
Cardiovascular and Cerebrovascular Implications of Growth Restriction: Mechanisms and Potential Treatments

 , ,
, , 
Abstract
1. Introduction
2. Fetal Growth Restriction
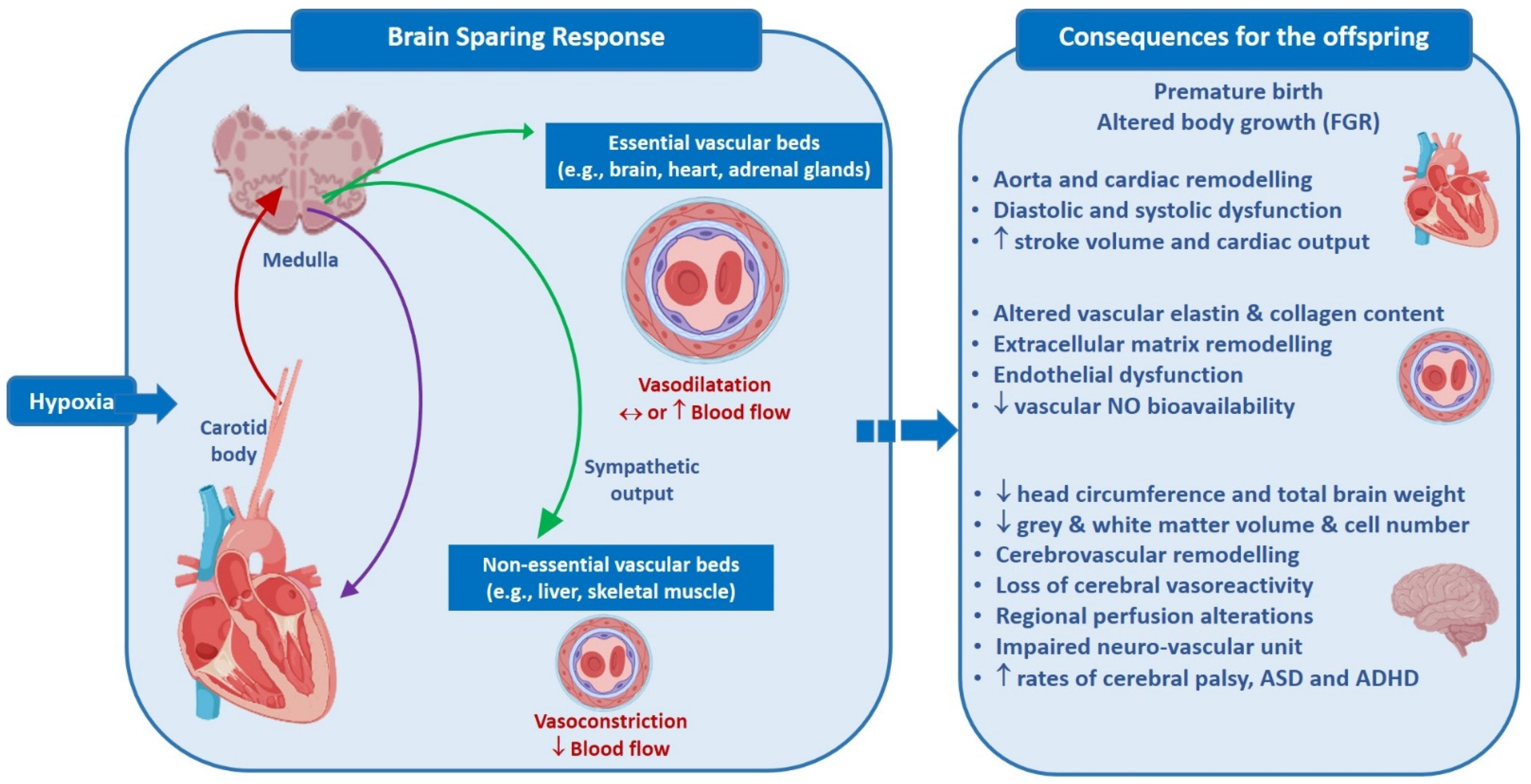
3. Fetal Cardiovascular Response to Acute Hypoxia
4. Fetal Cardiovascular Response to Chronic Hypoxia
5. Cardiovascular Disorders and FGR
6. Impact of FGR on Cardiac Function
7. Co-Morbidity of FGR and Prematurity
8. Extracellular Matrix Proteins: Elastin and Collagen in FGR
9. Vascular Reactivity and FGR
10. Vascular Endothelium and FGR
11. Vascular Homeostasis
12. Nitric Oxide (NO), Oxidative Stress and FGR
13. Cerebrovascular Changes in FGR
14. Detection of Brain Sparing In Vivo
15. Cerebrovascular circulation and the neurovascular unit in FGR
16. FGR and Neurodevelopment
17. Long-Term Neurological Implications of Brain Sparing
18. FGR and Neurodevelopmental Disorders
19. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Malhotra, A.; Ditchfield, M.; Fahey, M.C.; Castillo-Melendez, M.; Allison, B.; Polglase, G.; Wallace, E.; Hodges, R.; Jenkin, G.; Miller, S. Detection and assessment of brain injury in the growth-restricted fetus and neonate. Pediatr. Res. 2017, 82, 184–193. [Google Scholar] [CrossRef]
- Polglase, G.R.; Allison, B.J.; Coia, E.; Li, A.; Jenkin, G.; Malhotra, A.; Sehgal, A.; Kluckow, M.; Gill, A.W.; Hooper, S.B.; et al. Altered cardiovascular function at birth in growth-restricted preterm lambs. Pediatr. Res. 2016, 80, 538–546. [Google Scholar] [CrossRef]
- Cheong, J.N.; Cuffe, J.S.M.; Jefferies, A.J.; Moritz, K.M.; Wlodek, M.E. Adrenal, metabolic and cardio-renal dysfunction develops after pregnancy in rats born small or stressed by physiological measurements during pregnancy. J. Physiol. 2016, 594, 6055–6068. [Google Scholar] [CrossRef]
- Schreuder, M.; Delemarre-van de Waal, H.; van Wijk, A. Consequences of intrauterine growth restriction for the kidney. Kidney Blood Press. Res. 2006, 29, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Nardozza, L.M.M.; Caetano, A.C.R.; Zamarian, A.C.P.; Mazzola, J.B.; Silva, C.P.; Marçal, V.; Lobo, T.F.; Peixoto, A.B.; Júnior, E.A. Fetal growth restriction: Current knowledge. Arch. Gynecol. Obstet. 2017, 295, 1061–1077. [Google Scholar] [CrossRef]
- Lee, A.C.; Kozuki, N.; Cousens, S.; Stevens, G.A.; Blencowe, H.; Silveira, M.F.; Sania, A.; Rosen, H.E.; Schmiegelow, C.; Adair, L.S.; et al. Estimates of burden and consequences of infants born small for gestational age in low and middle income countries with INTERGROWTH-21(st) standard: Analysis of CHERG datasets. BMJ 2017, 358, j3677. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Hüppi, P.; Mallard, C. The consequences of fetal growth restriction on brain structure and neurodevelopmental outcome. J. Physiol. 2016, 594, 807–823. [Google Scholar] [CrossRef]
- Gordijn, S.J.; Beune, I.M.; Thilaganathan, B.; Papageorghiou, A.; Baschat, A.A.; Baker, P.N.; Silver, R.M.; Wynia, K.; Ganzevoort, W. Consensus definition of fetal growth restriction: A Delphi procedure. Ultrasound Obstet. Gynecol. 2016, 48, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Melendez, M.; Yawno, T.; Allison, B.J.; Jenkin, G.; Wallace, E.M.; Miller, S.L. Cerebrovascular adaptations to chronic hypoxia in the growth restricted lamb. Int. J. Dev. Neurosci. 2015, 45, 55–65. [Google Scholar] [CrossRef]
- De Boo, H.A.; Harding, J.E. The developmental origins of adult disease (Barker) hypothesis. Aust. N. Z. J. Obstet. Gynaecol. 2006, 46, 4–14. [Google Scholar] [CrossRef]
