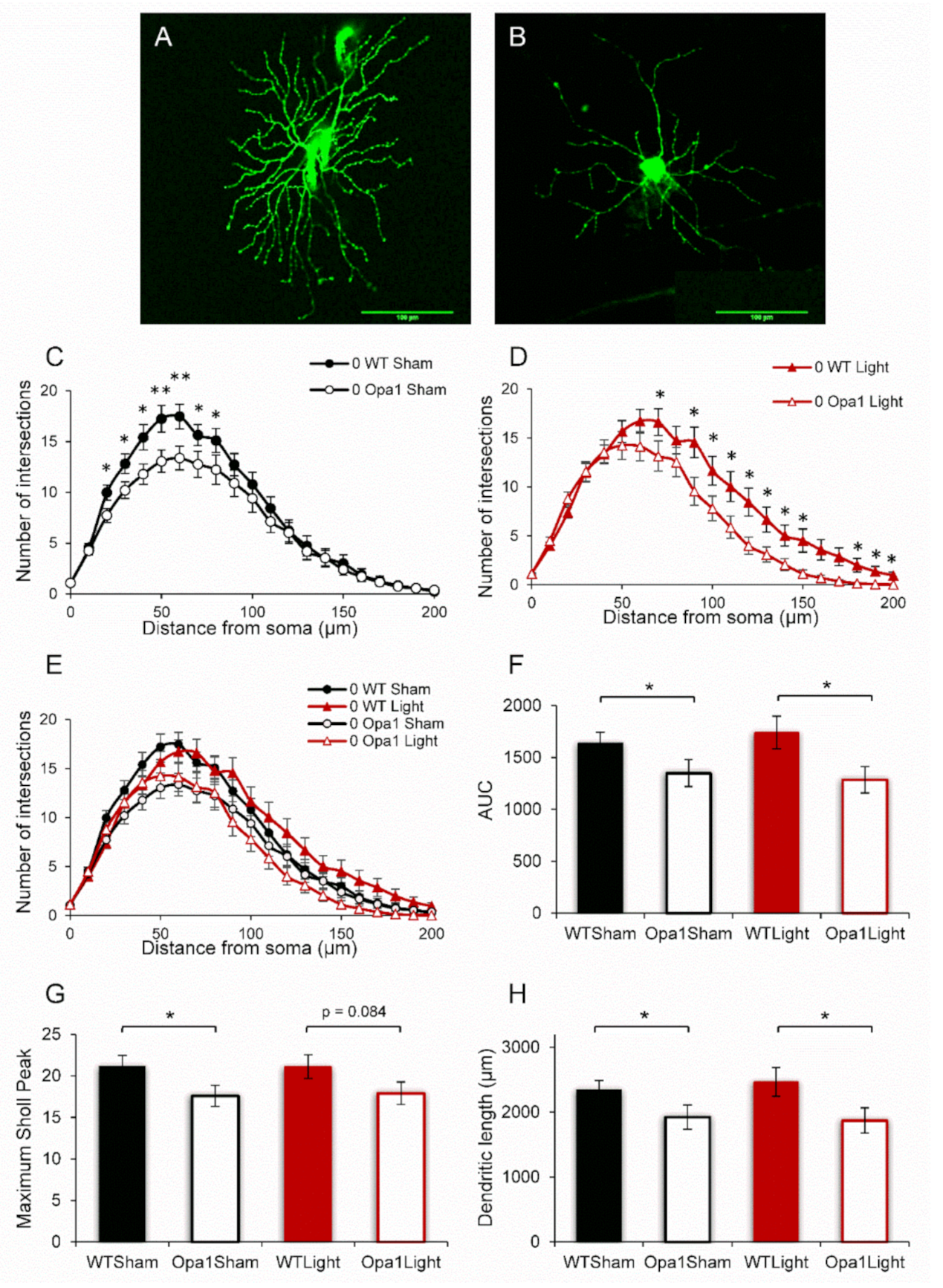
2.1. Aged Opa1 RGCs Exhibit Significant Degeneration in Comparison with RGCs in Age-Matched Wild-Type Retinas
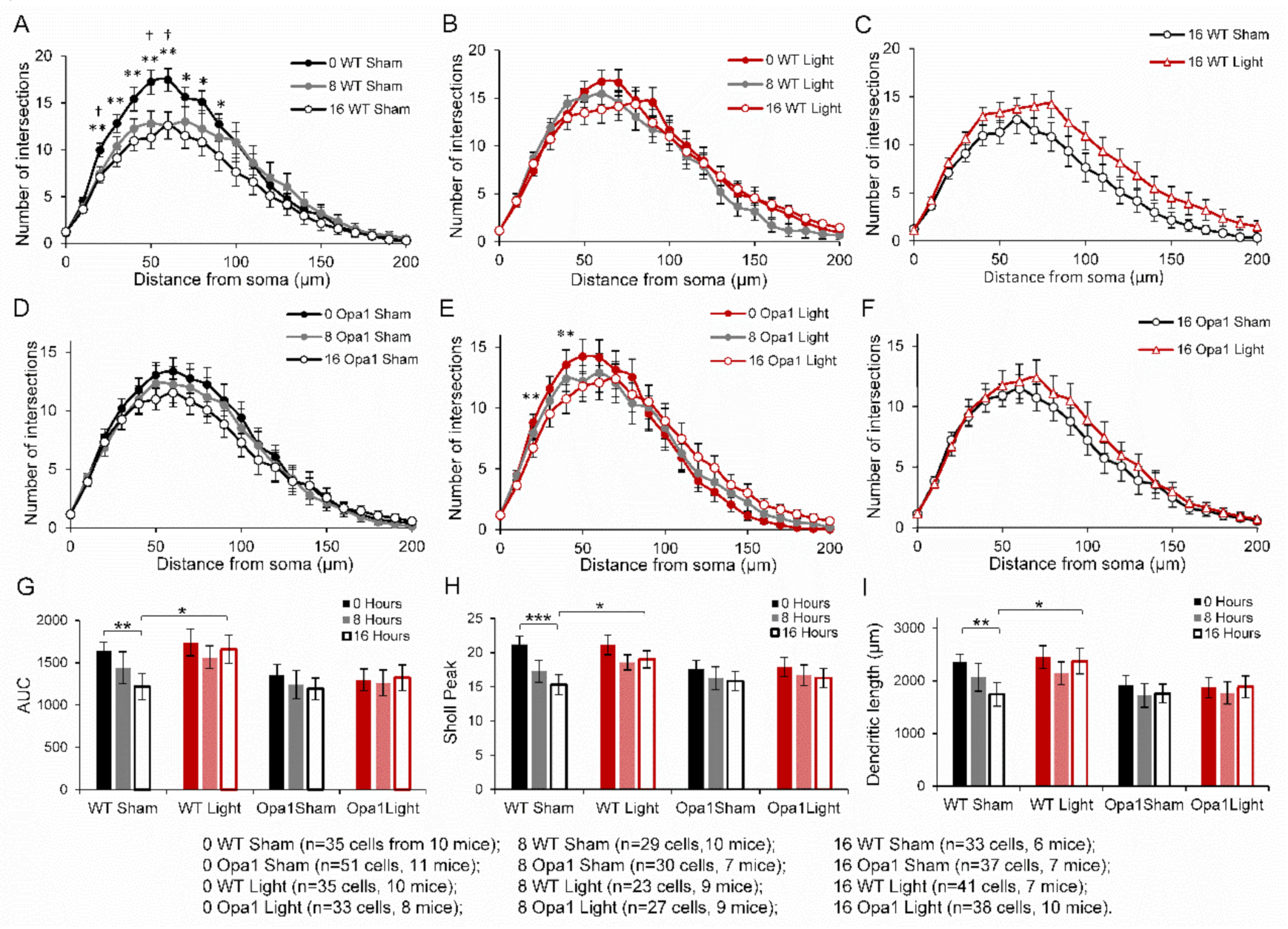
2.2. 670 nm PBM Inhibited the Axotomy-Induced Dendropathy in Aged WT RGCs
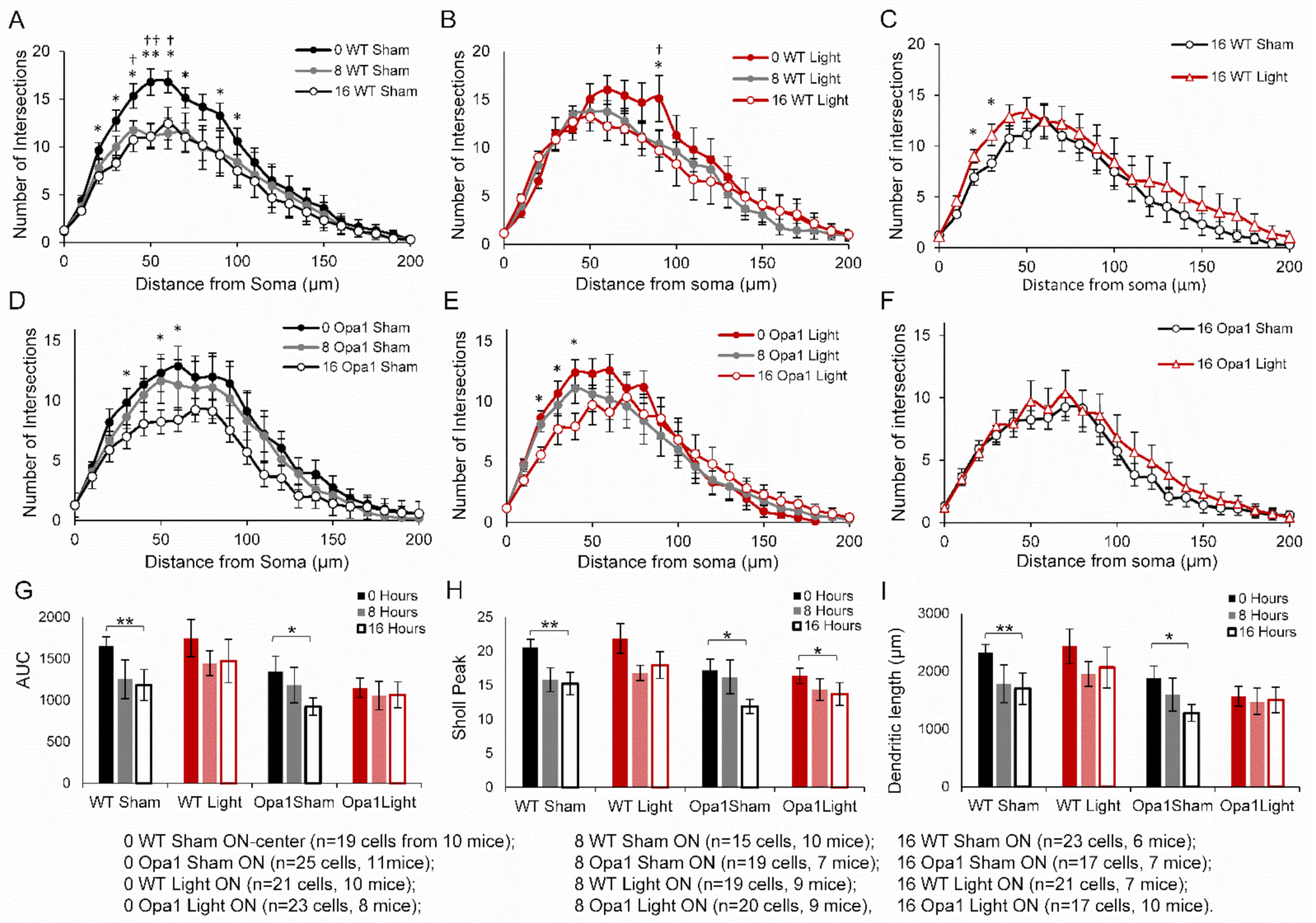
2.3. PBM Did Not Inhibit Axotomy-Induced Dendropathy in the ON-Centre RGC Subpopulation
2.4. IHC Analysis of Molecular Mechanisms
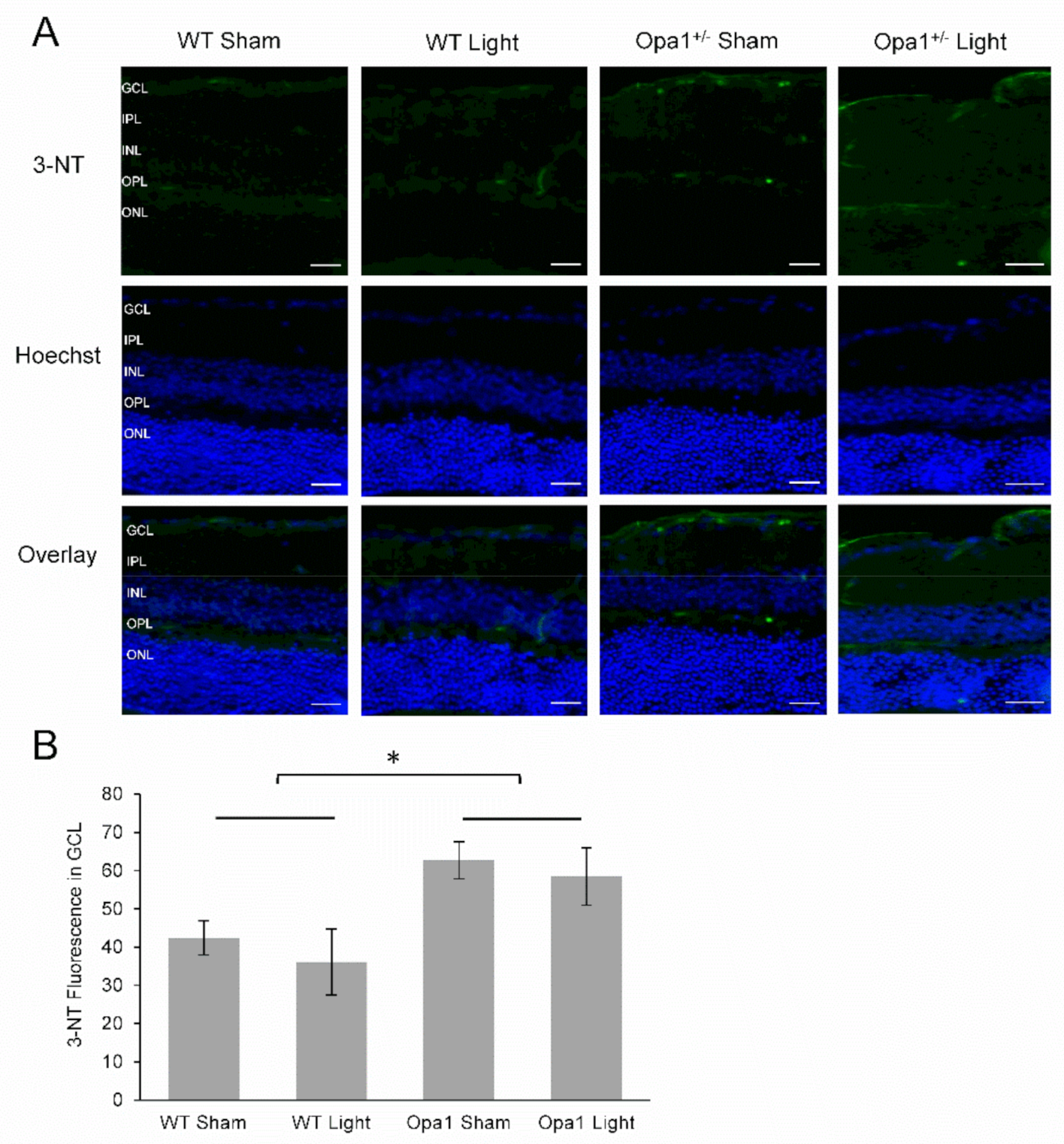
2.4.1. Aged Opa1+/− Mice Display Enhanced Nitrosative Stress in the GCL
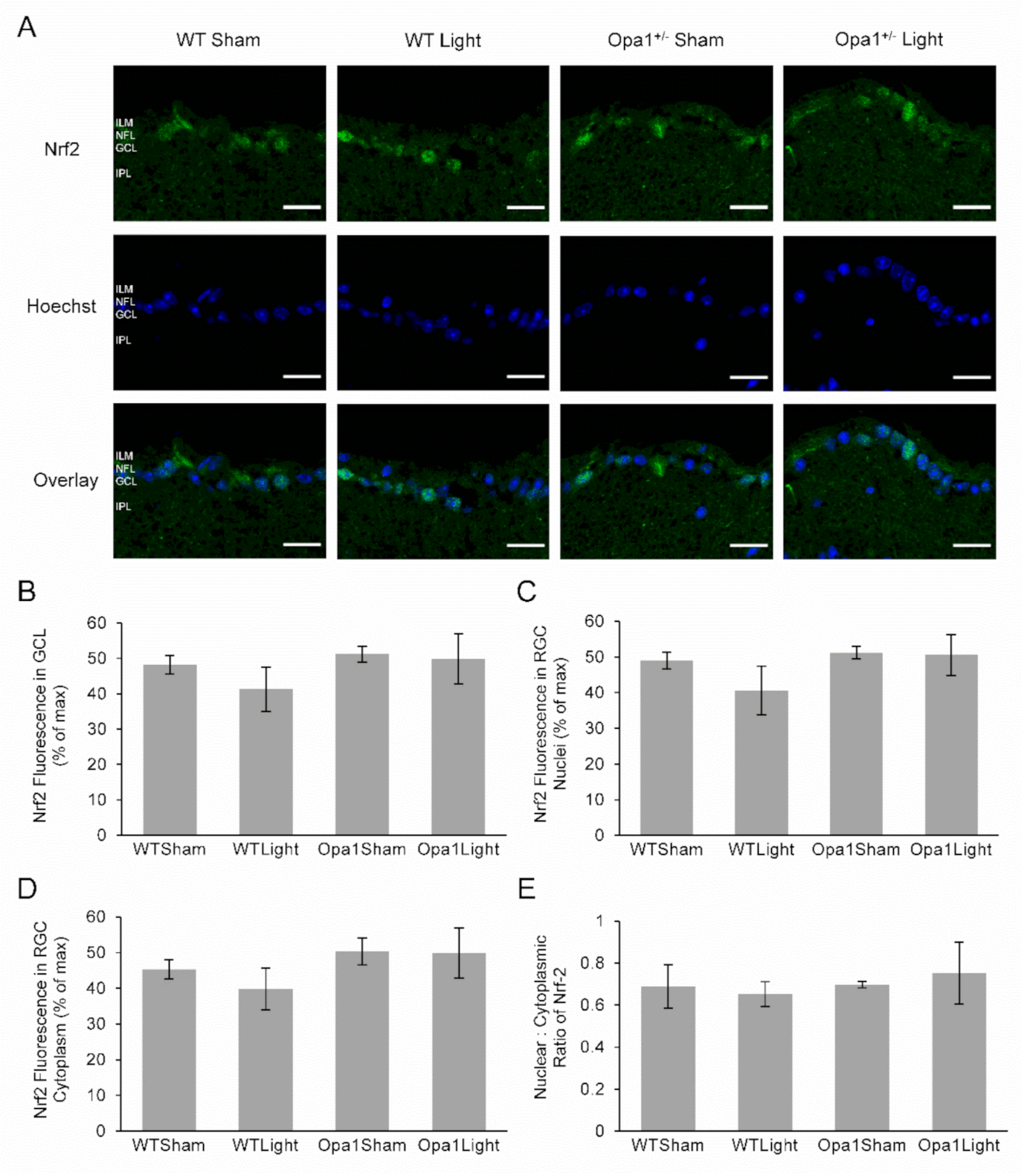
2.4.2. Neither Light Treatment nor the Opa1+/− Mutation Affected Nrf2 Expression or Activation in the GCL
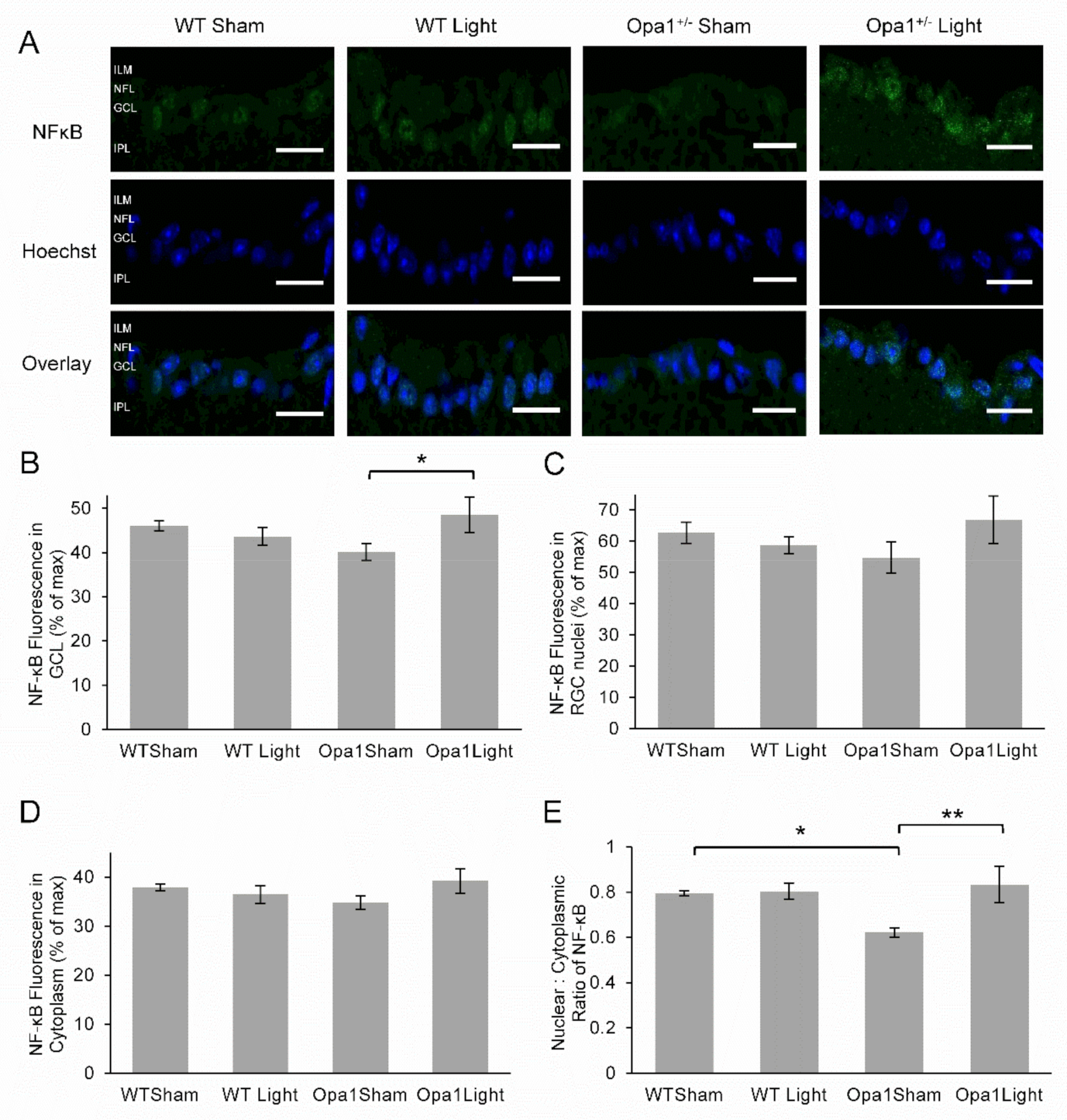
2.4.3. Aged Opa1+/− Retinae Exhibited Reduced NF-kB Activation in the GCL, which was Upregulated by 670 nm Light Treatment
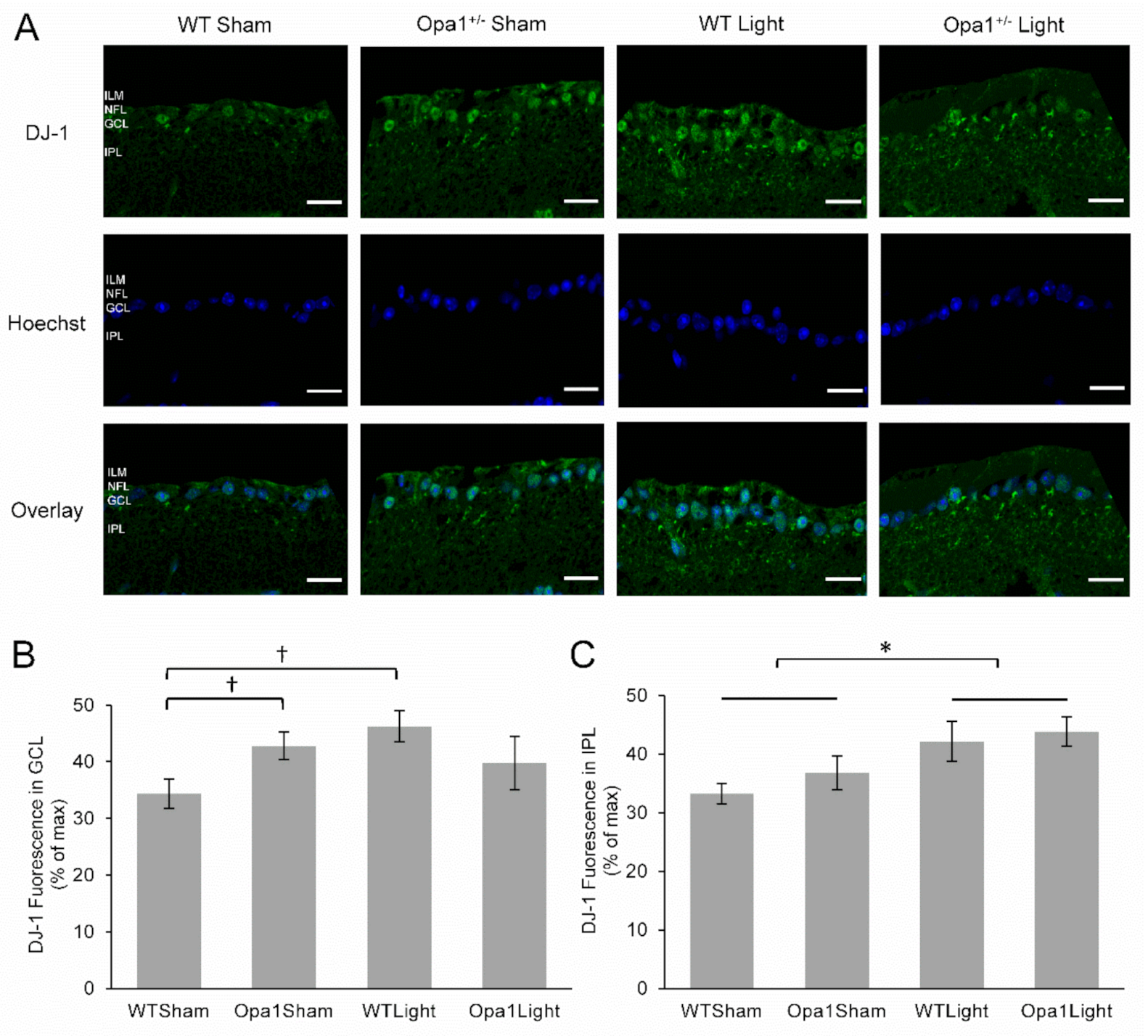
2.4.4. 670 nm Light Treatment Upregulated DJ-1 Expression in the IPL
3. Discussion
4. Materials and Methods
4.1. Animals
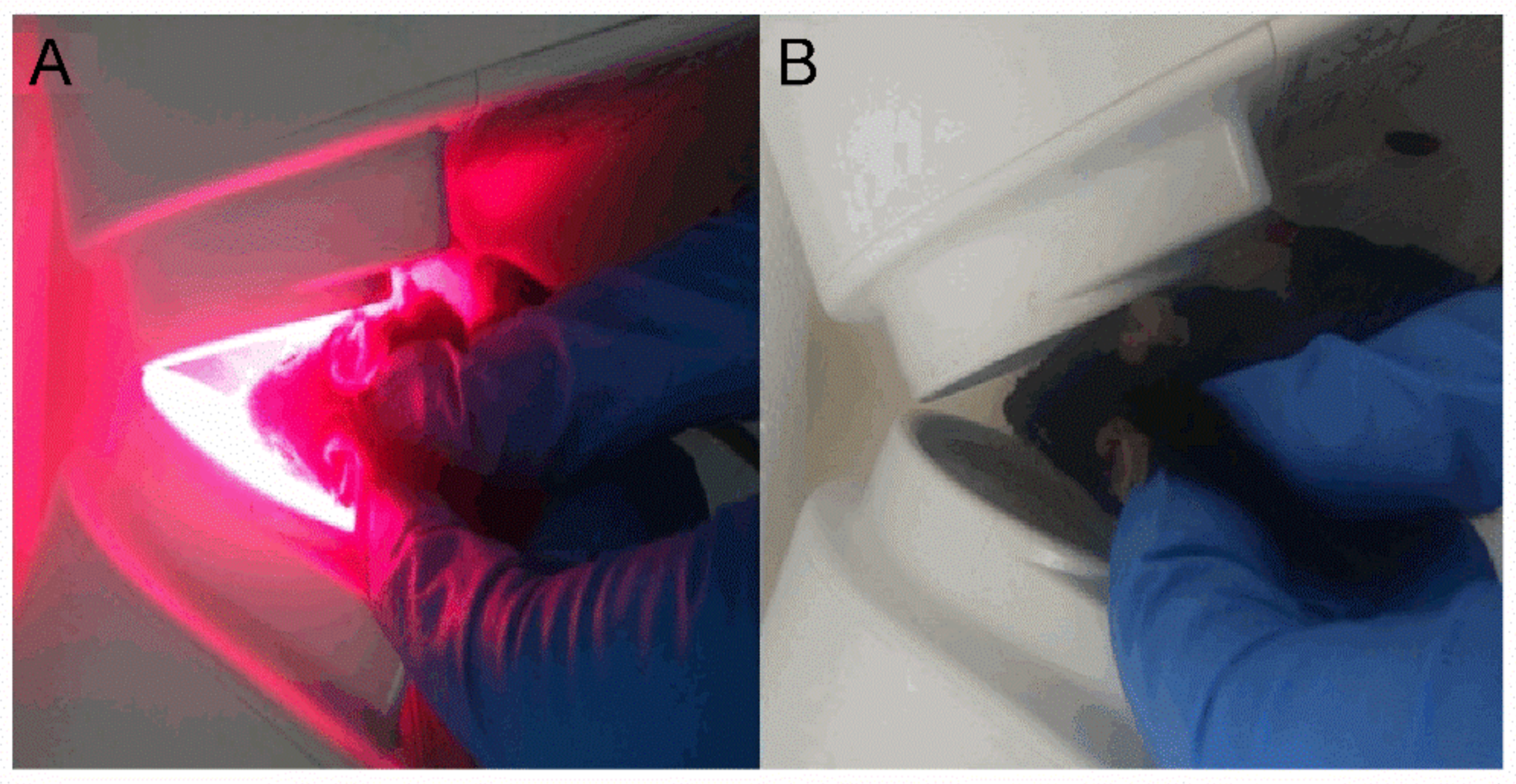
4.2. 670 nm Light Treatment
4.3. Preparation of Eyes for Analysis
4.4. DiOlistic Labeling and Analysis of RGCs
4.5. Immunohistochemistry
4.6. Imaging and Fluorescence Quantification
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Votruba, M.; Moore, A.T.; Bhattacharya, S.S. Clinical features, molecular genetics, and pathophysiology of dominant optic atrophy. J. Med. Genet. 1998, 35, 793–800. [Google Scholar] [CrossRef]
- Morgan, J.E. Retina ganglion cell degeneration in glaucoma: An opportunity missed? A review. Clin. Exp. Ophthalmol. 2012, 40, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Bevan, R.J.; Hughes, T.R.; Williams, P.A.; Good, M.A.; Morgan, B.P.; Morgan, J.E. Retinal ganglion cell degeneration correlates with hippocampal spine loss in experimental Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- La Morgia, C.; Di Vito, L.; Carelli, V.; Carbonelli, M. Patterns of retinal ganglion cell damage in neurodegenerative disorders: Parvocellular vs magnocellular degeneration in optical coherence tomography studies. Front. Neurol. 2017, 8, 710. