
Mitochondrial Metal Ion Transport in Cell Metabolism and Disease





Abstract
1. Introduction

{kind=link}
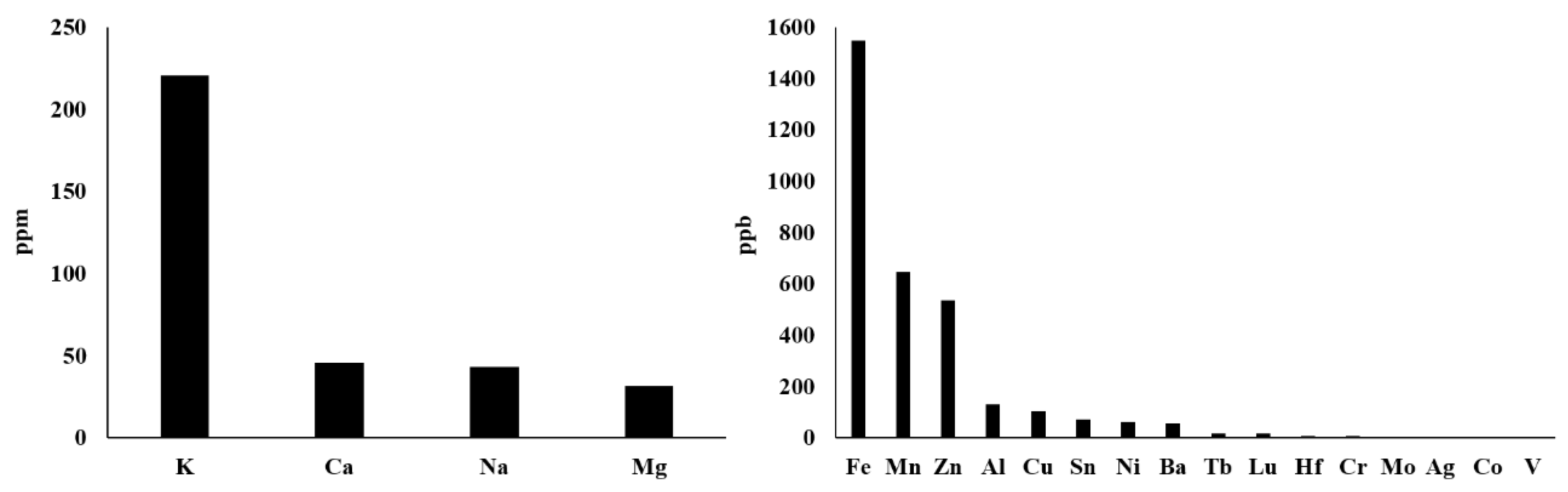{kind=link}
| Metal Ions | Mitochondrial Channels/Transporters | Related Diseases | References | |
|---|---|---|---|---|
| Importer/Influx | Exporter/Efflux | |||
| Ca2+ | VDAC, MCU, mRYR | Letm1, NCLX, mPTP | Insulin resistance, T2D, Diabetes-related cardiac disease, Heart failure, Ischemia, Reperfusion injury, Brain aging, Neurodegenerative diseases, Cancer | [17,20,24,25,26,27,28,29,30,31,32,33,34,35,36] |
| K+ | mitoKATP, KCa, Kv, mitoTASK-3 | KHE | Epilepsy, Diabetic cardiomyopathy, Ischemia, Reperfusion injury, Pulmonary artery hypertension, Neurodegeneration, Cancer, Schizophrenia, Sudden cardiac death | [37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53] |
| Na+ | NCLX | NHE | Heart failure, Sudden death, Neurodegenerative diseases | [18,35,54,55,56,57] |
| Mg2+ | MRS2 | SLC41A3, Mme1 | Cancer, Demyelination, Neurodegeneration | [58,59,60,61,62,63,64,65] |
| Zn2+ | MCU, ZnT4 | ZIP8, mitoKATP | Neurodegeneration | [66,67,68,69,70,71] |
| Fe2+/Fe3+ | MFRN, Tf/TfR2, DMT1 | —— | Anemia, Neurodegenerative diseases | [72,73,74,75,76,77,78,79,80,81] |
2. Mitochondrial Ca2+
3. Mitochondrial K+
4. Mitochondrial Na+
5. Mitochondrial Mg2+
6. Mitochondrial Zn2+
7. Mitochondrial Iron Ion
8. Mitochondrial Manganese Ion
9. Other Mitochondrial Metal Ions
10. Concluding Remarks and Prospects
Funding
Conflicts of Interest
Abbreviations
References
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. Steroid hormone synthesis in mitochondria. Mol. Cell. Endocrinol. 2013, 379, 62–73. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B. Mitochondrial ion channels. Annu. Rev. Physiol. 2007, 69, 19–49. [Google Scholar] [CrossRef]
- Passarella, S.; Atlante, A.; Valenti, D.; de Bari, L. The role of mitochondrial transport in energy metabolism. Mitochondrion 2003, 2, 319–343. [Google Scholar] [CrossRef]
- Lemeshko, S.V.; Lemeshko, V.V. Metabolically derived potential on the outer membrane of mitochondria: A computational model. Biophys. J. 2000, 79, 2785–2800. [Google Scholar] [CrossRef]
- De Marchi, U.; Fernandez-Martinez, S.; de la Fuente, S.; Wiederkehr, A.; Santo-Domingo, J. Mitochondrial ion channels in pancreatic Beta—Cells: Novel pharmacological targets for the treatment of Type 2 diabetes. Br. J. Pharmacol. 2020, 178, 2077–2095. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B.; Cortassa, S.; Aon, M.A. Mitochondrial ion channels: Gatekeepers of life and death. Physiology 2005, 20, 303–315. [Google Scholar] [CrossRef]
- Xia, M.; Zhang, Y.; Jin, K.; Lu, Z.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 27. [Google Scholar] [CrossRef]
- Nam, E.; Han, J.; Suh, J.M.; Yi, Y.; Lim, M.H. Link of impaired metal ion homeostasis to mitochondrial dysfunction in neurons. Curr. Opin. Chem. Biol. 2018, 43, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Pierrel, F.; Cobine, P.A.; Winge, D.R. Metal Ion availability in mitochondria. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2007, 20, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Matak, P.; Matak, A.; Moustafa, S.; Aryal, D.K.; Benner, E.J.; Wetsel, W.; Andrews, N.C. Disrupted iron homeostasis causes dopaminergic neurodegeneration in mice. Proc. Natl. Acad. Sci. USA 2016, 113, 3428–3435. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.P.; Greenamyre, J.T. Mitochondrial iron metabolism and its role in neurodegeneration. J. Alzheimer’s Dis. JAD 2010, 20, S551–S568. [Google Scholar] [CrossRef]
- Wang, W.; Fan, Y.; Wang, S.; Wang, L.; He, W.; Zhang, Q.; Li, X. Effects of voltage-gated K+ channel on cell proliferation in multiple myeloma. Sci. World J. 2014, 2014, 785140. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Wei, Y.H. Role of mitochondrial dysfunction and dysregulation of Ca(2+) homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J. Biomed. Sci. 2017, 24, 70. [Google Scholar] [CrossRef]
- Ummarino, D. Calcium: Mitochondrial calcium efflux essential for heart function. Nat. Rev. Cardiol. 2017, 14, 317. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Lederer, W.J. Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell. Cardiol. 2015, 78, 35–45. [Google Scholar] [CrossRef]
- Wang, W.; Karamanlidis, G.; Tian, R. Novel targets for mitochondrial medicine. Sci. Transl. Med. 2016, 8, 326rv3. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Costa, R.; Peruzzo, R.; Prosdocimi, E.; Checchetto, V.; Leanza, L. Targeting Mitochondrial Ion Channels to Fight Cancer. Int. J. Mol. Sci. 2018, 19, 2060. [Google Scholar] [CrossRef]