- Nardozza, L.M.M.; Júnior, E.A.; Barbosa, M.M.; Caetano, A.C.R.; Lee, D.J.R.; Moron, A.F. Fetal growth restriction: Current knowledge to the general Obs/Gyn. Arch. Gynecol. Obstet. 2012, 286, 1–13. [Google Scholar] [CrossRef]
- Economides, D.L.; Nicolaides, K.; Campbell, S. Metabolic and endocrine findings in appropriate and small for gestational age fetuses. J. Périnat. Med. 1991, 19, 97–105. [Google Scholar] [CrossRef]
- Morrison, J.L. Sheep models of intrauterine growth restriction: Fetal adaptations and consequences. Clin. Exp. Pharmacol. Physiol. 2008, 35, 730–743. [Google Scholar] [CrossRef]
- Giussani, D. The fetal brain sparing response to hypoxia: Physiological mechanisms. J. Physiol. 2016, 594, 1215–1230. [Google Scholar] [CrossRef]
- Kara, T.; Narkiewicz, K.; Somers, V.K. Chemoreflexes—Physiology and clinical implications. Acta Physiol. Scand. 2003, 177, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Freet, C.S.; Stoner, J.F.; Tang, X. Baroreflex and chemoreflex controls of sympathetic activity following intermittent hypoxia. Auton. Neurosci. 2013, 174, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Pipkin, F.B.; Lumbers, E.R.; Mott, J.C. Factors influencing plasma renin and angiotensin II in the conscious pregnant ewe and its foetus. J. Physiol. 1974, 243, 619–636. [Google Scholar] [CrossRef]
- Jones, C.T.; Robinson, R.O. Plasma catecholamines in foetal and adult sheep. J. Physiol. 1975, 248, 15–33. [Google Scholar] [CrossRef]
- Rurak, D.W. Plasma vasopressin levels during hypoxaemia and the cardiovascular effects of exogenous vasopressin in foetal and adult sheep. J. Physiol. 1978, 277, 341–357. [Google Scholar] [CrossRef]
- Sutherland, A.E.; Crossley, K.J.; Allison, B.J.; Jenkin, G.; Wallace, E.M.; Miller, S.L. The effects of intrauterine growth restriction and antenatal glucocorticoids on ovine fetal lung development. Pediatr. Res. 2012, 71, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Umezaki, H.; Kaushal, K.M.; Ducsay, C.A. Long-term hypoxia alters ovine fetal endocrine and physiological responses to hypotension. Am. J. Physiol. Integr. Comp. Physiol. 2004, 287, R209–R217. [Google Scholar] [CrossRef]
- Malhotra, A.; Allison, B.J.; Castillo-Melendez, M.; Jenkin, G.; Polglase, G.R.; Miller, S.L. Neonatal Morbidities of Fetal Growth Restriction: Pathophysiology and Impact. Front. Endocrinol. 2019, 10, 55. [Google Scholar] [CrossRef]
- Allison, B.; Brain, K.L.; Niu, Y.; Kane, A.D.; Herrera, E.A.; Thakor, A.S.; Botting, K.J.; Cross, C.M.; Itani, N.; Skeffington, K.; et al. Fetal in vivo continuous cardiovascular function during chronic hypoxia. J. Physiol. 2016, 594, 1247–1264. [Google Scholar] [CrossRef]
- Brain, K.L.; Allison, B.J.; Niu, Y.; Cross, C.M.; Itani, N.; Kane, A.D.; Herrera, E.A.; Giussani, D.A. Induction of controlled hypoxic pregnancy in large mammalian species. Physiol. Rep. 2015, 3, e12614. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.W.; Zhang, L.; McMillen, I.C.; Botting, K.J.; Duffield, J.A.; Zhang, S.; Suter, C.M.; Brooks, U.A.; Morrison, J.L. Fetal growth restriction and the programming of heart growth and cardiac insulin-like growth factor 2 expression in the lamb. J. Physiol. 2011, 589, 4709–4722. [Google Scholar] [CrossRef]
- Leon, D.A.; Lithell, H.O.; Vågerö, D.; Koupilová, I.; Mohsen, R.; Berglund, L.; Lithell, U.-B.; McKeigue, P.M. Reduced fetal growth rate and increased risk of death from ischaemic heart disease: Cohort study of 15 000 Swedish men and women born 1915-29. BMJ 1998, 317, 241–245. [Google Scholar] [CrossRef]