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C.; Beato, J.; Monteiro, A.; Costa, A.; Penas, S.; Guimarães, J.; Reis, F.F.; Garrett, C. Spectral-domain optical coherence tomography as a potential biomarker in Huntington’s disease. Mov. Disord. 2016, 31, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Del Dotto, V.; Fogazza, M.; Lenaers, G.; Rugolo, M.; Carelli, V.; Zanna, C. OPA1: How much do we know to approach therapy? Pharm. Res. 2018, 131, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Kjer, P.; Jensen, O.A.; Klinken, L. Histopathology of eye, optic nerve and brain in a case of dominant optic atrophy. Acta Ophthalmol. 1983, 61, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Pesch, U.E.A.; Fries, J.E.; Bette, S.; Kalbacher, H.; Wissinger, B.; Alexander, C.; Kohler, K. OPA1, the disease gene for autosomal dominant optic atrophy, is specifically expressed in ganglion cells and intrinsic neurons of the retina. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4217–4225. [Google Scholar] [CrossRef]
- Lenaers, G.; Neutzner, A.; Le Dantec, Y.; Jüschke, C.; Xiao, T.; Decembrini, S.; Swirski, S.; Kieninger, S.; Agca, C.; Kim, U.S.; et al. Dominant optic atrophy: Culprit mitochondria in the optic nerve. Prog. Retin. Eye Res. 2020, 83, 100935. [Google Scholar] [CrossRef]
- Davies, V.J.; Hollins, A.J.; Piechota, M.J.; Yip, W.; Davies, J.R.; White, K.E.; Nicols, P.P.; Boulton, M.E.; Votruba, M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 2007, 16, 1307–1318. [Google Scholar] [CrossRef]
- Williams, P.A.; Morgan, J.E.; Votruba, M. Opa1 deficiency in a mouse model of dominant optic atrophy leads to retinal ganglion cell dendropathy. Brain 2010, 133, 2942–2951. [Google Scholar] [CrossRef]
- Alavi, M.V.; Bette, S.; Schimpf, S.; Schuettauf, F.; Schraermeyer, U.; Wehrl, H.F.; Ruttiger, L.; Beck, S.C.; Tonagel, F.; Pichler, B.J.; et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 2007, 130, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Heiduschka, P.; Schnichels, S.; Fuhrmann, N.; Hofmeister, S.; Schraermeyer, U.; Wissinger, B.; Alavi, M.V. Electrophysiological and histologic assessment of retinal ganglion cell fate in a mouse model for OPA1-associated autosomal dominant optic atrophy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1424–1431. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sun, S.; Erchova, I.; Sengpiel, F.; Votruba, M. Opa1 deficiency leads to diminished mitochondrial bioenergetics with compensatory increased mitochondrial motility. Investig. Ophthalmol. Vis. Sci. 2020, 61, 42. [Google Scholar] [CrossRef]
- Beirne, K.; Rozanowska, M.; Votruba, M. Photostimulation of mitochondria as a treatment for retinal neurodegeneration. Mitochondrion 2017, 36, 85–95. [Google Scholar] [CrossRef]
- Begum, R.; Powner, M.B.; Hudson, N.; Hogg, C.; Jeffery, G. Treatment with 670 Nm light up regulates cytochrome C oxidase expression and reduces inflammation in an age-related macular degeneration model. PLoS ONE 2013, 8, e57828. [Google Scholar] [CrossRef]
- Samanta, A.; Aziz, A.A.; Jhingan, M.; Singh, S.R.; Khanani, A.M.; Chhablani, J. Emerging therapies in nonexudative age-related macular degeneration in 2020. Asia Pac. J. Ophthalmol. 2021. [Google Scholar] [CrossRef]
- Natoli, R.; Valter, K.; Barbosa, M.; Dahlstrom, J.; Rutar, M.; Kent, A.; Provis, J. 670nm photobiomodulation as a novel protection against retinopathy of prematurity: Evidence from oxygen induced retinopathy models. PLoS ONE 2013, 8, e72135. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Du, Y.; Liu, H.; Tang, J.; Veenstra, A.; Kern, T.S. Photobiomodulation inhibits long-term structural and functional lesions of diabetic retinopathy. Diabetes 2018, 67, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Sivapathasuntharam, C.; Sivaprasad, S.; Hogg, C.; Jeffery, G. Aging retinal function is improved by near infrared light (670 Nm) that is associated with corrected mitochondrial decline. Neurobiol. Aging 2017, 52, 66–70. [Google Scholar] [CrossRef]
- Núñez-Álvarez, C.; Osborne, N.N. Blue light exacerbates and red light counteracts negative insults to retinal ganglion cells in situ and R28 cells in vitro. Neurochem. Int. 2019, 125, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Albarracin, R.; Eells, J.; Valter, K. Photobiomodulation protects the retina from light-induced photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3582–3592. [Google Scholar] [CrossRef]