- Tajti, G.; Wai, D.C.C.; Panyi, G.; Norton, R.S. The Voltage—Gated potassium channel KV1.3 as a therapeutic target for venom-derived peptides. Biochem. Pharmacol. 2020, 181, 114146. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Y.; Hu, H.; Shi, L.; Chen, J.; Wang, B.; Chen, C.; Zhu, H.; Li, Y.; Li, Q.; et al. Potential therapeutic targets for hypoxia-induced pulmonary artery hypertension. J. Transl. Med. 2014, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Israelson, A.; Brdiczka, D.; Sheu, S.S. The Voltage—Dependent anion channel (VDAC): Function in intracellular signalling, cell life and cell death. Curr. Pharm. Des. 2006, 12, 2249–2270. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Roy Chowdhury, A.; Srinivasan, S.; Csordas, G.; Hajnoczky, G.; Avadhani, N.G. Dysregulation of RyR Calcium Channel Causes the Onset of Mitochondrial Retrograde Signaling. iScience 2020, 23, 101370. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome—Wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhai, D.; Huang, Y. Study on the relationship between calcium-induced calcium release from mitochondria and PTP opening. Mol. Cell. Biochem. 2000, 213, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Juarez, J.; Suarez, J.A.; Dillmann, W.H.; Suarez, J. Mitochondrial calcium handling and heart disease in diabetes mellitus. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 165984. [Google Scholar] [CrossRef]
- Xu, H.X.; Cui, S.M.; Zhang, Y.M.; Ren, J. Mitochondrial Ca(2+) regulation in the etiology of heart failure: Physiological and pathophysiological implications. Acta Pharmacol. Sin. 2020, 41, 1301–1309. [Google Scholar] [CrossRef]
- Jung, H.; Kim, S.Y.; Canbakis Cecen, F.S.; Cho, Y.; Kwon, S.K. Dysfunction of Mitochondrial Ca(2+) Regulatory Machineries in Brain Aging and Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 599792. [Google Scholar] [CrossRef]
- Naia, L.; Ferreira, I.L.; Ferreiro, E.; Rego, A.C. Mitochondrial Ca(2+) handling in Huntington’s and Alzheimer’s Diseases—Role of ER—Mitochondria crosstalk. Biochem. Biophys. Res. Commun. 2017, 483, 1069–1077. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Pratico, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Abramov, A.Y. Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci. Lett. 2018, 663, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Pathak, T.; Gueguinou, M.; Walter, V.; Delierneux, C.; Johnson, M.T.; Zhang, X.; Xin, P.; Yoast, R.E.; Emrich, S.M.; Yochum, G.S.; et al. Dichotomous role of the human mitochondrial Na(+)/Ca2(+)/Li(+) exchanger NCLX in colorectal cancer growth and metastasis. eLife 2020, 9, e59686. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Nagase, H.; Kishi, K.; Higuti, T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 1991, 352, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.O.; Jogini, V.; Borhani, D.W.; Leffler, A.E.; Dror, R.O.; Shaw, D.E. Mechanism of voltage gating in potassium channels. Science 2012, 336, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Berkefeld, H.; Fakler, B.; Schulte, U. Ca2+-activated K+ channels: From protein complexes to function. Physiol. Rev. 2010, 90, 1437–1459. [Google Scholar] [CrossRef]
- Bednarczyk, P.; Kowalczyk, J.E.; Beresewicz, M.; Dolowy, K.; Szewczyk, A.; Zablocka, B. Identification of a Voltage—Gated potassium channel in gerbil hippocampal mitochondria. Biochem. Biophys. Res. Commun. 2010, 397, 614–620. [Google Scholar] [CrossRef]
- Rusznak, Z.; Bakondi, G.; Kosztka, L.; Pocsai, K.; Dienes, B.; Fodor, J.; Telek, A.; Gonczi, M.; Szucs, G.; Csernoch, L. Mitochondrial expression of the Two—Pore domain TASK-3 channels in malignantly transformed and non-malignant human cells. Virchows Arch. Int. J. Pathol. 2008, 452, 415–426. [Google Scholar] [CrossRef]
- Brierley, G.P.; Jurkowitz, M.S.; Farooqui, T.; Jung, D.W. K+/H+ antiport in heart mitochondria. J. Biol. Chem. 1984, 259, 14672–14678. [Google Scholar] [CrossRef]
- Nikbakht, F.; Khanizadeh, A.M.; Golab, F.; Baluchnejadmojarad, T.; Vazifehkhah, S.; Moeinsadat, A. Mitochondrial ATP-sensitive potassium channel, MitoKATP, ameliorates mitochondrial dynamic disturbance induced by temporal lobe epilepsy. J. Chem. Neuroanat. 2020, 113, 101808. [Google Scholar] [CrossRef]
- Krylova, I.B.; Kachaeva, E.V.; Rodionova, O.M.; Negoda, A.E.; Evdokimova, N.R.; Balina, M.I.; Sapronov, N.S.; Mironova, G.D. The cardioprotective effect of uridine and uridine-5’-monophosphate: The role of the mitochondrial ATP—Dependent potassium channel. Exp. Gerontol. 2006, 41, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Wang, J.; Li, Y.; Wei, S.; Su, F.; Zhang, S.; Duan, Y.; Wang, L.; Zhu, Q. Opening of mitoKATP improves cardiac function and inhibits apoptosis via the AKT-Foxo1 signaling pathway in diabetic cardiomyopathy. Int. J. Mol. Med. 2018, 42, 2709–2719. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Hu, J.; Xiao, J.; Dan, G.; Yang, L.; Ye, F.; Zou, Z.; Cao, J.; Sai, Y. Mitochondrial ATP—Sensitive potassium channel regulates mitochondrial dynamics to participate in neurodegeneration of Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1086–1103. [Google Scholar] [CrossRef]
- Rameshrad, M.; Omidkhoda, S.F.; Razavi, B.M.; Hosseinzadeh, H. Evaluating the possible role of mitochondrial ATP—Sensitive potassium channels in the cardioprotective effects of morin in the isolated rat heart. Life Sci. 2021, 264, 118659. [Google Scholar] [CrossRef]