- Martyn, C.N.; Barker, D.J.; Jespersen, S.; Greenwald, S.; Osmond, C.; Berry, C. Growth in utero, adult blood pressure, and arterial compliance. Br. Heart J. 1995, 73, 116–121. [Google Scholar] [CrossRef]
- Arnold, L.W.; Hoy, W.E.; Wang, Z. Low birth weight and large adult waist circumference increase the risk of cardiovascular disease in remote indigenous Australians—An 18 year cohort study. Int. J. Cardiol. 2015, 186, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, A.; Allison, B.; Gwini, S.; Miller, S.; Polglase, G. Cardiac Morphology and Function in Preterm Growth Restricted Infants: Relevance for Clinical Sequelae. J. Pediatr. 2017, 188, 128–134.e2. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, A.; Allison, B.J.; Gwini, S.M.; Menahem, S.; Miller, S.L.; Polglase, G.R. Vascular aging and cardiac maladaptation in growth-restricted preterm infants. J. Perinatol. 2017, 38, 92–97. [Google Scholar] [CrossRef]
- Cruz-Lemini, M.; Crispi, F.; Valenzuela-Alcaraz, B.; Figueras, F.; Sitges, M.; Bijnens, B.; Gratacós, E. Fetal cardiovascular remodeling persists at 6 months in infants with intrauterine growth restriction. Ultrasound Obstet. Gynecol. 2016, 48, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Crispi, F.; Figueras, F.; Cruz-Lemini, M.; Bartrons, J.; Bijnens, B.; Gratacos, E. Cardiovascular programming in children born small for gestational age and relationship with prenatal signs of severity. Am. J. Obstet. Gynecol. 2012, 207, 121.e1–121.e9. [Google Scholar] [CrossRef] [PubMed]
- Morsing, E.; Liuba, P.; Fellman, V.; Maršál, K.; Brodszki, J. Cardiovascular function in children born very preterm after intrauterine growth restriction with severely abnormal umbilical artery blood flow. Eur. J. Prev. Cardiol. 2014, 21, 1257–1266. [Google Scholar] [CrossRef]
- Brodszki, J.; Länne, T.; Marsál, K.; Ley, D. Impaired vascular growth in late adolescence after intrauterine growth restriction. Circulation 2005, 111, 2623–2628. [Google Scholar] [CrossRef]
- Cocciolone, A.J.; Hawes, J.Z.; Staiculescu, M.C.; Johnson, E.O.; Murshed, M.; Wagenseil, J.E. Elastin, arterial mechanics, and cardiovascular disease. Am. J. Physiol. Circ. Physiol. 2018, 315, H189–H205. [Google Scholar] [CrossRef]
- Cecelja, M.; Chowienczyk, P. Role of arterial stiffness in cardiovascular disease. JRSM Cardiovasc. Dis. 2012, 1, 1–10. [Google Scholar] [CrossRef]
- Bendeck, M.P.; Keeley, F.W.; Langille, B.L. Perinatal accumulation of arterial wall constituents: Relation to hemodynamic changes at birth. Am. J. Physiol. Circ. Physiol. 1994, 267, H2268–H2279. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Sen, U.; Tyagi, N.; Tyagi, S.C. Blood flow interplays with elastin: Collagen and MMP: TIMP ratios to maintain healthy vascular structure and function. Vasc. Health Risk Manag. 2010, 6, 215. [Google Scholar] [PubMed]
- Thompson, J.A.; Richardson, B.S.; Gagnon, R.; Regnault, T. Chronic intrauterine hypoxia interferes with aortic development in the late gestation ovine fetus. J. Physiol. 2011, 589, 3319–3332. [Google Scholar] [CrossRef]
- Dodson, R.B.; Rozance, P.J.; Fleenor, B.S.; Petrash, C.C.; Shoemaker, L.; Hunter, K.S.; Ferguson, V.L. Increased arterial stiffness and extracellular matrix reorganization in intrauterine growth–restricted fetal sheep. Pediatr. Res. 2012, 73, 147–154. [Google Scholar] [CrossRef]
- Dodson, R.B.; Rozance, P.J.; Petrash, C.C.; Hunter, K.S.; Ferguson, V.L. Thoracic and abdominal aortas stiffen through unique extracellular matrix changes in intrauterine growth restricted fetal sheep. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H429–H437. [Google Scholar] [CrossRef] [PubMed]