- Lu, Y.-Z.; Fernando, N.; Natoli, R.; Madigan, M.; Valter, K. 670nm light treatment following retinal injury modulates Müller cell gliosis: Evidence from in vivo and in vitro stress models. Exp. Eye Res. 2018, 169, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Merry, G.F.; Munk, M.R.; Dotson, R.S.; Walker, M.G.; Devenyi, R.G. Photobiomodulation reduces drusen volume and improves visual acuity and contrast sensitivity in dry age-related macular degeneration. Acta Ophthalmol. 2017, 95, e270–e277. [Google Scholar] [CrossRef]
- Tang, J.; Herda, A.A.; Kern, T.S. Photobiomodulation in the treatment of patients with non-center-involving diabetic macular oedema. Br. J. Ophthalmol. 2014, 98, 1013–1015. [Google Scholar] [CrossRef]
- Grewal, M.K.; Sivapathasuntharam, C.; Chandra, S.; Gurudas, S.; Chong, V.; Bird, A.; Jeffery, G.; Sivaprasad, S. A pilot study evaluating the effects of 670 Nm photobiomodulation in healthy ageing and age-related macular degeneration. J. Clin. Med. 2020, 9, 1001. [Google Scholar] [CrossRef]
- Shinhmar, H.; Grewal, M.; Sivaprasad, S.; Hogg, C.; Chong, V.; Neveu, M.; Jeffery, G. Optically improved mitochondrial function redeems aged human visual decline. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, e49–e52. [Google Scholar] [CrossRef]
- Markowitz, S.N.; Devenyi, R.G.; Munk, M.R.; Croissant, C.L.; Tedford, S.E.; Rückert, R.; Walker, M.G.; Patino, B.E.; Chen, L.; Nido, M.; et al. A double-masked, randomized, sham-controlled, single-center study with photobiomodulation for the treatment of dry age-related macular degeneration. Retina 2020, 40, 1471–1482. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.R. Mechanisms and mitochondrial redox signaling in photobiomodulation. Photochem. Photobiol. 2018, 94, 199–212. [Google Scholar] [CrossRef]
- Karu, T.I. Mitochondrial signaling in mammalian cells activated by red and near-IR radiation. Photochem. Photobiol. 2008, 84, 1091–1099. [Google Scholar] [CrossRef]