- Hu, H.; Ding, Y.; Wang, Y.; Geng, S.; Liu, J.; He, J.; Lu, Y.; Li, X.; Yuan, M.; Zhu, S.; et al. MitoKATP channels promote the proliferation of hypoxic human pulmonary artery smooth muscle cells via the ROS/HIF/miR-210/ISCU signaling pathway. Exp. Ther. Med. 2017, 14, 6105–6112. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Trombetta-Lima, M.; Krabbendam, I.E.; Dolga, A.M. Calcium-activated potassium channels: Implications for aging and age-related neurodegeneration. Int. J. Biochem. Cell Biol. 2020, 123, 105748. [Google Scholar] [CrossRef]
- Burton, M.J.; Cresser-Brown, J.; Thomas, M.; Portolano, N.; Basran, J.; Freeman, S.L.; Kwon, H.; Bottrill, A.R.; Llansola-Portoles, M.J.; Pascal, A.A.; et al. Discovery of a Heme—Binding domain in a neuronal Voltage—Gated potassium channel. J. Biol. Chem. 2020, 295, 13277–13286. [Google Scholar] [CrossRef]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage—Gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef] [PubMed]
- Prosdocimi, E.; Checchetto, V.; Leanza, L. Targeting the Mitochondrial Potassium Channel Kv1.3 to Kill Cancer Cells: Drugs, Strategies, and New Perspectives. SLAS Discov. Adv. Life Sci. R D 2019, 24, 882–892. [Google Scholar] [CrossRef]
- Wrzosek, A.; Augustynek, B.; Zochowska, M.; Szewczyk, A. Mitochondrial Potassium Channels as Druggable Targets. Biomolecules 2020, 10, 1200. [Google Scholar] [CrossRef]
- Murphy, E.; Eisner, D.A. Regulation of intracellular and mitochondrial sodium in health and disease. Circ. Res. 2009, 104, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Nita, I.I.; Hershfinkel, M.; Kantor, C.; Rutter, G.A.; Lewis, E.C.; Sekler, I. Pancreatic beta-cell Na+ channels control global Ca2+ signaling and oxidative metabolism by inducing Na+ and Ca2+ responses that are propagated into mitochondria. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 3301–3312. [Google Scholar]
- Nita, I.I.; Hershfinkel, M.; Lewis, E.C.; Sekler, I. A crosstalk between Na(+) channels, Na(+)/K(+) pump and mitochondrial Na(+) transporters controls Glucose—Dependent cytosolic and mitochondrial Na(+) signals. Cell Calcium 2015, 57, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Takimoto, E.; Dimaano, V.L.; DeMazumder, D.; Kettlewell, S.; Smith, G.; Sidor, A.; Abraham, T.P.; O’Rourke, B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 2014, 115, 44–54. [Google Scholar] [CrossRef]
- Schindl, R.; Weghuber, J.; Romanin, C.; Schweyen, R.J. Mrs2p forms a high conductance Mg2+ selective channel in mitochondria. Biophys. J. 2007, 93, 3872–3883. [Google Scholar] [CrossRef] [PubMed]
- Moomaw, A.S.; Maguire, M.E. The unique nature of mg2+ channels. Physiology 2008, 23, 275–285. [Google Scholar] [CrossRef]
- Mastrototaro, L.; Smorodchenko, A.; Aschenbach, J.R.; Kolisek, M.; Sponder, G. Solute carrier 41A3 encodes for a mitochondrial Mg(2+) efflux system. Sci. Rep. 2016, 6, 27999. [Google Scholar] [CrossRef]
- Cui, Y.; Zhao, S.; Wang, X.; Zhou, B. A novel Drosophila mitochondrial carrier protein acts as a Mg(2+) exporter in fine-tuning mitochondrial Mg(2+) homeostasis. Biochim. Biophys. Acta 2016, 1863, 30–39. [Google Scholar] [CrossRef]
- Cui, Y.; Zhao, S.; Wang, J.; Wang, X.; Gao, B.; Fan, Q.; Sun, F.; Zhou, B. A novel mitochondrial carrier protein Mme1 acts as a yeast mitochondrial magnesium exporter. Biochim. Biophys. Acta 2015, 1853, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, T.; Kuwamura, M.; Tokuda, S.; Izawa, T.; Nakane, Y.; Kitada, K.; Akao, M.; Guenet, J.L.; Serikawa, T. A mutation in the gene encoding mitochondrial Mg(2)+ channel MRS2 results in demyelination in the rat. PLoS Genet. 2011, 7, e1001262. [Google Scholar] [CrossRef]
- Shindo, Y.; Yamanaka, R.; Suzuki, K.; Hotta, K.; Oka, K. Intracellular magnesium level determines cell viability in the MPP(+) model of Parkinson’s disease. Biochim. Biophys. Acta 2015, 1853, 3182–3191. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wei, X.; Yan, P.; Han, Y.; Sun, S.; Wu, K.; Fan, D. Human mitochondrial Mrs2 protein promotes multidrug resistance in gastric cancer cells by regulating p27, cyclin D1 expression and cytochrome C release. Cancer Biol. Ther. 2009, 8, 607–614. [Google Scholar] [CrossRef]
- Ji, S.G.; Medvedeva, Y.V.; Weiss, J.H. Zn(2+) entry through the mitochondrial calcium uniporter is a critical contributor to mitochondrial dysfunction and neurodegeneration. Exp. Neurol. 2020, 325, 113161. [Google Scholar] [CrossRef]
- Medvedeva, Y.V.; Weiss, J.H. Intramitochondrial Zn2+ accumulation via the Ca2+ uniporter contributes to acute ischemic neurodegeneration. Neurobiol. Dis. 2014, 68, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Saris, N.E.; Niva, K. Is Zn2+ transported by the mitochondrial calcium uniporter? FEBS Lett. 1994, 356, 195–198. [Google Scholar] [CrossRef]
- Besecker, B.; Bao, S.; Bohacova, B.; Papp, A.; Sadee, W.; Knoell, D.L. The human zinc transporter SLC39A8 (Zip8) is critical in zinc-mediated cytoprotection in lung epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1127–L1136. [Google Scholar] [CrossRef]