- Visentin, S.; Londero, A.P.; Calanducci, M.; Grisan, E.; Bongiorno, M.C.; Marin, L.; Cosmi, E. Fetal Abdominal Aorta: Doppler and Structural Evaluation of Endothelial Function in Intrauterine Growth Restriction and Controls. Ultraschall Med. 2018, 40, 55–63. [Google Scholar] [CrossRef]
- Martyn, C.; Greenwald, S. Impaired synthesis of elastin in walls of aorta and large conduit arteries during early development as an initiating event in pathogenesis of systemic hypertension. Lancet 1997, 350, 953–955. [Google Scholar] [CrossRef]
- Paz, A.A.; Arenas, G.A.; Castillo-Galán, S.; Peñaloza, E.; Cáceres-Rojas, G.; Suazo, J.; Herrera, E.A.; Krause, B.J. Premature Vascular Aging in Guinea Pigs Affected by Fetal Growth Restriction. Int. J. Mol. Sci. 2019, 20, 3474. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.H.; Li, C.; Huber, H.F.; Clarke, G.D.; Nathanielsz, P. Intrauterine growth restriction results in persistent vascular mismatch in adulthood. J. Physiol. 2017, 596, 5777–5790. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S. The Vascular Endothelium; Elsevier BV: Amsterdam, The Netherlands, 2018; pp. 5–10. [Google Scholar]
- Chan, C.K.; Vanhoutte, P.M. Hypoxia, vascular smooth muscles and endothelium. Acta Pharm. Sin. B 2013, 3, 1–7. [Google Scholar] [CrossRef]
- Geiger, M. Fundamentals of Vascular Biology, 1st ed.; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar]
- Laurindo, F.R.; Liberman, M.; Fernandes, D.C.; Leite, P.F. Endothelium-Dependent Vasodilation: Nitric Oxide and Other Mediators. In Endothelium and Cardiovascular Diseases; Academic Press: Cambridge, MA, USA, 2018; pp. 97–113. [Google Scholar]
- Consolim-Colombo, F.M.; Bortolotto, L.A. Endothelium and Arterial Hypertension. In Endothelium and Cardiovascular Diseases; Academic Press: Cambridge, MA, USA, 2018; pp. 429–437. [Google Scholar]
- Favarato, D.; Da Luz, P.L. Endothelial Function and Cardiovascular Risk Factors. In Endothelium and Cardiovascular Diseases; Academic Press: Cambridge, MA, USA, 2018; pp. 513–526. [Google Scholar]
- Polglase, G.R.; Hooper, S.B.; Gill, A.W.; Allison, B.J.; Crossley, K.J.; Moss, T.J.; Nitsos, I.; Pillow, J.J.; Kluckow, M. Intrauterine inflammation causes pulmonary hypertension and cardiovascular sequelae in preterm lambs. J. Appl. Physiol. 2010, 108, 1757–1765. [Google Scholar] [CrossRef]
- Thakor, A.S.; Richter, H.; Kane, A.; Dunster, C.; Kelly, F.J.; Poston, L.; Giussani, D.A. Redox modulation of the fetal cardiovascular defence to hypoxaemia. J. Physiol. 2010, 588, 4235–4247. [Google Scholar] [CrossRef]
- Kane, A.D.; Herrera, E.A.; Camm, E.J.; Giussani, D. Vitamin C Prevents Intrauterine Programming of in vivo Cardiovascular Dysfunction in the Rat. Circ. J. 2013, 77, 2604–2611. [Google Scholar] [CrossRef]
- Kane, A.D.; Hansell, J.A.; Herrera, E.A.; Allison, B.; Niu, Y.; Brain, K.L.; Kaandorp, J.J.; Derks, J.B.; Giussani, D. Xanthine oxidase and the fetal cardiovascular defence to hypoxia in late gestation ovine pregnancy. J. Physiol. 2013, 592, 475–489. [Google Scholar] [CrossRef]
- Peterside, I.E.; Selak, M.A.; Simmons, R.A. Impaired oxidative phosphorylation in hepatic mitochondria in growth-retarded rats. Am. J. Physiol. Metab. 2003, 285, E1258–E1266. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem. J. 2007, 405, 1–9. [Google Scholar] [CrossRef]
- Hancock, J.T.; Neill, S.J. Nitric Oxide: Its Generation and Interactions with Other Reactive Signaling Compounds. Plants 2019, 8, 41. [Google Scholar] [CrossRef]