- Mason, M.G.; Nicholls, P.; Cooper, C.E. Re-evaluation of the near infrared spectra of mitochondrial cytochrome c oxidase: Implications for non invasive in vivo monitoring of tissues. Biochim. Biophys. Acta 2014, 1837, 1882–1891. [Google Scholar] [CrossRef]
- Kaynezhad, P.; Tachtsidis, I.; Jeffery, G. Optical monitoring of retinal respiration in real time: 670 Nm light increases the redox state of mitochondria. Exp. Eye Res. 2016, 152, 88–93. [Google Scholar] [CrossRef]
- Amaroli, A.; Ravera, S.; Baldini, F.; Benedicenti, S.; Panfoli, I.; Vergani, L. Photobiomodulation with 808-Nm diode laser light promotes wound healing of human endothelial cells through increased reactive oxygen species production stimulating mitochondrial oxidative phosphorylation. Lasers Med. Sci. 2019, 34, 495–504. [Google Scholar] [CrossRef]
- Huang, Y.-Y.; Nagata, K.; Tedford, C.E.; McCarthy, T.; Hamblin, M.R. Low-level laser therapy (LLLT) reduces oxidative stress in primary cortical neurons in vitro. J. Biophotonics 2013, 6, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.-H.; Arany, P.R.; Huang, Y.-Y.; Tomkinson, E.M.; Sharma, S.K.; Kharkwal, G.B.; Saleem, T.; Mooney, D.; Yull, F.E.; Blackwell, T.S.; et al. Low-level laser therapy activates NF-KB via generation of reactive oxygen species in mouse embryonic fibroblasts. PLoS ONE 2011, 6, e22453. [Google Scholar] [CrossRef]
- Lev, N.; Ickowicz, D.; Melamed, E.; Offen, D. Oxidative insults induce DJ-1 upregulation and redistribution: Implications for neuroprotection. Neurotoxicology 2008, 29, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.C.; Lee, J.; John, J.M.; Gonzalez-Lima, F. Neuroprotective effects of near-infrared light in an in vivo model of mitochondrial optic neuropathy. J. Neurosci. 2008, 28, 13511–13521. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.L.; Whelan, H.T.; Eells, J.T.; Wong-Riley, M.T.T. Near-infrared light via light-emitting diode treatment is therapeutic against rotenone- and 1-methyl-4-phenylpyridinium ion-induced neurotoxicity. Neuroscience 2008, 153, 963–974. [Google Scholar] [CrossRef]
- Beirne, K.; Rozanowska, M.; Votruba, M. Red light treatment in an axotomy model of neurodegeneration. Photochem. Photobiol. 2016, 92, 624–631. [Google Scholar] [CrossRef]