- Sun, Q.; Zhong, W.; Zhang, W.; Li, Q.; Sun, X.; Tan, X.; Sun, X.; Dong, D.; Zhou, Z. Zinc deficiency mediates Alcohol—Induced apoptotic cell death in the liver of rats through activating ER and mitochondrial cell death pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G757–G766. [Google Scholar] [CrossRef]
- Yang, D.M.; Huang, C.C.; Chang, Y.F. Combinatorial roles of mitochondria and cGMP/PKG pathway in the generation of neuronal free Zn2+ under the presence of nitric oxide. JCMA 2020, 83, 357–366. [Google Scholar] [CrossRef]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xia, Z.; Wang, F. Zebrafish in the sea of mineral (iron, zinc, and copper) metabolism. Front. Pharmacol. 2014, 5, 33. [Google Scholar] [CrossRef]
- Finoshin, A.D.; Adameyko, K.I.; Mikhailov, K.V.; Kravchuk, O.I.; Georgiev, A.A.; Gornostaev, N.G.; Kosevich, I.A.; Mikhailov, V.S.; Gazizova, G.R.; Shagimardanova, E.I.; et al. Iron metabolic pathways in the processes of sponge plasticity. PLoS ONE 2020, 15, e0228722. [Google Scholar]
- Seguin, A.; Jia, X.; Earl, A.M.; Li, L.; Wallace, J.; Qiu, A.; Bradley, T.; Shrestha, R.; Troadec, M.B.; Hockin, M.; et al. The mitochondrial metal transporters mitoferrin1 and mitoferrin2 are required for liver regeneration and cell proliferation in mice. J. Biol. Chem. 2020, 295, 11002–11020. [Google Scholar] [CrossRef]
- Mastroberardino, P.G.; Hoffman, E.K.; Horowitz, M.P.; Betarbet, R.; Taylor, G.; Cheng, D.; Na, H.M.; Gutekunst, C.A.; Gearing, M.; Trojanowski, J.Q.; et al. A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson’s disease. Neurobiol. Dis. 2009, 34, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thevenod, F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci. Rep. 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhou, Q.; Wu, D.; Chen, L. Mitochondrial iron metabolism and its role in diseases. Clin. Chim. Acta Int. J. Clin. Chem. 2021, 513, 6–12. [Google Scholar] [CrossRef]
- Mena, N.P.; Urrutia, P.J.; Lourido, F.; Carrasco, C.M.; Nunez, M.T. Mitochondrial iron homeostasis and its dysfunctions in neurodegenerative disorders. Mitochondrion 2015, 21, 92–105. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Mena, N.P.; Nunez, M.T. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 2014, 5, 38. [Google Scholar] [CrossRef]
- Isaya, G. Mitochondrial iron-sulfur cluster dysfunction in neurodegenerative disease. Front. Pharmacol. 2014, 5, 29. [Google Scholar] [CrossRef]
- Boyman, L.; Karbowski, M.; Lederer, W.J. Regulation of Mitochondrial ATP Production: Ca(2+) Signaling and Quality Control. Trends Mol. Med. 2020, 26, 21–39. [Google Scholar] [CrossRef]
- Szabadkai, G.; Duchen, M.R. Mitochondria: The hub of cellular Ca2+ signaling. Physiology 2008, 23, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Sisalli, M.J.; Feliciello, A.; Della Notte, S.; Di Martino, R.; Borzacchiello, D.; Annunziato, L.; Scorziello, A. Nuclear-encoded NCX3 and AKAP121: Two novel modulators of mitochondrial calcium efflux in normoxic and hypoxic neurons. Cell Calcium 2020, 87, 102193. [Google Scholar] [CrossRef] [PubMed]
- Dejos, C.; Gkika, D.; Cantelmo, A.R. The Two—Way Relationship Between Calcium and Metabolism in Cancer. Front. Cell Dev. Biol. 2020, 8, 573747. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Guy, H.R.; Mannella, C.A. Molecular genetics of the VDAC ion channel: Structural model and sequence analysis. J. Bioenerg. Biomembr. 1987, 19, 341–350. [Google Scholar] [CrossRef]
- Paschen, S.A.; Waizenegger, T.; Stan, T.; Preuss, M.; Cyrklaff, M.; Hell, K.; Rapaport, D.; Neupert, W. Evolutionary conservation of biogenesis of beta-barrel membrane proteins. Nature 2003, 426, 862–866. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage—Dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Fan, C.; Fan, M.; Orlando, B.J.; Fastman, N.M.; Zhang, J.; Xu, Y.; Chambers, M.G.; Xu, X.; Perry, K.; Liao, M.; et al. X-ray and cryo-EM structures of the mitochondrial calcium uniporter. Nature 2018, 559, 575–579. [Google Scholar] [CrossRef]
- Fan, M.; Zhang, J.; Tsai, C.W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.F.; Feng, L. Structure and mechanism of the mitochondrial Ca(2+) uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Vais, H.; Payne, R.; Paudel, U.; Li, C.; Foskett, J.K. Coupled transmembrane mechanisms control MCU-mediated mitochondrial Ca(2+) uptake. Proc. Natl. Acad. Sci. USA 2020, 117, 21731–21739. [Google Scholar] [CrossRef]
- Csordas, G.; Golenar, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; Perez, S.F.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Raffaello, A.; Granatiero, V.; Tosatto, A.; Merli, G.; De Stefani, D.; Wright, L.; Pallafacchina, G.; Terrin, A.; Mammucari, C.; et al. The mitochondrial calcium uniporter (MCU): Molecular identity and physiological roles. J. Biol. Chem. 2013, 288, 10750–10758. [Google Scholar] [CrossRef]
- Beutner, G.; Sharma, V.K.; Giovannucci, D.R.; Yule, D.I.; Sheu, S.S. Identification of a ryanodine receptor in rat heart mitochondria. J. Biol. Chem. 2001, 276, 21482–21488. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhao, L.; Clish, C.B.; Clapham, D.E. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc. Natl. Acad. Sci. USA 2013, 110, E2249–E2254. [Google Scholar] [CrossRef]