- Farrow, K.N.; Lakshminrusimha, S.; Reda, W.J.; Wedgwood, S.; Czech, L.; Gugino, S.F.; Davis, J.M.; Russell, J.A.; Steinhorn, R. Superoxide dismutase restores eNOS expression and function in resistance pulmonary arteries from neonatal lambs with persistent pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L979–L987. [Google Scholar] [CrossRef] [PubMed]
- Krause, B.; Carrasco-Wong, I.; Caniuguir, A.; Carvajal, J.; Faras, M.; Casanello, P. Endothelial eNOS/arginase imbalance contributes to vascular dysfunction in IUGR umbilical and placental vessels. Placenta 2013, 34, 20–28. [Google Scholar] [CrossRef]
- Leeson, C.P.; Whincup, P.H.; Cook, D.G.; Donald, A.E.; Papacosta, O.; Lucas, A.; Deanfield, J.E. Flow-mediated dilation in 9- to 11-year-old children: The influence of intrauterine and childhood factors. Circulation 1997, 96, 2233–2238. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, J.; Bellamy, M.F.; Gorman, S.T.; Brownlee, M.; Ramsey, M.W.; Lewis, M.J.; Davies, D.P.; Henderson, A.H. Endothelial function is impaired in fit young adults of low birth weight. Cardiovasc. Res. 1998, 40, 600–606. [Google Scholar] [CrossRef]
- Miller, M.; Voelker, C.; Olister, S.; Thompson, J.; Zhang, X.-J.; Rivera, D.; Eloby-Childress, S.; Liu, X.; Clark, D.; Pierce, M. Fetal growth retardation in rats may result from apoptosis: Role of peroxynitrite. Free Radic. Biol. Med. 1996, 21, 619–629. [Google Scholar] [CrossRef]
- Steinback, C.D.; Poulin, M.J.; Roach, R.C.; Hackett, P.H.; Wagner, P.D. Influence of Hypoxia on Cerebral Blood Flow Regulation in Humans. Adv. Exp. Med. Biol. 2016, 903, 131–144. [Google Scholar] [CrossRef]
- Geipel, A.; Gembruch, U. Evaluation of fetal and uteroplacental blood flow. In Ultrasound in Obstetrics and Gynaecology; Elsevier: Amsterdam, The Netherlands, 2009; pp. 209–227. [Google Scholar]
- Rossi, A.; Romanello, I.; Forzano, L.; Fachechi, G.; Marchesoni, D. Evaluation of fetal cerebral blood flow perfusion using power Doppler ultrasound angiography (3D-PDA) in growth-restricted fetuses. Facts Views Vis. ObGyn 2011, 3, 175–180. [Google Scholar] [PubMed]
- Akalin-Sel, T.; Nicolaides, K.; Peacock, J.; Campbell, S. Doppler dynamics and their complex interrelation with fetal oxygen pressure, carbon dioxide pressure, and pH in growth-retarded fetuses. Obstet. Gynecol. 1994, 84, 439–444. [Google Scholar] [PubMed]
- Spinillo, A.; Montanari, L.; Roccio, M.; Zanchi, S.; Tzialla, C.; Stronati, M. Prognostic significance of the interaction between abnormal umbilical and middle cerebral artery Doppler velocimetry in pregnancies complicated by fetal growth restriction. Acta Obstet. Gynecol. Scand. 2009, 88, 159–166. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, T.T.; Wu, C.X.; Lan, X.; Du, G.H. Targeting the neurovascular unit: Development of a new model and consideration for novel strategy for Alzheimer’s disease. Brain Res. Bull. 2011, 86, 13–21. [Google Scholar] [CrossRef]
- Chou, C.-H.; Sinden, J.D.; Couraud, P.-O.; Modo, M. In Vitro Modeling of the Neurovascular Environment by Coculturing Adult Human Brain Endothelial Cells with Human Neural Stem Cells. PLoS ONE 2014, 9, e106346. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.H.; Miller, S.L.; Castillo-Melendez, M.; Malhotra, A. The neurovascular unit: Effects of brain insults during the perinatal period. Front. Neurosci. 2020, 13, 1452. [Google Scholar] [CrossRef]
- Rees, S.; Mallard, C.; Breen, S.; Stringer, M.; Cock, M.; Harding, R.; Rees, S.; Mallard, C.; Breen, S.; Stringer, M.; et al. Fetal Brain Injury Following Prolonged Hypoxemia and Placental Insufficiency: A Review. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 1998, 119, 653–660. [Google Scholar] [CrossRef]