- Magharious, M.M.; D’Onofrio, P.M.; Koeberle, P.D. Optic nerve transection: A model of adult neuron apoptosis in the central nervous system. J. Vis. Exp. 2011. [Google Scholar] [CrossRef]
- Liang, H.L.; Whelan, H.T.; Eells, J.T.; Meng, H.; Buchmann, E.; Lerch-Gaggl, A.; Wong-Riley, M. Photobiomodulation partially rescues visual cortical neurons from cyanide-induced apoptosis. Neuroscience 2006, 139, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Dong, Y.; Wu, C.; Li, Y.; Guo, Y.; Yang, B.; Zong, X.; Hamblin, M.R.; Liu, T.C.-Y.; Zhang, Q. Photobiomodulation preconditioning prevents cognitive impairment in a neonatal rat model of hypoxia-ischemia. J. Biophotonics 2019, 12, e201800359. [Google Scholar] [CrossRef]
- Campolo, N.; Issoglio, F.M.; Estrin, D.A.; Bartesaghi, S.; Radi, R. 3-nitrotyrosine and related derivatives in proteins: Precursors, radical intermediates and impact in function. Essays Biochem. 2020, 64, 111–133. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Velichkova, M.; Hasson, T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol. Cell. Biol. 2005, 25, 4501–4513. [Google Scholar] [CrossRef]
- Rizzi, M.; Migliario, M.; Rocchetti, V.; Tonello, S.; Renò, F. Near-infrared laser increases MDPC-23 odontoblast-like cells proliferation by activating redox sensitive pathways. J. Photochem. Photobiol. B 2016, 164, 283–288. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Baltimore, D. I kappa B: A specific inhibitor of the NF-kappa B transcription factor. Science 1988, 242, 540–546. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef]
- Tang, S.; Le, P.K.; Tse, S.; Wallace, D.C.; Huang, T. Heterozygous mutation of Opa1 in drosophila shortens lifespan mediated through increased reactive oxygen species production. PLoS ONE 2009, 4, e4492. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, B.E.; Stephens, E.; Bartlett, C.A.; Serghiou, S.; Giacci, M.K.; Williams, A.; Hart, N.S.; Fitzgerald, M. Comparative assessment of phototherapy protocols for reduction of oxidative stress in partially transected spinal cord slices undergoing secondary degeneration. BMC Neurosci. 2016, 17, 21. [Google Scholar] [CrossRef]