- Tsai, M.F.; Jiang, D.; Zhao, L.; Clapham, D.; Miller, C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J. Gen. Physiol. 2014, 143, 67–73. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochem. Biokhimiia 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Huser, C.A.; Davies, M.E. Calcium signaling leads to mitochondrial depolarization in impact-induced chondrocyte death in equine articular cartilage explants. Arthritis Rheum. 2007, 56, 2322–2334. [Google Scholar] [CrossRef]
- Vercesi, A.E.; Kowaltowski, A.J.; Oliveira, H.C.; Castilho, R.F. Mitochondrial Ca2+ transport, permeability transition and oxidative stress in cell death: Implications in cardiotoxicity, neurodegeneration and dyslipidemias. Front. Biosci. A J. Virtual Libr. 2006, 11, 2554–2564. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Alevriadou, B.R.; Patel, A.; Noble, M.; Ghosh, S.; Gohil, V.M.; Stathopulos, P.B.; Madesh, M. Molecular nature and physiological role of the mitochondrial calcium uniporter channel. Am. J. Physiol. Cell Physiol. 2021, 320, C465–C482. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Regulation of Mitochondrial Ca(2+) Uptake. Annu. Rev. Physiol. 2021, 83, 107–126. [Google Scholar] [CrossRef]
- Gherardi, G.; Di Marco, G.; Rizzuto, R.; Mammucari, C. Crosstalk between Mitochondrial Ca(2+) Uptake and Autophagy in Skeletal Muscle. Oxidative Med. Cell. Longev. 2019, 2019, 1845321. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.; Kubo, H.; Harris, D.M.; Mills, G.D.; Moyer, J.; Berretta, R.; Potts, S.T.; Marsh, J.D.; Houser, S.R. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ. Res. 2005, 97, 1009–1017. [Google Scholar] [CrossRef]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca(2+) and apoptosis. Cell calcium 2012, 52, 36–43. [Google Scholar] [CrossRef]
- Gutierrez, T.; Parra, V.; Troncoso, R.; Pennanen, C.; Contreras-Ferrat, A.; Vasquez-Trincado, C.; Morales, P.E.; Lopez-Crisosto, C.; Sotomayor-Flores, C.; Chiong, M.; et al. Alteration in mitochondrial Ca(2+) uptake disrupts insulin signaling in hypertrophic cardiomyocytes. CCS 2014, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B.; Ashok, D.; Liu, T. Mitochondrial Ca(2+) in heart failure: Not enough or too much? J. Mol. Cell. Cardiol. 2021, 151, 126–134. [Google Scholar] [CrossRef]
- Dey, K.; Bazala, M.A.; Kuznicki, J. Targeting mitochondrial calcium pathways as a potential treatment against Parkinson’s disease. Cell Calcium 2020, 89, 102216. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Paucek, P. Mitochondrial potassium transport: The K(+) cycle. Biochim. Biophys. Acta 2003, 1606, 23–41. [Google Scholar] [CrossRef]
- Liu, D.; Slevin, J.R.; Lu, C.; Chan, S.L.; Hansson, M.; Elmer, E.; Mattson, M.P. Involvement of mitochondrial K+ release and cellular efflux in ischemic and apoptotic neuronal death. J. Neurochem. 2003, 86, 966–979. [Google Scholar] [CrossRef]
- Rotko, D.; Kunz, W.S.; Szewczyk, A.; Kulawiak, B. Signaling pathways targeting mitochondrial potassium channels. Int. J. Biochem. Cell Biol. 2020, 125, 105792. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ. Res. 2004, 94, 420–432. [Google Scholar] [CrossRef]
- Garlid, K.D.; Paucek, P.; Yarov-Yarovoy, V.; Murray, H.N.; Darbenzio, R.B.; D’Alonzo, A.J.; Lodge, N.J.; Smith, M.A.; Grover, G.J. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP—Sensitive K+ channels. Possible mechanism of cardioprotection. Circ. Res. 1997, 81, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Grover, G.J.; Burkett, D.E.; Parham, C.S.; Scalese, R.J.; Sadanaga, K.K. Protective effect of mitochondrial KATP activation in an isolated gracilis model of ischemia and reperfusion in dogs. J. Cardiovasc. Pharmacol. 2003, 42, 790–792. [Google Scholar] [CrossRef] [PubMed]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabo, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP—Sensitive potassium channel in mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef]
- Siemen, D.; Loupatatzis, C.; Borecky, J.; Gulbins, E.; Lang, F. Ca2+—Activated K channel of the BK-type in the inner mitochondrial membrane of a human glioma cell line. Biochem. Biophys. Res. Commun. 1999, 257, 549–554. [Google Scholar] [CrossRef]
- Kohler, R. Single—Nucleotide polymorphisms in vascular Ca2+-activated K+—Channel genes and cardiovascular disease. Pflug. Arch. Eur. J. Physiol. 2010, 460, 343–351. [Google Scholar] [CrossRef]
- Wang, X.; Yin, C.; Xi, L.; Kukreja, R.C. Opening of Ca2+—Activated K+ channels triggers early and delayed preconditioning against I/R injury independent of NOS in mice. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2070–H2077. [Google Scholar] [CrossRef]
- Dai, H.; Wang, M.; Patel, P.N.; Kalogeris, T.; Liu, Y.; Durante, W.; Korthuis, R.J. Preconditioning with the BKCa channel activator NS-1619 prevents ischemia-reperfusion-induced inflammation and mucosal barrier dysfunction: Roles for ROS and heme oxygenase-1. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H988–H999. [Google Scholar] [CrossRef]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Manago, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct Pharmacological Targeting of a Mitochondrial Ion Channel Selectively Kills Tumor Cells In Vivo. Cancer Cell 2017, 31, 516–531. [Google Scholar] [CrossRef]
