
A New N-Substituted 1H-Isoindole-1,3(2H)-Dione Derivative—Synthesis, Structure and Affinity for Cyclooxygenase Based on In Vitro Studies and Molecular Docking

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
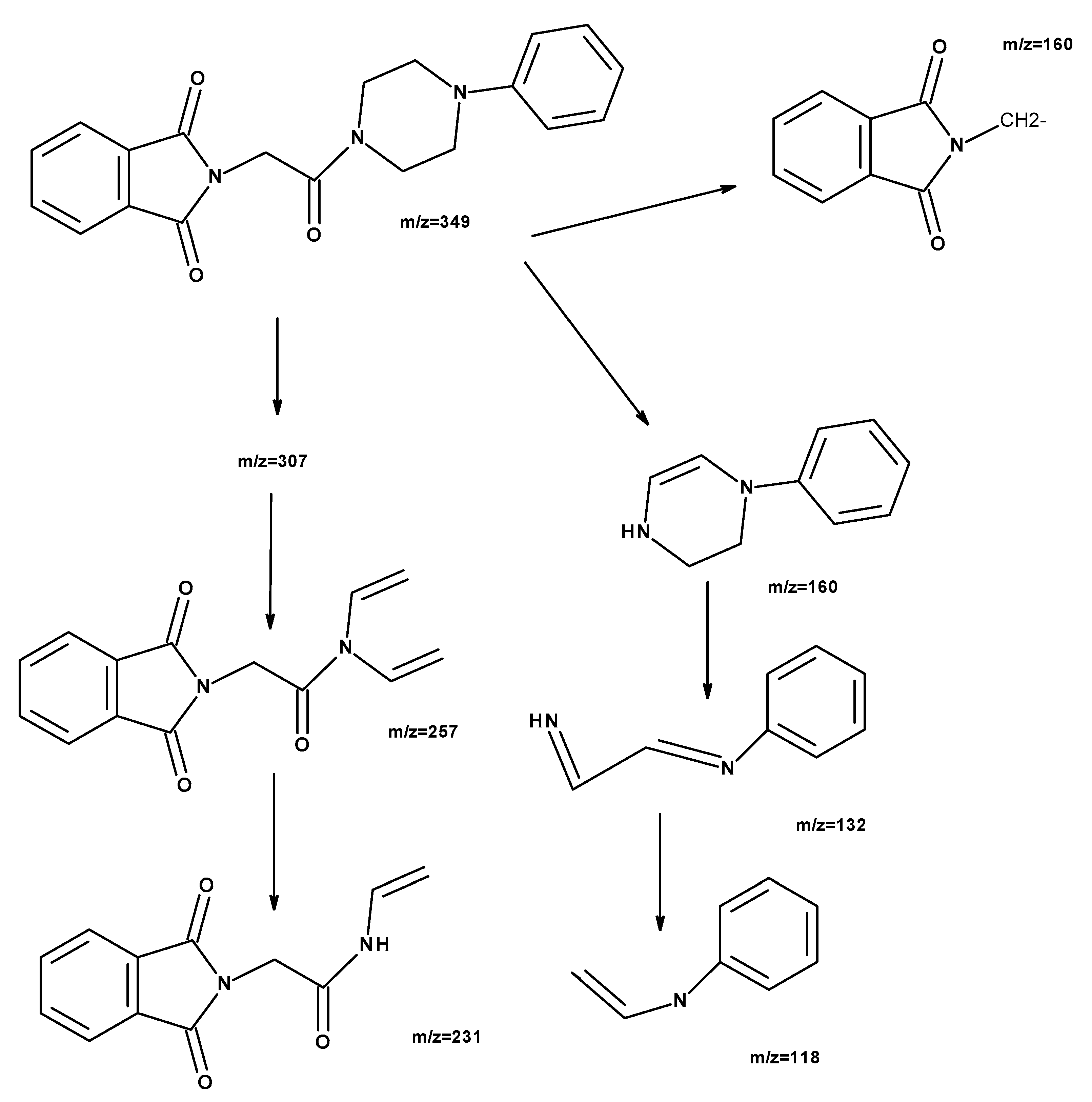
2.1. Chemistry
2.2. Molecular Properties
2.3. Biological Evaluation
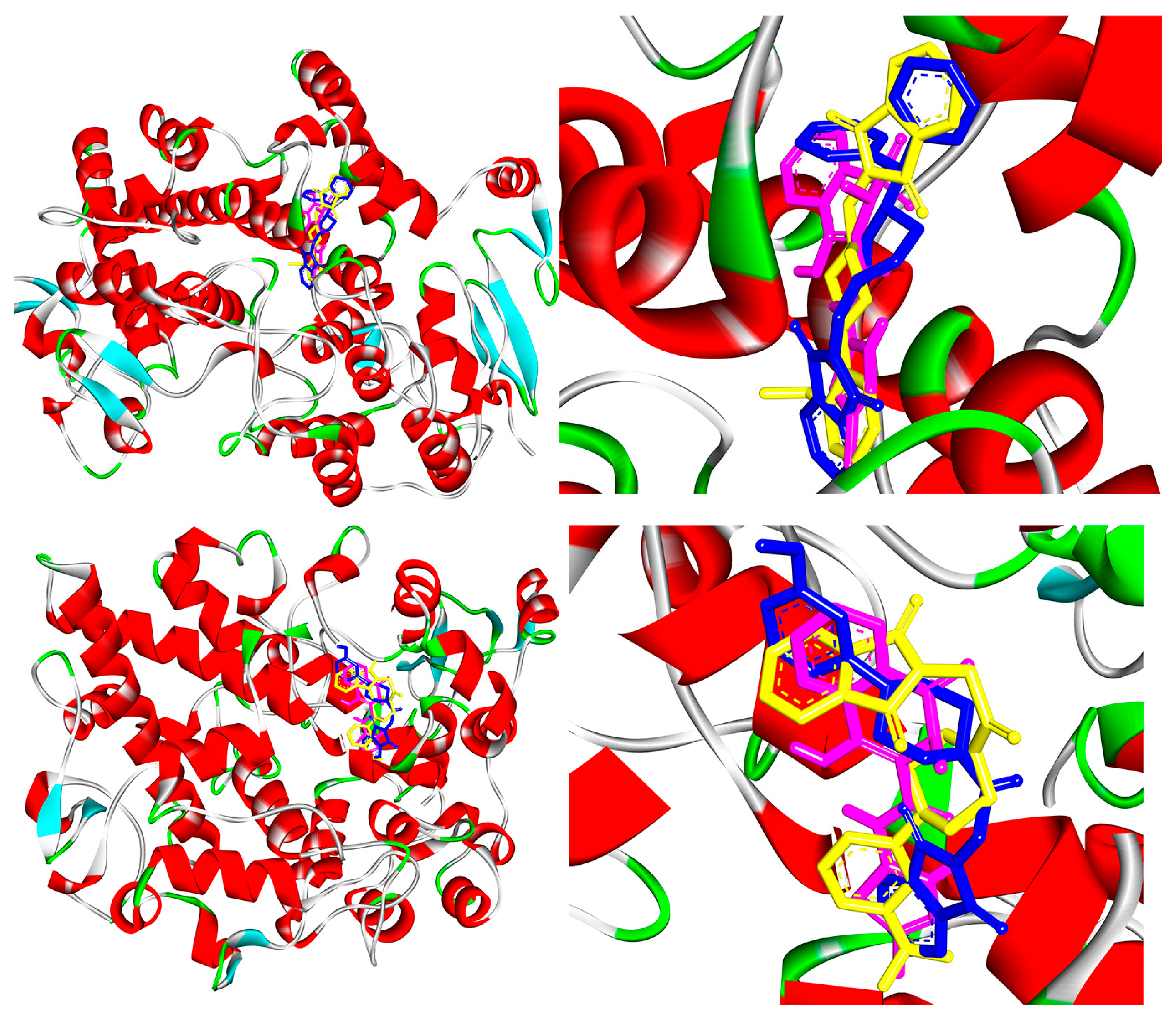
2.4. COX-1 and COX-2 Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
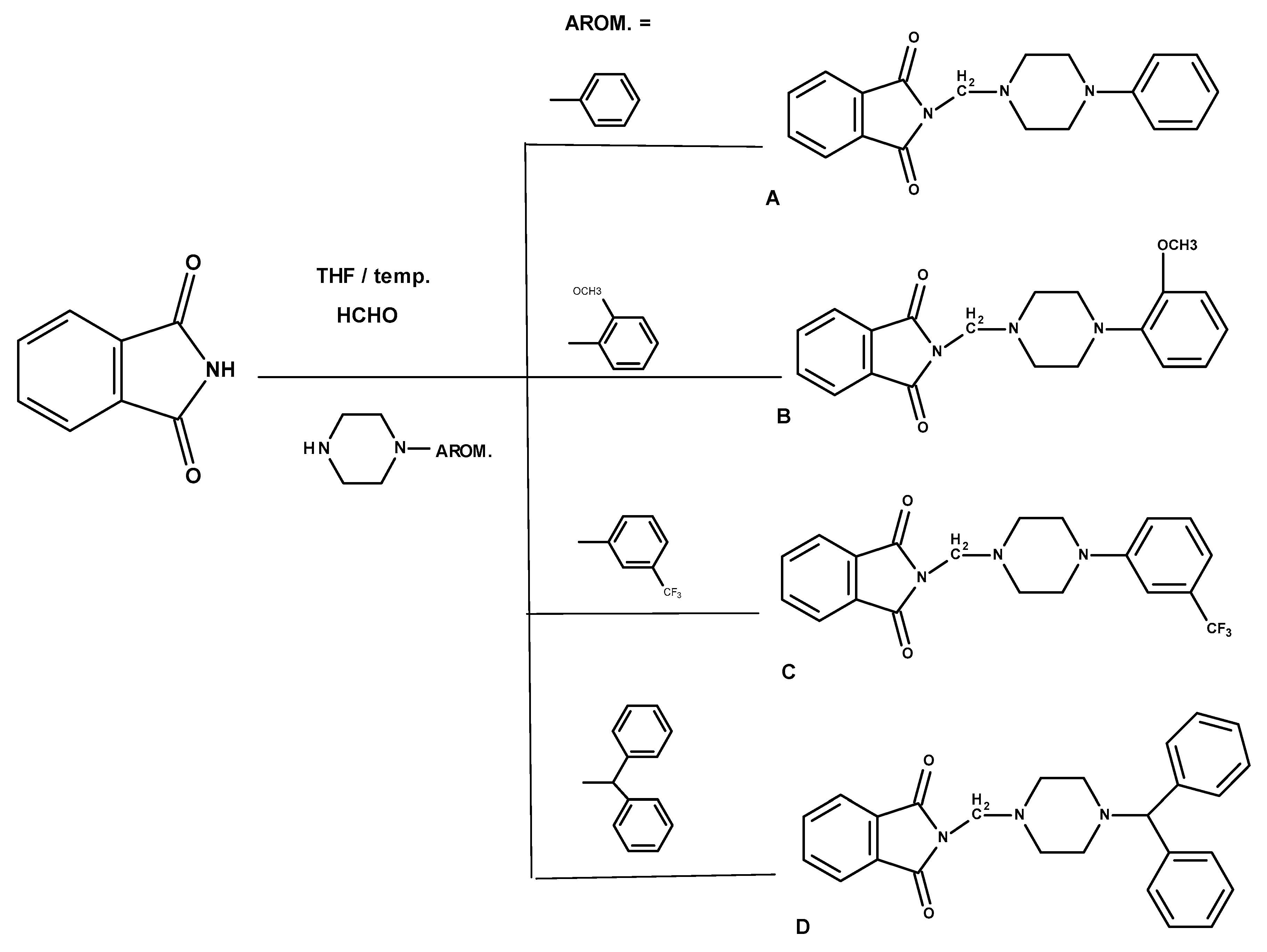
3.1.1. General Method for the Preparation of the Mannich Bases A–D
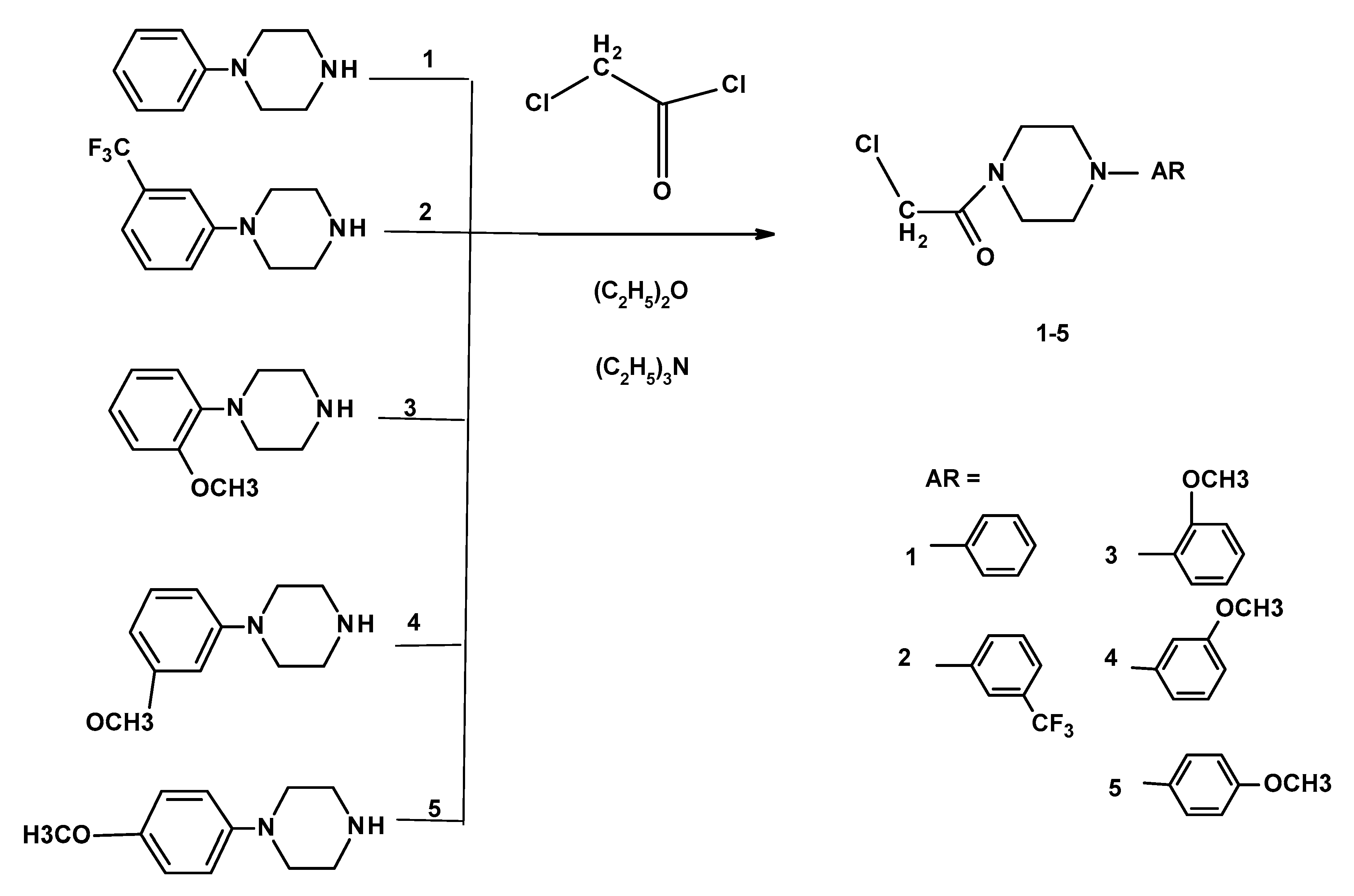
3.1.2. General Method for the Preparation of the 2-chloro-1-(N-arylopiperazinyl)ethanone 1–5
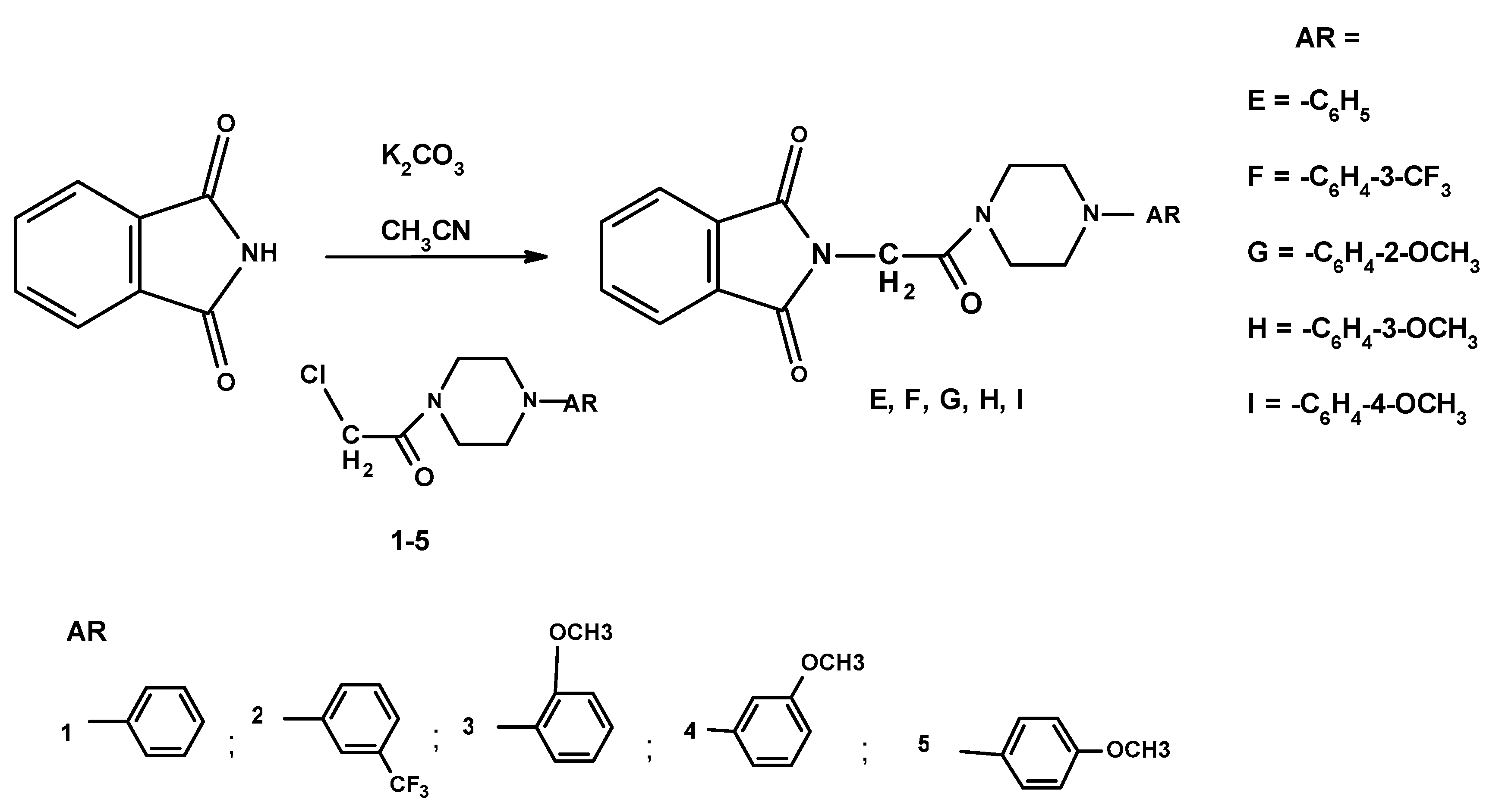
3.1.3. General Method for the Preparation of the 2-[2-oxo-2-[4-arylphenyl-1-piperazinyl]]ethyl-1H-isoindole-1,3(2H)-diones E–I
3.2. Cell Line and Condition
3.3. Tested Compounds
3.4. Cell Viability
3.5. ROS and RNS Level and DNA Damage
3.6. Cyclooxygenase Inhibition Assay
3.7. Statistical Analysis
3.8. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abu-Hashem, A.A.; Gouda, M.A. Synthesis, anti-inflammatory and analgesic evaluation of certain new 3a,4,9,9a-tetrahydro-4,9-benzenobenz[f]isoindole-1,3-diones. Arch. Pharm. 2011, 344, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Cao, J.; Huang, J.; Liu, S.; Im, D.S.; Yoo, J.W.; Jung, J.H. The in vitro and in vivo anti-inflammatory effects of a phthalimide PPAR-γ agonist. Mar. Drugs 2017, 15, 7. [Google Scholar] [CrossRef] [Green Version]
- Ignasik, M.; Bajda, M.; Guzior, N.; Prinz, M.; Holzgrabe, U.; Malawska, B. Design, synthesis and evaluation of novel 2-(Aminoalkyl)-isoindoline-1,3- dione derivatives as dual-binding site acetylcholinesterase inhibitors. Arch. Pharm. 2012, 345, 509–516. [Google Scholar] [CrossRef]
- Panek, D.; Wiȩckowska, A.; Pasieka, A.; Godyń, J.; Jończyk, J.; Bajda, M.; Knez, D.; Gobec, S.; Malawska, B. Design, synthesis, and biological evaluation of 2-(benzylamino-2-hydroxyalkyl)isoindoline-1,3-diones derivatives as potential disease-modifying multifunctional anti-Alzheimer agents. Molecules 2018, 23, 347. [Google Scholar] [CrossRef] [Green Version]