- Williams, J.M.; Pearce, W.J. Age-dependent modulation of endothelium-dependent vasodilatation by chronic hypoxia in ovine cranial arteries. J. Appl. Physiol. 2006, 100, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Tolcos, M.; Markwick, R.; O’Dowd, R.; Martin, V.; Turnley, A.; Rees, S. Intrauterine Growth Restriction: Effects on Neural Precursor Cell Proliferation and Angiogenesis in the Foetal Subventricular Zone. Dev. Neurosci. 2015, 37, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, I.; Shibuya, M.; Wennström, S. Differential activation of vascular genes by hypoxia in primary endothelial cells. Exp. Cell Res. 2004, 299, 476–485. [Google Scholar] [CrossRef]
- Castillo-Melendez, M.; Yawno, T.; Sutherland, A.; Jenkin, G.; Wallace, E.M.; Miller, S.L. Effects of Antenatal Melatonin Treatment on the Cerebral Vasculature in an Ovine Model of Fetal Growth Restriction. Dev. Neurosci. 2017, 39, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of Pericytes Leads to Endothelial Hyperplasia and Abnormal Vascular Morphogenesis. J. Cell Biol. 2001, 153, 543–554. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncology 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Roza, S.J.; Steegers, E.A.; Verburg, B.O.; Jaddoe, V.W.; Moll, H.A.; Hofman, A.; Verhulst, F.C.; Tiemeier, H. What is spared by fetal brain-sparing? Fetal circulatory redistribution and behavioral problems in the general population. Am. J. Epidemiol. 2008, 168, 1145–1152. [Google Scholar] [CrossRef]
- Yiallourou, S.R.; Wallace, E.; Miller, S.; Horne, R. Effects of intrauterine growth restriction on sleep and the cardiovascular system: The use of melatonin as a potential therapy? Sleep Med. Rev. 2016, 26, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Yawno, T.; Alers, N.O.; Castillo-Melendez, M.; Supramaniam, V.G.; Vanzyl, N.; Sabaretnam, T.; Loose, J.M.; Drummond, G.; Walker, D.W.; et al. Antenatal antioxidant treatment with melatonin to decrease newborn neurodevelopmental deficits and brain injury caused by fetal growth restriction. J. Pineal Res. 2014, 56, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Geva, R.; Yosipof, R.; Eshel, R.; Leitner, Y.; Valevski, A.F.; Harel, S. Readiness and Adjustments to School for Children with Intrauterine Growth Restriction (IUGR): An Extreme Test Case Paradigm. Except. Child. 2009, 75, 211–230. [Google Scholar] [CrossRef]
- Eixarch, E.; Muñoz-Moreno, E.; Bargallo, N.; Batalle, D.; Gratacos, E. Motor and cortico-striatal-thalamic connectivity alterations in intrauterine growth restriction. Am. J. Obstet. Gynecol. 2016, 214, 725.e1–725.e9. [Google Scholar] [CrossRef] [PubMed]
- Padilla, N.; Perapoch, J.; Carrascosa, A.; Acosta-Rojas, R.; Botet, F.; Gratacós, E. Twelve-month neurodevelopmental outcome in preterm infants with and without intrauterine growth restriction. Acta Paediatr. 2010, 99, 1498–1503. [Google Scholar] [CrossRef]
- Tolsa, C.B.; Zimine, S.; Warfield, S.K.; Freschi, M.; Rossignol, A.S.; Lazeyras, F.; Hanquinet, S.; Pfizenmaier, M.; Hüppi, P.S. Early Alteration of Structural and Functional Brain Development in Premature Infants Born with Intrauterine Growth Restriction. Pediatr. Res. 2004, 56, 132–138. [Google Scholar] [CrossRef]
- De Bie, H.M.A.; Oostrom, K.J.; Boersma, M.; Veltman, D.J.; Barkhof, F.; De Waal, H.A.D.-V.; Heuvel, M.V.D. Global and regional differences in brain anatomy of young children born small for gestational age. PLoS ONE 2011, 6, e24116. [Google Scholar] [CrossRef]