- Zaccagnino, A.; Manago, A.; Leanza, L.; Gontarewitz, A.; Linder, B.; Azzolini, M.; Biasutto, L.; Zoratti, M.; Peruzzo, R.; Legler, K.; et al. Tumor—Reducing effect of the clinically used drug clofazimine in a SCID mouse model of pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 38276–38293. [Google Scholar] [CrossRef]
- Chow, L.W.; Cheng, K.S.; Wong, K.L.; Leung, Y.M. Voltage-gated K(+) channels promote BT-474 breast cancer cell migration. Chin. J. Cancer Res. 2018, 30, 613–622. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef] [PubMed]
- Checchetto, V.; Azzolini, M.; Peruzzo, R.; Capitanio, P.; Leanza, L. Mitochondrial potassium channels in cell death. Biochem. Biophys. Res. Commun. 2018, 500, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Rossa, A.; Antoniazzi, G.; Biasutto, L.; Carrer, A.; Campagnaro, M.; Leanza, L.; Gonczi, M.; Csernoch, L.; Paradisi, C.; et al. Synthesis and cellular effects of a mitochondria-targeted inhibitor of the Two—Pore potassium channel TASK-3. Pharmacol. Res. 2021, 164, 105326. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.C.; Bonini, M.G.; Dudley, S.C., Jr. Mitochondria and arrhythmias. Free. Radic. Biol. Med. 2014, 71, 351–361. [Google Scholar] [CrossRef]
- Zotova, L.; Aleschko, M.; Sponder, G.; Baumgartner, R.; Reipert, S.; Prinz, M.; Schweyen, R.J.; Nowikovsky, K. Novel components of an active mitochondrial K(+)/H(+) exchange. J. Biol. Chem. 2010, 285, 14399–14414. [Google Scholar] [CrossRef] [PubMed]
- Hernansanz-Agustin, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Pina, T.; Moreno, L.; Izquierdo-Alvarez, A.; Cabrera-Garcia, J.D.; Cortes, A.; et al. Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020, 586, 287–291. [Google Scholar] [CrossRef]
- Azarias, G.; Van de Ville, D.; Unser, M.; Chatton, J.Y. Spontaneous NA+ transients in individual mitochondria of intact astrocytes. Glia 2008, 56, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Bohm, M.; O’Rourke, B.; Maack, C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.W.; Apel, L.; Brierley, G.P. Matrix free Mg2+ changes with metabolic state in isolated heart mitochondria. Biochemistry 1990, 29, 4121–4128. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Tabata, S.; Shindo, Y.; Hotta, K.; Suzuki, K.; Soga, T.; Oka, K. Mitochondrial Mg(2+) homeostasis decides cellular energy metabolism and vulnerability to stress. Sci. Rep. 2016, 6, 30027. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, R.K.; Qi, F.; Beard, D.A.; Dash, R.K. Characterization of Mg2+ inhibition of mitochondrial Ca2+ uptake by a mechanistic model of mitochondrial Ca2+ uniporter. Biophys. J. 2011, 101, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Gout, E.; Rebeille, F.; Douce, R.; Bligny, R. Interplay of Mg2+, ADP, and ATP in the cytosol and mitochondria: Unravelling the role of Mg2+ in cell respiration. Proc. Natl. Acad. Sci. USA 2014, 111, E4560–E4567. [Google Scholar] [CrossRef]
- Behrens, V.A.; Walter, W.J.; Peters, C.; Wang, T.; Brenner, B.; Geeves, M.A.; Scholz, T.; Steffen, W. Mg(2+)—Free ATP regulates the processivity of native cytoplasmic dynein. FEBS Lett. 2019, 593, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Niermann, K.J.; Olsen, N.J.; Park, J.H. Magnesium abnormalities of skeletal muscle in dermatomyositis and juvenile dermatomyositis. Arthritis Rheum. 2002, 46, 475–488. [Google Scholar] [CrossRef]
- Iotti, S.; Malucelli, E. In vivo assessment of Mg2+ in human brain and skeletal muscle by 31P-MRS. Magnes. Res. 2008, 21, 157–162. [Google Scholar]
- Pilchova, I.; Klacanova, K.; Tatarkova, Z.; Kaplan, P.; Racay, P. The Involvement of Mg(2+) in Regulation of Cellular and Mitochondrial Functions. Oxidative Med. Cell. Longev. 2017, 2017, 6797460. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Funato, Y.; Imamura, H.; Miki, H.; Mizukami, S.; Kikuchi, K. Visualization of Long—Term Mg(2+) dynamics in apoptotic cells using a novel targetable fluorescent probe. Chem. Sci. 2017, 8, 8255–8264. [Google Scholar] [CrossRef]
- Cappadone, C.; Merolle, L.; Marraccini, C.; Farruggia, G.; Sargenti, A.; Locatelli, A.; Morigi, R.; Iotti, S. Intracellular magnesium content decreases during mitochondria-mediated apoptosis induced by a new indole-derivative in human colon cancer cells. Magnes. Res. 2012, 25, 104–111. [Google Scholar] [CrossRef]
- Piskacek, M.; Zotova, L.; Zsurka, G.; Schweyen, R.J. Conditional knockdown of hMRS2 results in loss of mitochondrial Mg(2+) uptake and cell death. J. Cell. Mol. Med. 2009, 13, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Merolle, L.; Sponder, G.; Sargenti, A.; Mastrototaro, L.; Cappadone, C.; Farruggia, G.; Procopio, A.; Malucelli, E.; Parisse, P.; Gianoncelli, A.; et al. Overexpression of the mitochondrial Mg channel MRS2 increases total cellular Mg concentration and influences sensitivity to apoptosis. Met. Integr. Biometal Sci. 2018, 10, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Kuwamura, M.; Tanimura, S.; Hasegawa, Y.; Hoshiai, R.; Moriyama, Y.; Tanaka, M.; Takenaka, S.; Nagayoshi, H.; Izawa, T.; Yamate, J.; et al. Downregulation of aspartoacylase during the progression of myelin breakdown in the dmy mutant rat with mitochondrial magnesium channel MRS2 defect. Brain Res. 2019, 1718, 169–175. [Google Scholar] [CrossRef]