- Guzior, N.; Bajda, M.; Skrok, M.; Kurpiewska, K.; Lewiński, K.; Brus, B.; Pišlar, A.; Kos, J.; Gobec, S.; Malawska, B. Development of multifunctional, heterodimeric isoindoline-1,3-dione derivatives as cholinesterase and β-amyloid aggregation inhibitors with neuroprotective properties. Eur. J. Med. Chem. 2015, 92, 738–749. [Google Scholar] [CrossRef]
- Godin, A.M.; Araújo, D.P.; Menezes, R.R.; Brito, A.M.S.; Melo, I.S.F.; Coura, G.M.E.; Soares, D.G.; Bastos, L.F.S.; Amaral, F.A.; Ribeiro, L.S.; et al. Activities of 2-phthalimidethanol and 2-phthalimidethyl nitrate, phthalimide analogs devoid of the glutarimide moiety, in experimental models of inflammatory pain and edema. Pharmacol. Biochem. Behav. 2014, 122, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Godin, A.M.; Araújo, D.P.; César, I.C.; Menezes, R.R.; Brito, A.M.S.; Melo, I.S.F.; Coura, G.M.E.; Bastos, L.F.S.; Almeida, M.O.; Byrro, R.M.D.; et al. Activities of 2-phthalimidethyl nitrate and 2-phthalimidethanol in the models of nociceptive response and edema induced by formaldehyde in mice and preliminary investigation of the underlying mechanisms. Eur. J. Pharmacol. 2015, 756, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Kumar, P.; Kumar, N.; Singh, B. Recent Advances in the Chemistry of Phthalimide Analogues and their Therapeutic Potential. Mini-Rev. Med. Chem. 2010, 10, 678–704. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.H.; Liu, J.Y.; Kim, W.K.; Hong, J.Y.; Park, S.H.; Kim, D.; Qin, S.N.; Luu, T.T.T.; Park, H.J.; Xu, Y.N.; et al. Synthesis and biological activity of new phthalimides as potential anti-inflammatory agents. Bioorg. Med. Chem. 2017, 25, 3396–3405. [Google Scholar] [CrossRef] [PubMed]
- Godin, A.M.; Araújo, D.P.; Menezes, R.R.; de Brito, A.M.S.; Melo, I.S.F.; Coura, G.M.E.; Bastos, L.F.S.; Amaral, F.A.; Teixeira, M.M.; de Fátima, Â.; et al. 2-Phthalimidethanol and 2-phthalimidethyl nitrate inhibit mechanical allodynia, neutrophil recruitment and cytokine and chemokine production in a murine model of articular inflammation. Pharmacol. Rep. 2017, 69, 691–695. [Google Scholar] [CrossRef]
