
The Potential of OMICs Technologies for the Treatment of Immune-Mediated Inflammatory Diseases

 , and
, and
Abstract
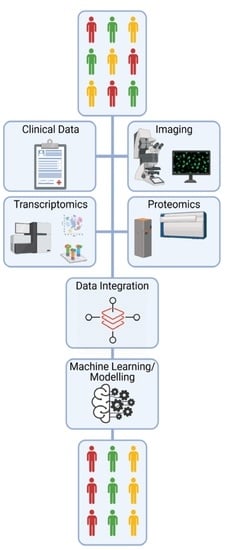
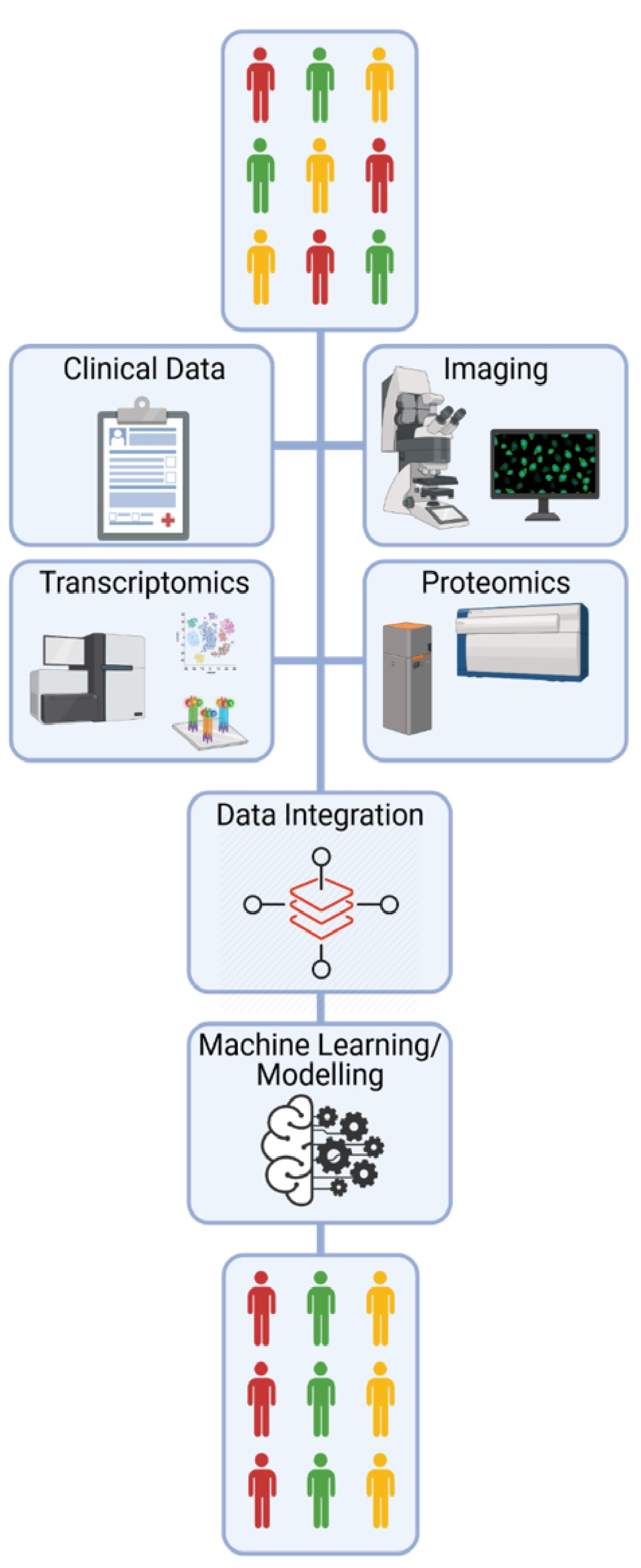
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