- Sun, Q.; Zhong, W.; Zhang, W.; Zhou, Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: Role of zinc deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G205–G214. [Google Scholar] [CrossRef]
- Adebayo, O.L.; Adenuga, G.A.; Sandhir, R. Selenium and zinc protect brain mitochondrial antioxidants and electron transport chain enzymes following postnatal protein malnutrition. Life Sci. 2016, 152, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Ton-That, D.; Sullivan, P.G.; Jonas, E.A.; Gee, K.R.; Kaczmarek, L.K.; Weiss, J.H. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc. Natl. Acad. Sci. USA 2003, 100, 6157–6162. [Google Scholar] [CrossRef]
- Medvedeva, Y.V.; Lin, B.; Shuttleworth, C.W.; Weiss, J.H. Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 1105–1114. [Google Scholar] [CrossRef]
- Ji, S.G.; Weiss, J.H. Zn(2+)-induced disruption of neuronal mitochondrial function: Synergism with Ca(2+), critical dependence upon cytosolic Zn(2+) buffering, and contributions to neuronal injury. Exp. Neurol. 2018, 302, 181–195. [Google Scholar] [CrossRef]
- Dineley, K.E.; Votyakova, T.V.; Reynolds, I.J. Zinc inhibition of cellular energy production: Implications for mitochondria and neurodegeneration. J. Neurochem. 2003, 85, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Pochwat, B.; Nowak, G.; Szewczyk, B. Relationship between Zinc (Zn (2+)) and Glutamate Receptors in the Processes Underlying Neurodegeneration. Neural Plast. 2015, 2015, 591563. [Google Scholar] [CrossRef]
- Ji, S.G.; Medvedeva, Y.V.; Wang, H.L.; Yin, H.Z.; Weiss, J.H. Mitochondrial Zn(2+) Accumulation: A Potential Trigger of Hippocampal Ischemic Injury. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2019, 25, 126–138. [Google Scholar]
- Ward, D.M.; Cloonan, S.M. Mitochondrial Iron in Human Health and Disease. Annu. Rev. Physiol. 2019, 81, 453–482. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Muhlenhoff, U. Iron-sulfur-protein biogenesis in eukaryotes. Trends Biochem. Sci. 2005, 30, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Tong, W.H. Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat. Rev. Mol. Cell Biol. 2005, 6, 345–351. [Google Scholar] [CrossRef]
- Taher, A.T.; Saliba, A.N. Iron overload in thalassemia: Different organs at different rates. Hematol. Am. Soc. Hematology. Educ. Program 2017, 2017, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Yanatori, I.; Tanaka, A.; Kishi, F.; Lemasters, J.J.; Nishina, S.; Sasaki, K.; Hino, K. Iron loss triggers mitophagy through induction of mitochondrial ferritin. EMBO Rep. 2020, 21, e50202. [Google Scholar] [CrossRef]
- Muhlenhoff, U.; Stadler, J.A.; Richhardt, N.; Seubert, A.; Eickhorst, T.; Schweyen, R.J.; Lill, R.; Wiesenberger, G. A specific role of the yeast mitochondrial carriers MRS3/4p in mitochondrial iron acquisition under iron-limiting conditions. J. Biol. Chem. 2003, 278, 40612–40620. [Google Scholar] [CrossRef]
- Zhang, Y.; Lyver, E.R.; Knight, S.A.; Lesuisse, E.; Dancis, A. Frataxin and mitochondrial carrier proteins, Mrs3p and Mrs4p, cooperate in providing iron for heme synthesis. J. Biol. Chem. 2005, 280, 19794–19807. [Google Scholar] [CrossRef]
- Hamdi, A.; Roshan, T.; Sheftel, A.; Ponka, P. Interaction of Transferrin-Endosomes with Mitochondria: Implications for Iron Transport to Ferrochelatase in Erythroid Cells. Blood 2015, 126, 407. [Google Scholar] [CrossRef]
- Gao, G.; Chang, Y.Z. Mitochondrial ferritin in the regulation of brain iron homeostasis and neurodegenerative diseases. Front. Pharmacol. 2014, 5, 19. [Google Scholar] [CrossRef]
- Yang, H.; Yang, M.; Guan, H.; Liu, Z.; Zhao, S.; Takeuchi, S.; Yanagisawa, D.; Tooyama, I. Mitochondrial ferritin in neurodegenerative diseases. Neurosci. Res. 2013, 77, 1–7. [Google Scholar] [CrossRef]
- Ito, H.; Kurokawa, H.; Matsui, H. Mitochondrial reactive oxygen species and heme, non-heme iron metabolism. Arch. Biochem. Biophys. 2021, 700, 108695. [Google Scholar] [CrossRef]
- Karnati, S.; Luers, G.; Pfreimer, S.; Baumgart-Vogt, E. Mammalian SOD2 is exclusively located in mitochondria and not present in peroxisomes. Histochem. Cell Biol. 2013, 140, 105–117. [Google Scholar] [CrossRef]
- Boycott, K.M.; Beaulieu, C.L.; Kernohan, K.D.; Gebril, O.H.; Mhanni, A.; Chudley, A.E.; Redl, D.; Qin, W.; Hampson, S.; Kury, S.; et al. Autosomal—Recessive Intellectual Disability with Cerebellar Atrophy Syndrome Caused by Mutation of the Manganese and Zinc Transporter Gene SLC39A8. Am. J. Hum. Genet. 2015, 97, 886–893. [Google Scholar] [CrossRef]