- Batista, C.R.A.; Godin, A.M.; Melo, I.S.F.; Coura, G.M.E.; Matsui, T.C.; Dutra, M.M.G.B.; Brito, A.M.S.; Canhestro, W.G.; Alves, R.J.; Araújo, D.P.; et al. The phthalimide analogues N-3-hydroxypropylphthalimide and N-carboxymethyl-3-nitrophthalimide exhibit activity in experimental models of inflammatory and neuropathic pain. Pharmacol. Rep. 2019, 71, 1177–1183. [Google Scholar] [CrossRef]
- Magli, E.; Kędzierska, E.; Kaczor, A.A.; Bielenica, A.; Severino, B.; Gibuła-Tarłowska, E.; Kotlińska, J.H.; Corvino, A.; Sparaco, R.; Esposito, G.; et al. Synthesis, docking studies, and pharmacological evaluation of 2-hydroxypropyl-4-arylpiperazine derivatives as serotoninergic ligands. Arch. Pharm. 2021, 354, 2000414. [Google Scholar] [CrossRef]
- Wójcicka, A.; Redzicka, A. An overview of the biological activity of pyrrolo[3,4-c]pyridine derivatives. Pharmaceuticals 2021, 14, 354. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M.; El-Azab, A.S.; Ghiaty, A.H.; Gratteri, P.; Supuran, C.T.; Nocentini, A. 4-Substituted benzenesulfonamides featuring cyclic imides moieties exhibit potent and isoform-selective carbonic anhydrase II/IX inhibition. Bioorg. Chem. 2019, 83, 198–204. [Google Scholar] [CrossRef]
- Stiz, D.; Corrêa, R.; D’Auria, F.D.; Simonetti, G.; Cechinel-Filho, V. Synthesis of cyclic imides (methylphtalimides, carboxylic acid phtalimides and itaconimides) and evaluation of their antifungal potential. Med. Chem. 2016, 12, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh, F.; Jafari, E. Cyclic imide derivatives: As promising scaffold for the synthesis of antimicrobial agents. J. Res. Med. Sci. 2018, 23. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Krzyżak, E.; Zborowska, A.; Zając, P.; Potyrak, K.; Peregrym, K.; Wiatrak, B.; Marciniak, A.; Świątek, P. Design, synthesis and comprehensive investigations of pyrrolo[3,4-d]pyridazinone-based 1,3,4-oxadiazole as new class of selective cox-2 inhibitors. Int. J. Mol. Sci. 2020, 21, 9623. [Google Scholar] [CrossRef] [PubMed]