- Muili, K.A.; Gopalakrishnan, S.; Eells, J.T.; Lyons, J.-A. Photobiomodulation induced by 670 Nm light ameliorates MOG35-55 induced EAE in female C57BL/6 mice: A role for remediation of nitrosative stress. PLoS ONE 2013, 8, e67358. [Google Scholar] [CrossRef] [PubMed]
- de Freitas, L.F.; Hamblin, M.R. Proposed mechanisms of photobiomodulation or low-level light therapy. IEEE J. Sel. Top. Quantum Electron. 2016, 22, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Park, H.-A.; Mnatsakanyan, N.; Niu, Y.; Licznerski, P.; Wu, J.; Miranda, P.; Graham, M.; Tang, J.; Boon, A.J.W.; et al. Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Almikhlafi, M.A.; Stauch, K.L.; Villeneuve, L.M.; Purnell, P.R.; Lamberty, B.G.; Fox, H.S. Deletion of DJ-1 in rats affects protein abundance and mitochondrial function at the synapse. Sci. Rep. 2020, 10, 13719. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Aredo, B.; Chen, B.; Zhao, C.X.; He, Y.-G.; Ufret-Vincenty, R.L. Mice with a combined deficiency of superoxide dismutase 1 (Sod1), DJ-1 (Park7), and parkin (Prkn) develop spontaneous retinal degeneration with aging. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3740–3751. [Google Scholar] [CrossRef]
- Upadhyay, M.; Milliner, C.; Bell, B.A.; Bonilha, V.L. Oxidative stress in the retina and retinal pigment epithelium (RPE): Role of aging, and DJ-1. Redox Biol. 2020, 37. [Google Scholar] [CrossRef]
- Zeng, J.; Zhao, H.; Chen, B. DJ-1/PARK7 inhibits high glucose-induced oxidative stress to prevent retinal pericyte apoptosis via the PI3K/AKT/MTOR signaling pathway. Exp. Eye Res. 2019, 189, 107830. [Google Scholar] [CrossRef]
- Martín-Nieto, J.; Uribe, M.L.; Esteve-Rudd, J.; Herrero, M.T.; Campello, L. A role for DJ-1 against oxidative stress in the mammalian retina. Neurosci. Lett. 2019, 708, 134361. [Google Scholar] [CrossRef]
- Lingappan, K. NF-ΚB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Wu, M.; Bian, Q.; Liu, Y.; Fernandes, A.F.; Taylor, A.; Pereira, P.; Shang, F. Sustained oxidative stress inhibits NF-ΚB activation partially via inactivating the proteasome. Free Radic. Biol. Med. 2009, 46, 62–69. [Google Scholar] [CrossRef]