- Østgård, H.F.; Løhaugen, G.C.; Bjuland, K.J.; Rimol, L.M.; Brubakk, A.-M.; Martinussen, M.; Vik, T.; Håberg, A.K.; Skranes, J. Brain Morphometry and Cognition in Young Adults Born Small for Gestational Age at Term. J. Pediatr. 2014, 165, 921–927.e1. [Google Scholar] [CrossRef]
- Batalle, D.; Eixarch, E.; Figueras, F.; Muñoz-Moreno, E.; Bargallo, N.; Illa, M.; Acosta-Rojas, R.; Amat-Roldan, I.; Gratacos, E. Altered small-world topology of structural brain networks in infants with intrauterine growth restriction and its association with later neurodevelopmental outcome. NeuroImage 2012, 60, 1352–1366. [Google Scholar] [CrossRef] [PubMed]
- Ramenghi, L.A.; Martinelli, A.; De Carli, A.; Brusati, V.; Mandia, L.; Fumagalli, M.; Triulzi, F.; Mosca, F.; Cetin, I. Cerebral Maturation in IUGR and Appropriate for Gestational Age Preterm Babies. Reprod. Sci. 2011, 18, 469–475. [Google Scholar] [CrossRef]
- Eixarch, E.; Meler, E.; Iraola, A.; Illa, M.; Crispi, F.; Hernandez-Andrade, E.; Gratacos, E.; Figueras, F. Neurodevelopmental outcome in 2-year-old infants who were small-for-gestational age term fetuses with cerebral blood flow redistribution. Ultrasound Obstet. Gynecol. 2008, 32, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, S.; Glinianaia, S.V.; Torrioli, M.-G.; Platt, M.-J.; Miceli, M.; Jouk, P.-S.; Johnson, A.; Hutton, J.; Hemming, K.; Hagberg, G.; et al. Cerebral palsy and intrauterine growth in single births: European collaborative study. Lancet 2003, 362, 1106–1111. [Google Scholar] [CrossRef]
- Leitner, Y.; Fattal-Valevski, A.; Geva, R.; Eshel, R.; Toledano-Alhadef, H.; Rotstein, M.; Bassan, H.; Radianu, B.; Bitchonsky, O.; Jaffa, A.J.; et al. Neurodevelopmental outcome of children with intrauterine growth retardation: A longitudinal, 10-year prospective study. J. Child Neurol. 2007, 22, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Padilla, N.; Fransson, P.; Donaire, A.; Figueras, F.; Arranz, A.; Sanz-Cortés, M.; Tenorio, V.; Bargallo, N.; Junqué, C.; Lagercrantz, H.; et al. Intrinsic Functional Connectivity in Preterm Infants with Fetal Growth Restriction Evaluated at 12 Months Corrected Age. Cereb. Cortex 2017, 27, 4750–4758. [Google Scholar] [CrossRef]
- Heinonen, K.; Räikkönen, K.; Pesonen, A.-K.; Andersson, S.; Kajantie, E.; Eriksson, J.G.; Wolke, D.; Lano, A. Behavioural symptoms of attention deficit/hyperactivity disorder in preterm and term children born small and appropriate for gestational age: A longitudinal study. BMC Pediatr. 2010, 10, 91. [Google Scholar] [CrossRef]
- Sparks, F.B.; Friedman, S.D.; Shaw, D.W.; Aylward, E.H.; Echelard, D.; Artru, A.A.; Maravilla, K.R.; Giedd, J.N.; Munson, J.; Dawson, G.; et al. Brain structural abnormalities in young children with autism spectrum disorder. Neurology 2002, 59, 184–192. [Google Scholar] [CrossRef]
- Shariat, M.; Gharaee, J.; Dalili, H.; Mohammadzadeh, Y.; Ansari, S.; Farahani, Z. Association between small for gestational age and low birth weight with attention deficit and impaired executive functions in 3–6 years old children. J. Matern. Neonatal Med. 2017, 32, 1474–1477. [Google Scholar] [CrossRef]
- Tan, Y.-W.; Liu, L.; Wang, Y.-F.; Li, H.-M.; Pan, M.-R.; Zhao, M.-J.; Huang, F.; Wang, Y.-F.; He, Y.; Liao, X.-H.; et al. Alterations of cerebral perfusion and functional brain connectivity in medication-naïve male adults with attention-deficit/hyperactivity disorder. CNS Neurosci. Ther. 2019, 26, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, H.; Nakato, E.; Kanazawa, S.; Shimamura, K.; Sakuta, Y.; Sakuta, R.; Yamaguchi, M.K.; Kakigi, R. Hemodynamic response of children with attention-deficit and hyperactive disorder (ADHD) to emotional facial expressions. Neuropsychologia 2014, 63, 51–58. [Google Scholar] [CrossRef]