- Jaafar, A.-H.; Zuhair, M.-E.; Sadeq, A.-T.; Rand, A.-Q. Synthesis of isoindoline-1,3-dione derivatives as cyclooxygenase (Cox) S inhibitors. Int. J. Pharma Bio Sci. 2021, 11. [Google Scholar] [CrossRef]
- Szkatuła, D.; Krzyżak, E.; Mogilski, S.; Sapa, J.; Filipek, B.; Świątek, P. Bioresearch of new 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-diones. Molecules 2020, 25, 5883. [Google Scholar] [CrossRef] [PubMed]
- Krzyżak, E.; Szkatuła, D.; Wiatrak, B.; Gębarowski, T.; Marciniak, A. Synthesis, cyclooxygenases inhibition activities and interactions with BSA of N-substituted 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-diones derivatives. Molecules 2020, 25, 2934. [Google Scholar] [CrossRef]
- Dogruer, D.S.; Kupeli, E.; Yesilada, E.; Sahin, M.F. Synthesis of new 2-[1(2H)-phthalazinon-2-yl]-acetamide and 3-[1(2H)-phthalazinon-2-yl]-propanamide derivatives as antinociceptive and anti-inflammatory agents. Arch. Pharm. 2004, 337, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Zarghi, A.; Arfaei, S. Selective COX-2 inhibitors: A review of their structure-activity relationships. Iran. J. Pharm. Res. 2011, 10, 655. [Google Scholar] [PubMed]
- Vardeny, O.; Solomon, S.D. Cyclooxygenase-2 inhibitors, nonsteroidal anti-inflammatory drugs, and cardiovascular risk. Cardiol. Clin. 2008, 26, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Śladowska, H.; Filipek, B.; Szkatuła, D.; Sabiniarz, A.; Kardasz, M.; Potoczek, J.; Sieklucka-Dziuba, M.; Rajtar, G.; Kleinrok, Z.; Lis, T. Investigations on the synthesis and pharmacological properties of 4-alkoxy-2-[2-hydroxy-3-(4-aryl-1-piperazinyl)propyl]-6-methyl-1H-pyrrolo[3,4-c]pyridine-1,3(2H)-diones. Farmaco 2002, 57, 897–908. [Google Scholar] [CrossRef]
- Sladowska, H.; Szkatuła, D.; Filipek, B.; Maciag, D.; Sapa, J.; Zygmunt, M. Synthesis and properties of 2-(4-substituted)butyl derivatives of some 2,3-dihydro-1,3-dioxo-1H-pyrrolo[3,4-c]pyridines. Pharmazie 2001, 56, 133–138. [Google Scholar] [CrossRef]