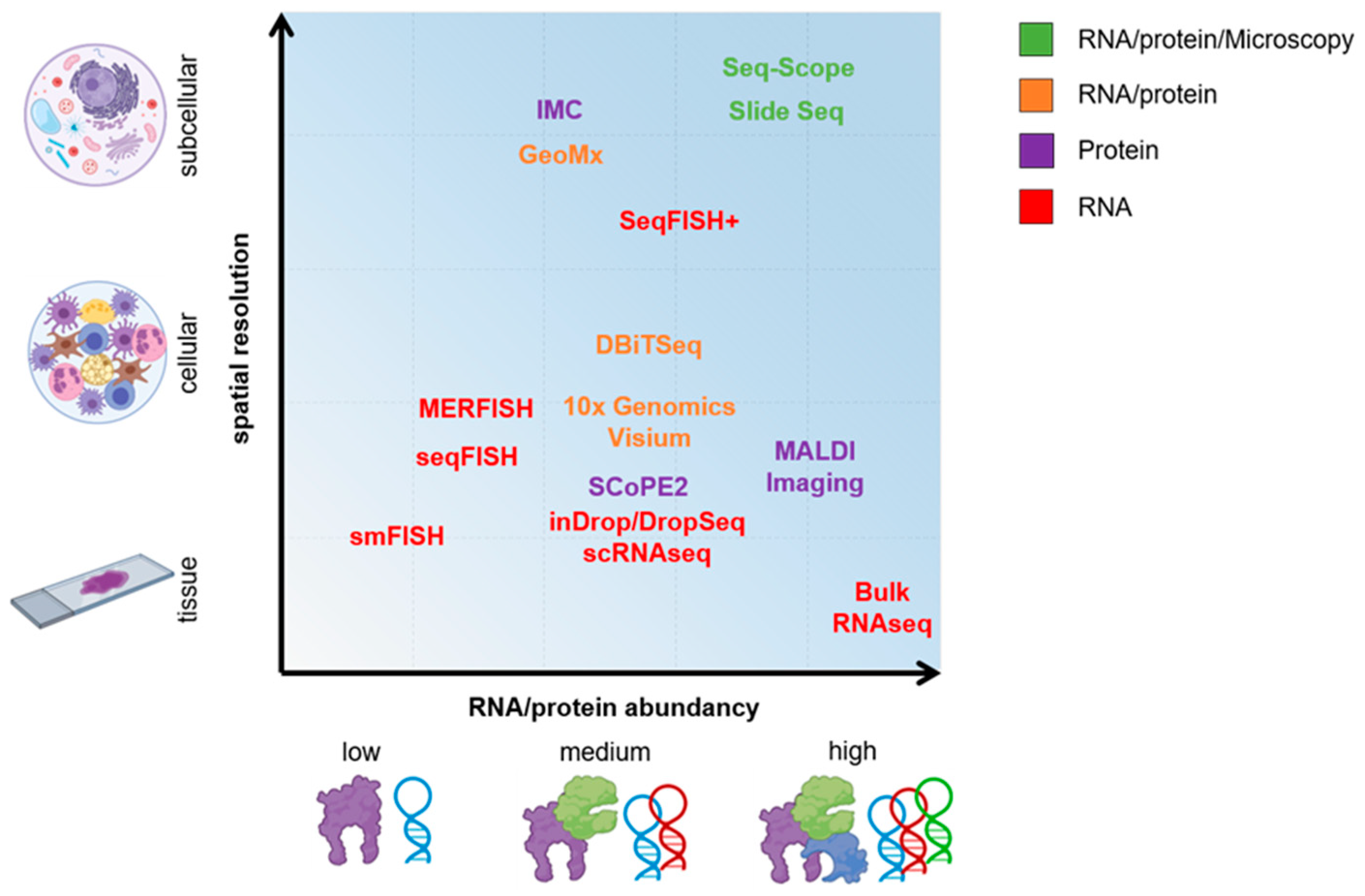
2. On the Path of Single-Cell Omics
3. Single-Cell RNA Sequencing (scRNA-Seq)
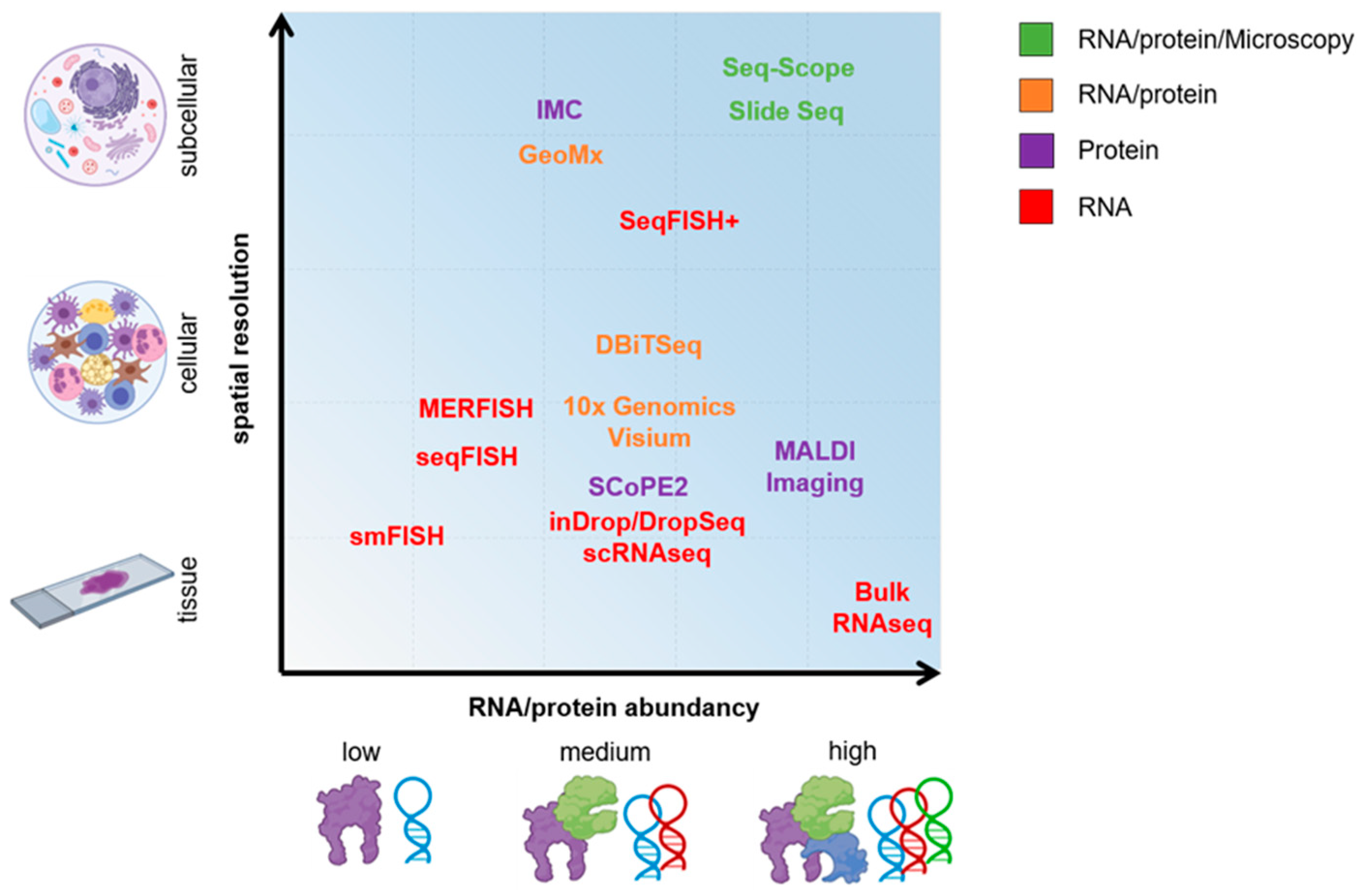
4. Spatial Sequencing