- Rennie, J.M.; Coughtrey, H.; Morley, R.; Evans, D.H. Comparison of cerebral blood flow velocity estimation with cranial ultrasound imaging for early prediction of outcome in preterm infants. J. Clin. Ultrasound 1995, 23, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.C.; Henriksen, L.; Bruhn, P. Focal cerebral dysfunction in developmental learning disabilities. Lancet 1990, 335, 8–11. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, B.N.; Kang, E.; Lee, D.S.; Kim, Y.K.; Chung, J.-K.; Lee, M.C.; Cho, S.C. Regional cerebral blood flow in children with attention deficit hyperactivity disorder: Comparison before and after methylphenidate treatment. Hum. Brain Mapp. 2005, 24, 157–164. [Google Scholar] [CrossRef]
- Bjørklund, G.; Kern, J.K.; Urbina, M.A.; Saad, K.; El-Houfey, A.A.; Geier, D.A.; Chirumbolo, S.; Geier, M.R.; Mehta, J.A.; Aaseth, J. Cerebral hypoperfusion in autism spectrum disorder. Acta Neurobiol. Exp. 2018, 78, 21–29. [Google Scholar] [CrossRef]
- Abazov, V.M.; Abbott, B.; Abolins, M.; Acharya, B.S.; Adams, M.; Adams, T.; Agelou, M.; Agram, J.-L.; Ahn, S.H.; Ahsan, M.; et al. Measurement of the lifetime difference in the B0(s) system. Phys. Rev. Lett. 2005, 95, 171801. [Google Scholar] [CrossRef]
- Kay, V.R.; Ratsep, M.T.; Cahill, L.S.; Hickman, A.F.; Zavan, B.; Newport, M.E.; Ellegood, J.; Laliberte, C.L.; Reynolds, J.N.; Carmeliet, P.; et al. Effects of placental growth factor deficiency on behavior, neuroanatomy, and cerebrovasculature of mice. Physiol. Genom. 2018, 50, 862–875. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Early FGR | Late FGR | |
|---|---|---|
| Gestation | <32 weeks | >32 weeks |
| Congenital anomalies | absent | absent |
| Estimated fetal weight or abdominal circumference | <3rd percentile or absent end-diastolic flow in the umbilical artery | <3rd percentile |
| OR | OR 2 out of | |
| Estimated fetal weight or abdominal circumference | <10th percentile | <10th percentile or crossing > 2 quartiles on growth percentiles |
| Uterine artery pulsatility index | >95th percentile | |
| Umbilical artery pulsatility index | >95th percentile | >95th percentile |
| Cerebroplacental ratio | <5th percentile | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rock, C.R.; White, T.A.; Piscopo, B.R.; Sutherland, A.E.; Miller, S.L.; Camm, E.J.; Allison, B.J. Cardiovascular and Cerebrovascular Implications of Growth Restriction: Mechanisms and Potential Treatments. Int. J. Mol. Sci. 2021, 22, 7555. https://doi.org/10.3390/ijms22147555
Rock CR, White TA, Piscopo BR, Sutherland AE, Miller SL, Camm EJ, Allison BJ. Cardiovascular and Cerebrovascular Implications of Growth Restriction: Mechanisms and Potential Treatments. International Journal of Molecular Sciences. 2021; 22(14):7555. https://doi.org/10.3390/ijms22147555
Chicago/Turabian StyleRock, Charmaine R., Tegan A. White, Beth R. Piscopo, Amy E. Sutherland, Suzanne L. Miller, Emily J. Camm, and Beth J. Allison. 2021. "Cardiovascular and Cerebrovascular Implications of Growth Restriction: Mechanisms and Potential Treatments" International Journal of Molecular Sciences 22, no. 14: 7555. https://doi.org/10.3390/ijms22147555
APA StyleRock, C. R., White, T. A., Piscopo, B. R., Sutherland, A. E., Miller, S. L., Camm, E. J., & Allison, B. J. (2021). Cardiovascular and Cerebrovascular Implications of Growth Restriction: Mechanisms and Potential Treatments. International Journal of Molecular Sciences, 22(14), 7555. https://doi.org/10.3390/ijms22147555