- Roman, G. Mannich bases in medicinal chemistry and drug design. Eur. J. Med. Chem. 2015, 89, 743–816. [Google Scholar] [CrossRef] [PubMed]
- Muszalska, I.; Ciemniejewski, M.P.; Lesniewska, M.A.; Szkatuła, D.; Malinka, W. Forced degradation and photodegradation studies of pyrrolo[3,4-c]pyridine-1,3-dione derivatives as analgesic active compounds using HPLC, UV and IR spectrometry, and HPLC/MS methods. J. AOAC Int. 2015, 98, 1248–1259. [Google Scholar] [CrossRef]
- Świątek, P.; Strzelecka, M.; Urniaz, R.; Gębczak, K.; Gębarowski, T.; Gąsiorowski, K.; Malinka, W. Synthesis, COX-1/2 inhibition activities and molecular docking study of isothiazolopyridine derivatives. Bioorganic Med. Chem. 2017, 25, 316–326. [Google Scholar] [CrossRef]
- Top 12 Software & Tools for Data Analysts. 2021. Available online: www.datapine.com/articles/data-analyst-tools-software (accessed on 3 June 2021).
- Clapham, D. Stability testing. In ICH Quality Guidelines; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 45–72. [Google Scholar]
- Jensen, A.W. Drugs: Photochemistry and Photostability Edited by A. Albini and E. Fasani (Dell’ Universita Di Pavia). Royal Society of Chemistry: Cambridge. 1998. viii + 330 pp. ISBN 0-85404-743-3. J. Am. Chem. Soc. 1999, 121, 8678. [Google Scholar] [CrossRef]
- Boreen, A.L.; Arnold, W.A.; McNeill, K. Photodegradation of pharmaceuticals in the aquatic environment: A review. Aquat. Sci. 2003, 65, 320–341. [Google Scholar] [CrossRef]
- De Vries, H.; van Henegouwen, G.M.J.B. Photochemical decomposition of Lomefloxacin in vitro and in vivo. J. Photochem. Photobiol. B Biol. 2000, 58, 6–12. [Google Scholar] [CrossRef]
- Hjorth Tønnesen, H. Photostability of Drugs and Drug Formulation, 2nd ed.; Taylor Francis Group: Abingdon, UK, 2004. [Google Scholar]