- Hemish, J.; Nakaya, N.; Mittal, V.; Enikolopov, G. Nitric oxide activates diverse signaling pathways to regulate gene expression. J. Biol. Chem. 2003, 278, 42321–42329. [Google Scholar] [CrossRef]
- Tribble, J.R.; Williams, P.A.; Caterson, B.; Sengpiel, F.; Morgan, J.E. Digestion of the glycosaminoglycan extracellular matrix by chondroitinase ABC supports retinal ganglion cell dendritic preservation in a rodent model of experimental glaucoma. Mol. Brain 2018, 11, 69. [Google Scholar] [CrossRef]
- Longair, M.H.; Baker, D.A.; Armstrong, J.D. Simple Neurite Tracer: Open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 2011, 27, 2453–2454. [Google Scholar] [CrossRef] [PubMed]
- Sholl, D.A. Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat. 1953, 87, 387–406. [Google Scholar] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beirne, K.; Freeman, T.J.; Rozanowska, M.; Votruba, M. Red Light Irradiation In Vivo Upregulates DJ-1 in the Retinal Ganglion Cell Layer and Protects against Axotomy-Related Dendritic Pruning. Int. J. Mol. Sci. 2021, 22, 8380. https://doi.org/10.3390/ijms22168380
Beirne K, Freeman TJ, Rozanowska M, Votruba M. Red Light Irradiation In Vivo Upregulates DJ-1 in the Retinal Ganglion Cell Layer and Protects against Axotomy-Related Dendritic Pruning. International Journal of Molecular Sciences. 2021; 22(16):8380. https://doi.org/10.3390/ijms22168380
Chicago/Turabian StyleBeirne, Kathy, Thomas J. Freeman, Malgorzata Rozanowska, and Marcela Votruba. 2021. "Red Light Irradiation In Vivo Upregulates DJ-1 in the Retinal Ganglion Cell Layer and Protects against Axotomy-Related Dendritic Pruning" International Journal of Molecular Sciences 22, no. 16: 8380. https://doi.org/10.3390/ijms22168380
APA StyleBeirne, K., Freeman, T. J., Rozanowska, M., & Votruba, M. (2021). Red Light Irradiation In Vivo Upregulates DJ-1 in the Retinal Ganglion Cell Layer and Protects against Axotomy-Related Dendritic Pruning. International Journal of Molecular Sciences, 22(16), 8380. https://doi.org/10.3390/ijms22168380