- Park, J.H.; Hogrebe, M.; Gruneberg, M.; DuChesne, I.; von der Heiden, A.L.; Reunert, J.; Schlingmann, K.P.; Boycott, K.M.; Beaulieu, C.L.; Mhanni, A.A.; et al. SLC39A8 Deficiency: A Disorder of Manganese Transport and Glycosylation. Am. J. Hum. Genet. 2015, 97, 894–903. [Google Scholar] [CrossRef]
- Tuschl, K.; Meyer, E.; Valdivia, L.E.; Zhao, N.; Dadswell, C.; Abdul-Sada, A.; Hung, C.Y.; Simpson, M.A.; Chong, W.K.; Jacques, T.S.; et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat. Commun. 2016, 7, 11601. [Google Scholar] [CrossRef] [PubMed]
- Hutchens, S.; Liu, C.; Jursa, T.; Shawlot, W.; Chaffee, B.K.; Yin, W.; Gore, A.C.; Aschner, M.; Smith, D.R.; Mukhopadhyay, S. Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice. J. Biol. Chem. 2017, 292, 9760–9773. [Google Scholar] [CrossRef]
- Mercadante, C.J.; Prajapati, M.; Conboy, H.L.; Dash, M.E.; Herrera, C.; Pettiglio, M.A.; Cintron-Rivera, L.; Salesky, M.A.; Rao, D.B.; Bartnikas, T.B. Manganese transporter Slc30a10 controls physiological manganese excretion and toxicity. J. Clin. Investig. 2019, 129, 5442–5461. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Barker, S.; Knutson, M.D. Iron and manganese transport in mammalian systems. Biochim. Biophys. Acta. Mol. Cell Res. 2021, 1868, 118890. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Sancak, Y.; Fomina, Y.; Meisel, J.D.; Chaudhuri, D.; Grabarek, Z.; Mootha, V.K. MICU1 imparts the mitochondrial uniporter with the ability to discriminate between Ca(2+) and Mn(2+). Proc. Natl. Acad. Sci. USA 2018, 115, E7960–E7969. [Google Scholar] [CrossRef]
- Bowman, A.B.; Kwakye, G.F.; Herrero Hernandez, E.; Aschner, M. Role of manganese in neurodegenerative diseases. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. 2011, 25, 191–203. [Google Scholar] [CrossRef]
- Mezzaroba, L.; Alfieri, D.F.; Colado Simao, A.N.; Vissoci Reiche, E.M. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Liu, K.; Liu, Z.F.; Cong, L.; Lei, M.Y.; Ma, Z.; Li, J.; Deng, Y.; Liu, W.; Xu, B. Manganese-induced alpha-synuclein overexpression aggravates mitochondrial damage by repressing PINK1/Parkin-mediated mitophagy. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2021, 152, 112213. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Freinkman, E.; Sabatini, D.M. Rapid immunopurification of mitochondria for metabolite profiling and absolute quantification of matrix metabolites. Nat. Protoc. 2017, 12, 2215–2231. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Freinkman, E.; Wang, T.; Birsoy, K.; Sabatini, D.M. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 2016, 166, 1324–1337. [Google Scholar] [CrossRef]
- Figueroa, J.A.; Stiner, C.A.; Radzyukevich, T.L.; Heiny, J.A. Metal ion transport quantified by ICP-MS in intact cells. Sci. Rep. 2016, 6, 20551. [Google Scholar] [CrossRef] [PubMed]
- Nicolli, A.; Trevisan, A.; Bortoletti, I.; Pozzuoli, A.; Ruggieri, P.; Martinelli, A.; Gambalunga, A.; Carrieri, M. Biological Monitoring of Metal Ions Released from Hip Prostheses. Int. J. Environ. Res. Public Health 2020, 17, 3223. [Google Scholar] [CrossRef] [PubMed]
- Toninello, A.; Clari, G.; Mancon, M.; Tognon, G.; Zatta, P. Aluminum as an inducer of the mitochondrial permeability transition. J. Biol. Inorg. Chem. JBIC A Publ. Soc. Biol. Inorg. Chem. 2000, 5, 612–623. [Google Scholar] [CrossRef]
- Khezri, S.; Sabzalipour, T.; Jahedsani, A.; Azizian, S.; Atashbar, S.; Salimi, A. Chrysin ameliorates aluminum phosphide-induced oxidative stress and mitochondrial damages in rat cardiomyocytes and isolated mitochondria. Environ. Toxicol. 2020, 35, 1114–1124. [Google Scholar] [CrossRef]
- Zischka, H.; Einer, C. Mitochondrial copper homeostasis and its derailment in Wilson disease. Int. J. Biochem. Cell Biol. 2018, 102, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Jeyaraju, D.V.; Voisin, V.; Hurren, R.; Xu, C.; Hawley, J.R.; Barghout, S.H.; Khan, D.H.; Gronda, M.; Wang, X.; et al. Disrupting Mitochondrial Copper Distribution Inhibits Leukemic Stem Cell Self-Renewal. Cell Stem Cell 2020, 26, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Gouw, A.M.; LaGory, E.L.; Guo, S.; Attarwala, N.; Tang, Y.; Qi, J.; Chen, Y.S.; Gao, Z.; Casey, K.M.; et al. Mitochondrial copper depletion suppresses Triple—Negative breast cancer in mice. Nat. Biotechnol. 2021, 39, 357–367. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; An, P.; Gu, Z.; Luo, Y.; Luo, J. Mitochondrial Metal Ion Transport in Cell Metabolism and Disease. Int. J. Mol. Sci. 2021, 22, 7525. https://doi.org/10.3390/ijms22147525
Wang X, An P, Gu Z, Luo Y, Luo J. Mitochondrial Metal Ion Transport in Cell Metabolism and Disease. International Journal of Molecular Sciences. 2021; 22(14):7525. https://doi.org/10.3390/ijms22147525
Chicago/Turabian StyleWang, Xuan, Peng An, Zhenglong Gu, Yongting Luo, and Junjie Luo. 2021. "Mitochondrial Metal Ion Transport in Cell Metabolism and Disease" International Journal of Molecular Sciences 22, no. 14: 7525. https://doi.org/10.3390/ijms22147525
APA StyleWang, X., An, P., Gu, Z., Luo, Y., & Luo, J. (2021). Mitochondrial Metal Ion Transport in Cell Metabolism and Disease. International Journal of Molecular Sciences, 22(14), 7525. https://doi.org/10.3390/ijms22147525