- Vasquez, M.I.; Hapeshi, E.; Fatta-Kassinos, D.; Kümmerer, K. Biodegradation potential of ofloxacin and its resulting transformation products during photolytic and photocatalytic treatment. Environ. Sci. Pollut. Res. 2013, 20, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Tønnesen, H.H.; Brunsvik, A.; Løseth, K.; Bergh, K.; Gederaas, O.A. Photoreactivity of biologically active compounds. XVIII. Photostability of ofloxacin in the solid state and in a tablet formulation. Pharmazie 2007, 62, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; de Guidi, G.; Giuffrida, S.; Sortino, S.; Chillemi, R.; Sciuto, S. Molecular mechanisms of photosensitization induced by drugs XII. Photochemistry and photosensitization of rufloxacin: An unusual photodegradation path for the antibacterials containing a fluoroquinolone-like chromophore. Photochem. Photobiol. 1999, 70, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration Cheminformatics. Available online: http://www.molispiration.com (accessed on 3 June 2021).
- El-Gohary, N.S.; Shaaban, M.I. Synthesis, antimicrobial, antiquorum-sensing, and cytotoxic activities of new series of isoindoline-1,3-dione, Pyrazolo[5,1-a]isoindole, and pyridine derivatives. Arch. Pharm. 2015, 348, 666–680. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated h-bonding network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [Green Version]
- Grajzer, M.; Wiatrak, B.; Gębarowski, T.; Boba, A.; Rój, E.; Gorczyca, D.; Prescha, A. Bioactive compounds of raspberry oil emulsions induced oxidative stress via stimulating the accumulation of reactive oxygen species and NO in cancer cells. Oxidative Med. Cell. Longev. 2021, 2021, 5561672. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785. [Google Scholar] [CrossRef] [Green Version]
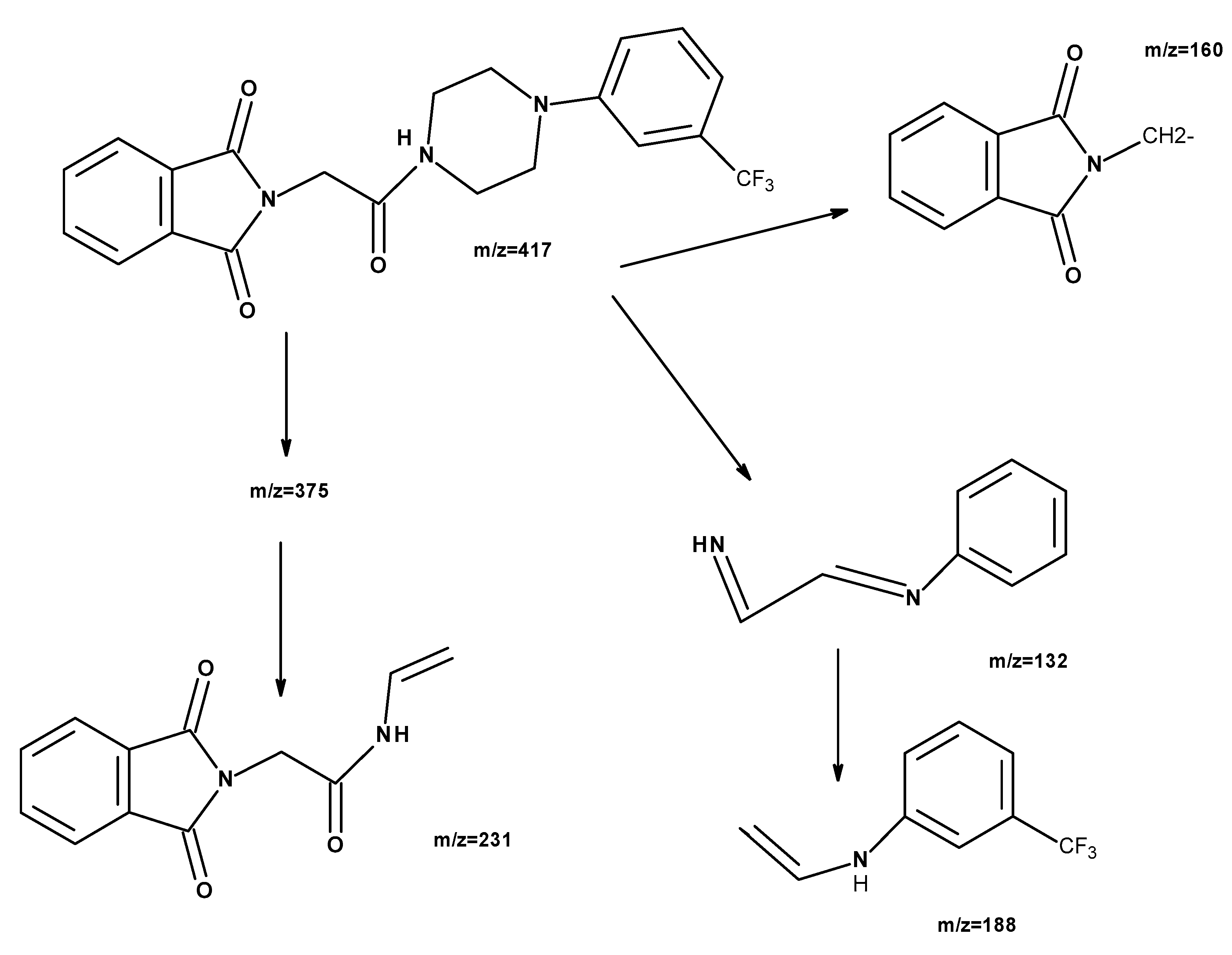
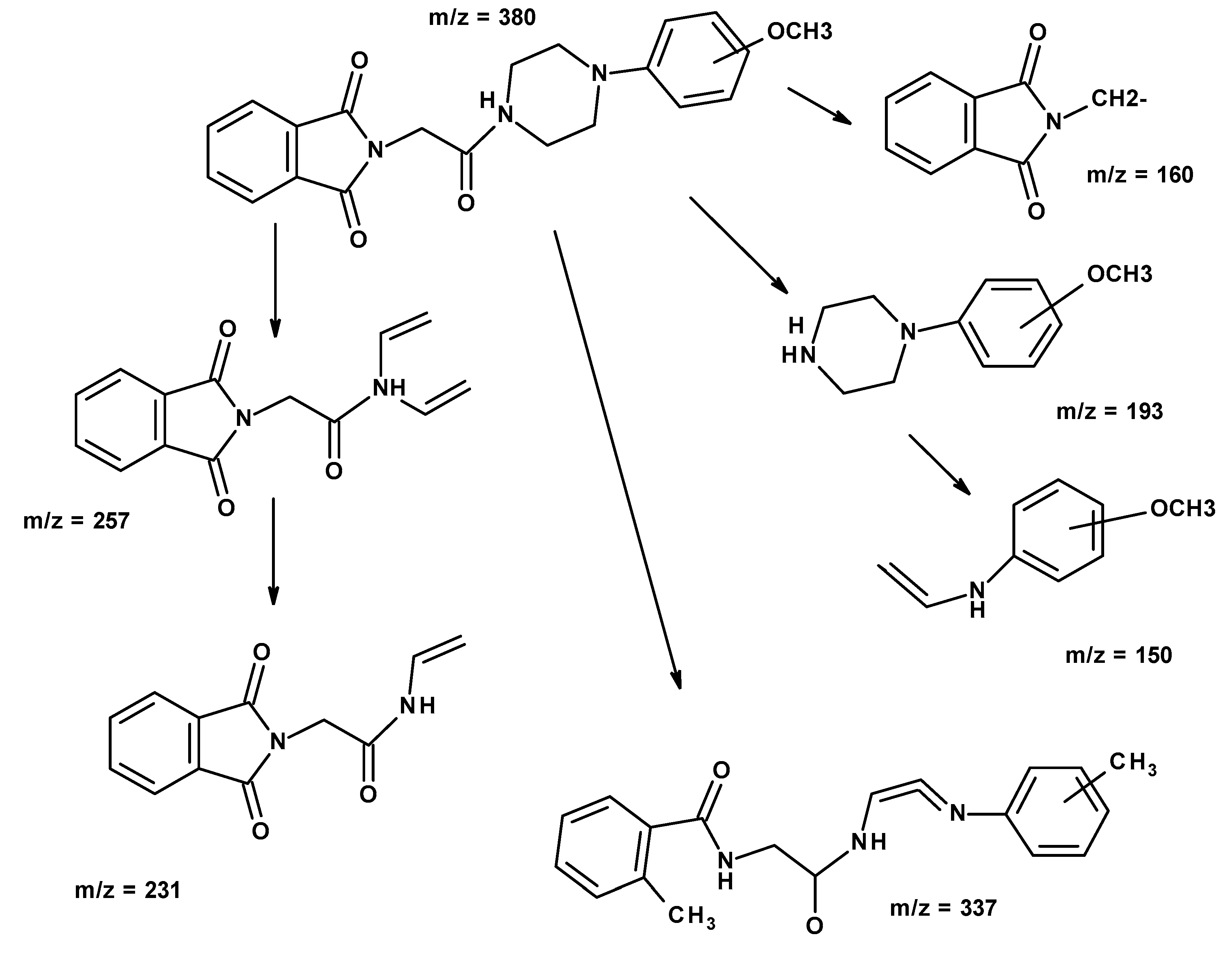
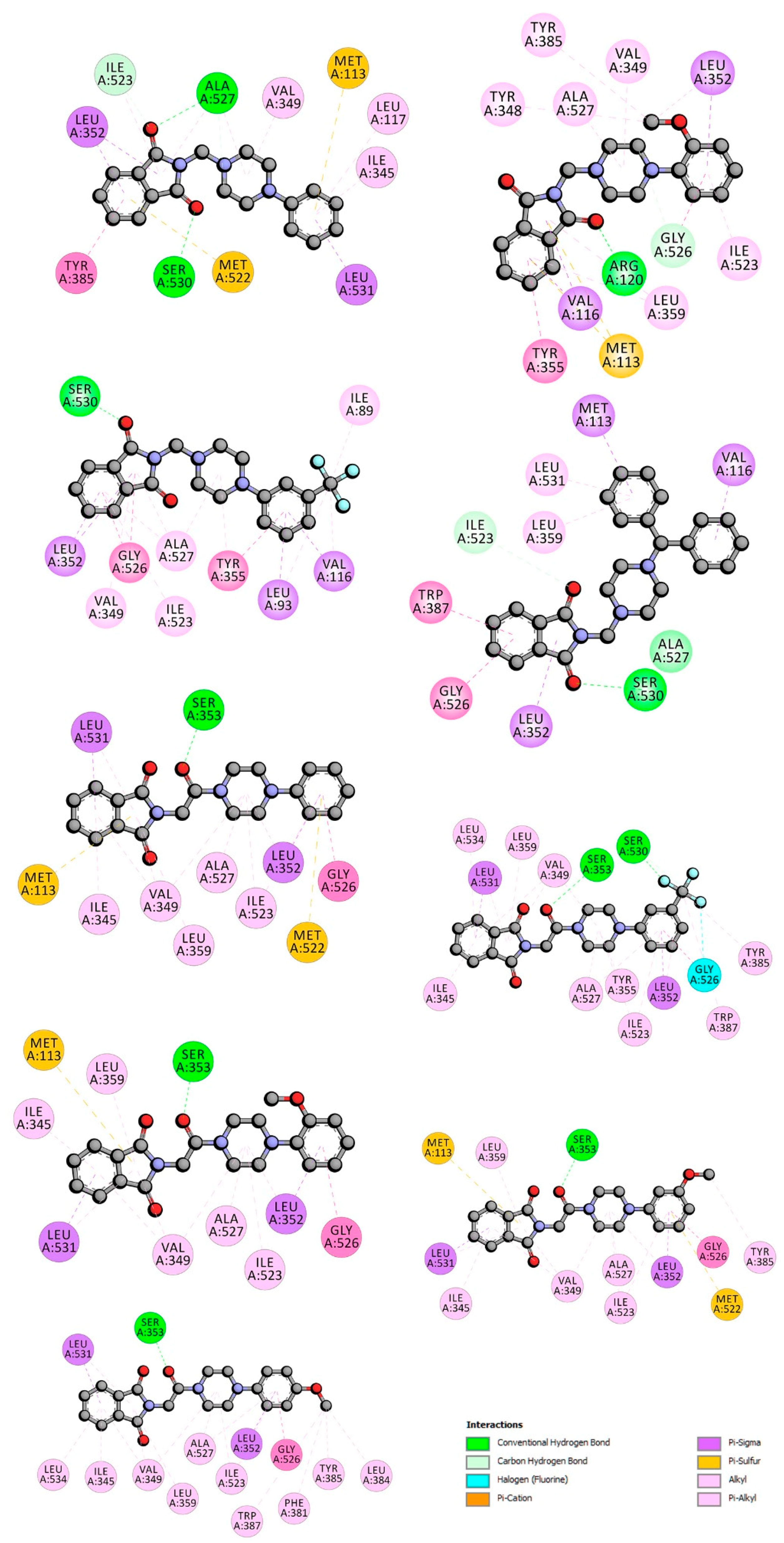
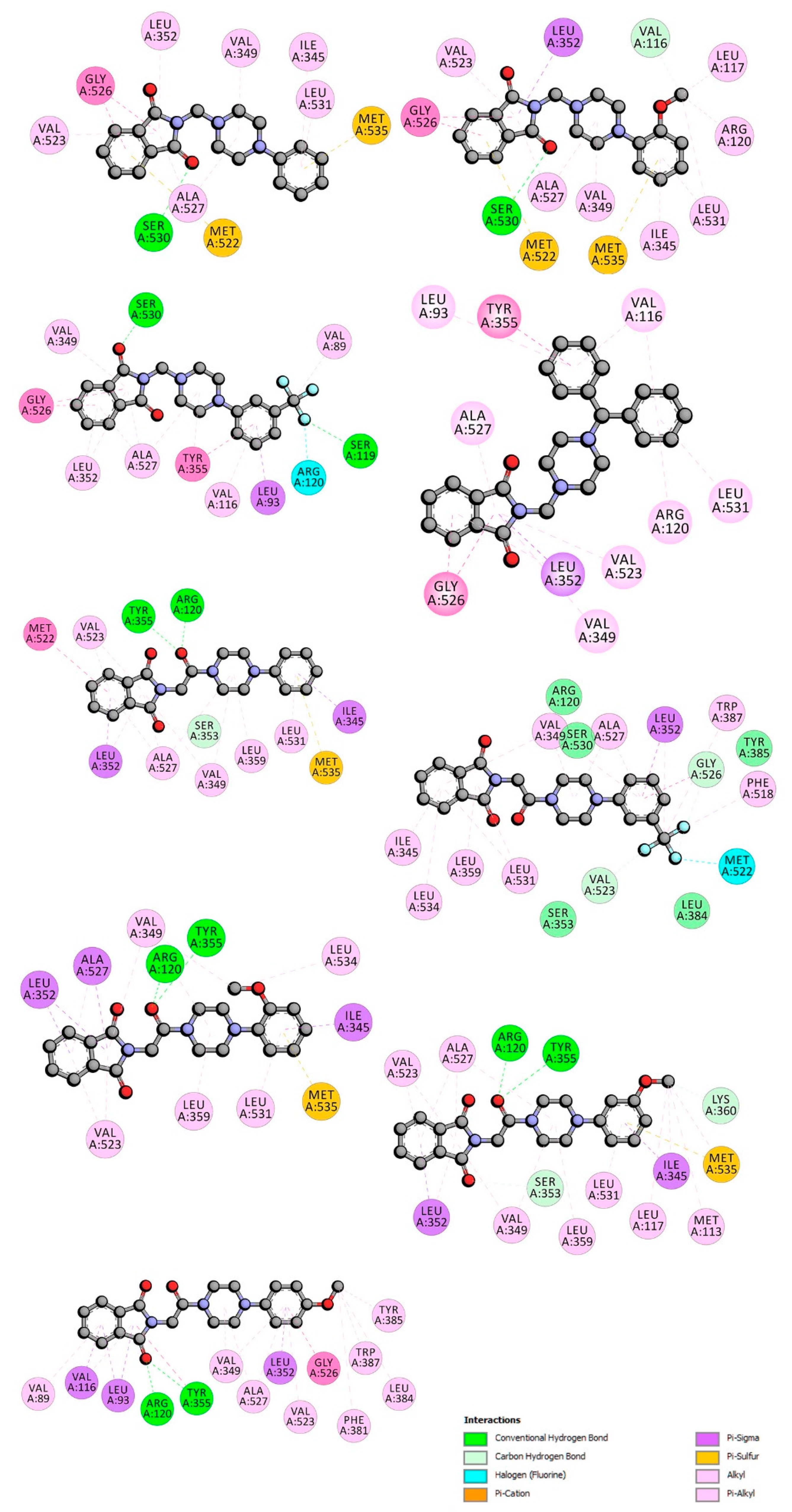

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MW (g/mol) | TPSA (Å2) | logP | H-d | H-a |
|---|---|---|---|---|---|
| A | 321.37 | 45.55 | 3.13 | 0 | 5 |
| B | 351.40 | 54.79 | 3.14 | 0 | 6 |
| C | 389.37 | 45.55 | 4.0 | 0 | 5 |
| D | 411.50 | 45.55 | 4.61 | 0 | 5 |
| E | 349.14 | 60.93 | 2.37 | 0 | 4 |
| F | 417.13 | 60.93 | 3.25 | 0 | 7 |
| G | 379.15 | 70.16 | 2.11 | 0 | 5 |
| H | 379.15 | 70.16 | 2.11 | 0 | 5 |
| I | 379.15 | 70.16 | 2.11 | 0 | 5 |
| Compound | IC50 (µM) (SD) | Ratio: COX-2/COX-1 | Cell Viability IC50 (µM) (SD) | |
|---|---|---|---|---|
| COX-1 | COX-2 | |||
| A | 238.3 (0.04) * | 107.2 (0.09) | 0.45 | non-toxic |
| B | 71.0 (0.03) * | 68.5 (0.05) | 0.96 | non-toxic |
| C | 104.1 (0.02) * | 70.3 (0.06) | 0.67 | non-toxic |
| D | 66.2 (0.09) | 72.8 (0.07) | 1.10 | 90.28 (0.07) |
| E | 76.7 (0.09) | 53.4 (0.08) | 0.70 | non-toxic |
| F | 171.0 (0.06) * | 47.6 (0.10) | 0.28 | non-toxic |
| G | 100.7 (0.07) * | 65.8(0.04) * | 0.65 | 96.78 (0.02) |
| H | 176.4 (0.07) * | 43.8 (0.09) | 0.25 | 94.12 (0.03) |
| I | 94.4 (0.05) * | 46.2 (0.10) | 0.49 | 93.32 (0.05) |
| Meloxicam | 83.7 (0.03) | 59.2 (0.07) | 0.71 | - |
| Compound | ROS Level (E/E0) | NO Level (pg/mL) | DNA Damage |
|---|---|---|---|
| Control [50 µM LPS] | 1.0 (0.03) | 32.0 (0.1) | 26.4 (0.08) |
| A | −18.6 (0.02) * | 6.4 (0.14) | 15.2 (0.04) * |
| B | −22.9 (0.04) * | 4.3 (0.05) * | 19.6 (0.09) * |
| C | −18.2 (0.02) * | 8.2 (0.03) * | 23.2 (0.07) * |
| D | −14.2 (0.08) * | 7.4 (0.12) | 17.3 (0.04) * |
| E | −26.2 (0.03) * | 5.3 (0.07) * | 18.4 (0.07) * |
| F | −22.1 (0.04) * | 9.1 (0.07) | 20.1 (0.03) * |
| G | −29.1 (0.09) * | 4.5 (0.08) | 18.7 (0.07) * |
| H | −27.6 (0.05) * | 4.0 (0.05) * | 17.1 (0.04) * |
| I | −28.5 (0.06) * | 4.8 (0.06) * | 19.2 (0.08) * |
| ΔG° (kJmol−1) | Ki (μM) | ΔG° (kJmol−1) | Ki (μM) | |
|---|---|---|---|---|
| COX-1 | COX-2 | |||
| A | −25.33 | 35.94 | −33.10 | 1.56 |
| B | −22.65 | 106.45 | −31.68 | 2.76 |
| C | −23.78 | 67.41 | −28.05 | 12.13 |
| D | −26.88 | 19.34 | −30.35 | 4.74 |
| E | −29.80 | 5.98 | −35.20 | 0.68 |
| F | −19.39 | 420.72 | −30.98 | 3.68 |
| G | −30.55 | 4.37 | −35.74 | 0.54 |
| H | −27.80 | 13.39 | −32.27 | 2.18 |
| I | −22.90 | 96.16 | −30.47 | 4.50 |
| Meloxicam | −33.35 | 1.42 | −34.02 | 1.09 |
| Comp. | Formula (MW) | M.p. (°C) Solvent | Yield (%) | TLC (Rf) | FT–IR | |
|---|---|---|---|---|---|---|
| OE: MeOH 7:3 | C=O | Mono-and Disubstituted Benzene | ||||
| A | C19H19N3O2 321.37 | 160–161 ethanol | 76.87 | 0.92 | 1701 1770 | 705, 770 |
| B | C20H21N3O3 351.40 | 134–135 ethanol | 92.91 | 0.78 | 1705 1768 | 727 |
| C | C20H18N3O2F3 389.37 | 147–148 ethanol | 47.26 | 0.77 | 1711 1771 | 696, 755 |
| D | C26H25N3O2 411.50 | 206–207 ethanol | 58.93 | 0.89 | 1705 1765 | 690, 750 |
| E | C20H19N3O3 349.40 | 180–180 ethanol | 51.87 | 0.74 | 1670 1710 1750 | 700, 750 |
| F | C21H18N3O3F3 417.38 | 206–208 ethanol | 53.59 | 0.79 | 1680 1705 1760 | 690, 720 |
| G | C21H21N3O4 379.41 | 152–154 ethanol | 61.43 | 0.47 | 1640 1689 1720 | 700 |
| H | C21H21N3O4 379.41 | 155–156 ethanol | 69.29 | 0.58 | 1650 1700 1730 | 710, 780 |
| I | C21H21N3O4 379.41 | 204–207 ethanol | 90.71 | 0.53 | 1620 1680 1710 | 770 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szkatuła, D.; Krzyżak, E.; Stanowska, P.; Duda, M.; Wiatrak, B. A New N-Substituted 1H-Isoindole-1,3(2H)-Dione Derivative—Synthesis, Structure and Affinity for Cyclooxygenase Based on In Vitro Studies and Molecular Docking. Int. J. Mol. Sci. 2021, 22, 7678. https://doi.org/10.3390/ijms22147678
Szkatuła D, Krzyżak E, Stanowska P, Duda M, Wiatrak B. A New N-Substituted 1H-Isoindole-1,3(2H)-Dione Derivative—Synthesis, Structure and Affinity for Cyclooxygenase Based on In Vitro Studies and Molecular Docking. International Journal of Molecular Sciences. 2021; 22(14):7678. https://doi.org/10.3390/ijms22147678
Chicago/Turabian StyleSzkatuła, Dominika, Edward Krzyżak, Paulina Stanowska, Magdalena Duda, and Benita Wiatrak. 2021. "A New N-Substituted 1H-Isoindole-1,3(2H)-Dione Derivative—Synthesis, Structure and Affinity for Cyclooxygenase Based on In Vitro Studies and Molecular Docking" International Journal of Molecular Sciences 22, no. 14: 7678. https://doi.org/10.3390/ijms22147678
APA StyleSzkatuła, D., Krzyżak, E., Stanowska, P., Duda, M., & Wiatrak, B. (2021). A New N-Substituted 1H-Isoindole-1,3(2H)-Dione Derivative—Synthesis, Structure and Affinity for Cyclooxygenase Based on In Vitro Studies and Molecular Docking. International Journal of Molecular Sciences, 22(14), 7678. https://doi.org/10.3390/ijms22147678