5. On the Road of Single-Cell Proteomics
6. New-Generation Microscopy
6.1. Confocal Laser Scanning Microscopy (CLSM)
6.2. Multi-Photon Laser Scanning Microscopy (MPLSM)
6.3. Light-Sheet Fluorescence Microscopy (LSFM)
6.4. Super-Resolution Microscopy (SRM)
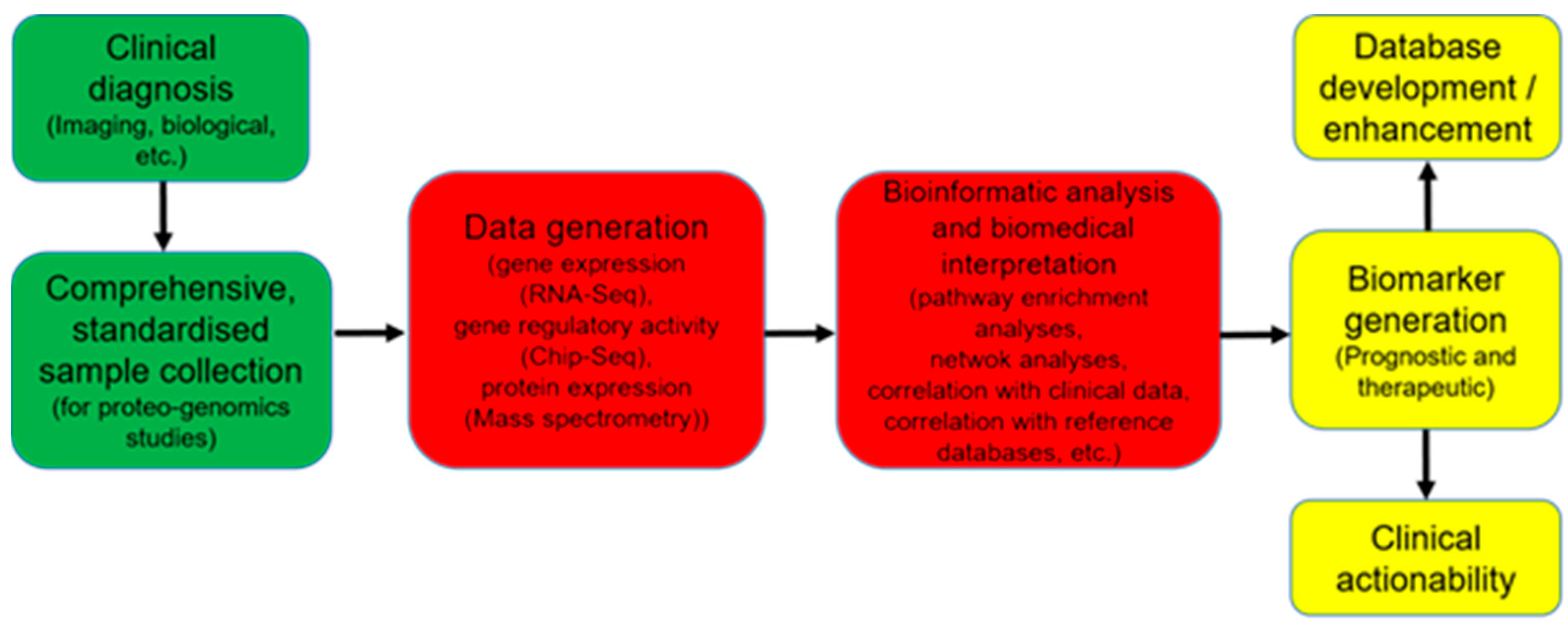
6.5. Limitations in the Direct Translation of Omics Data into Clinical Use
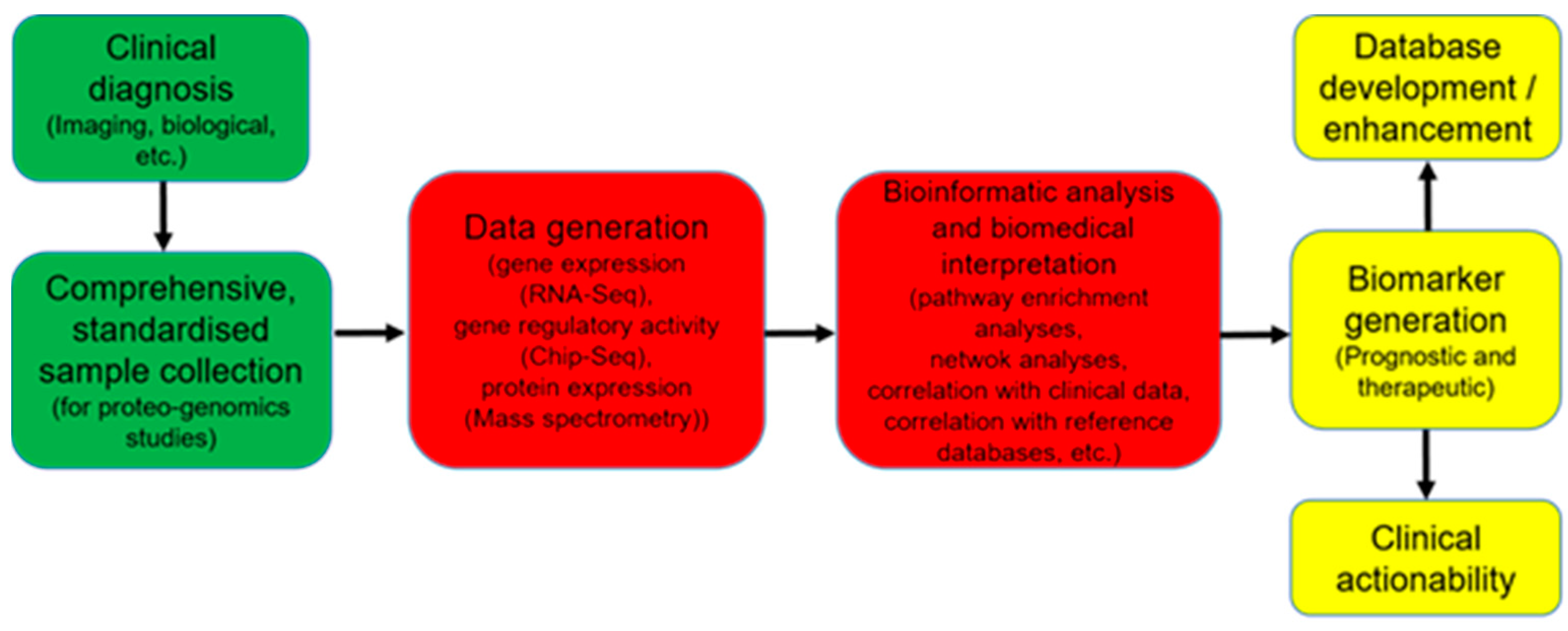
7. Systems Biology Approach to Mine through Complex Omics Data
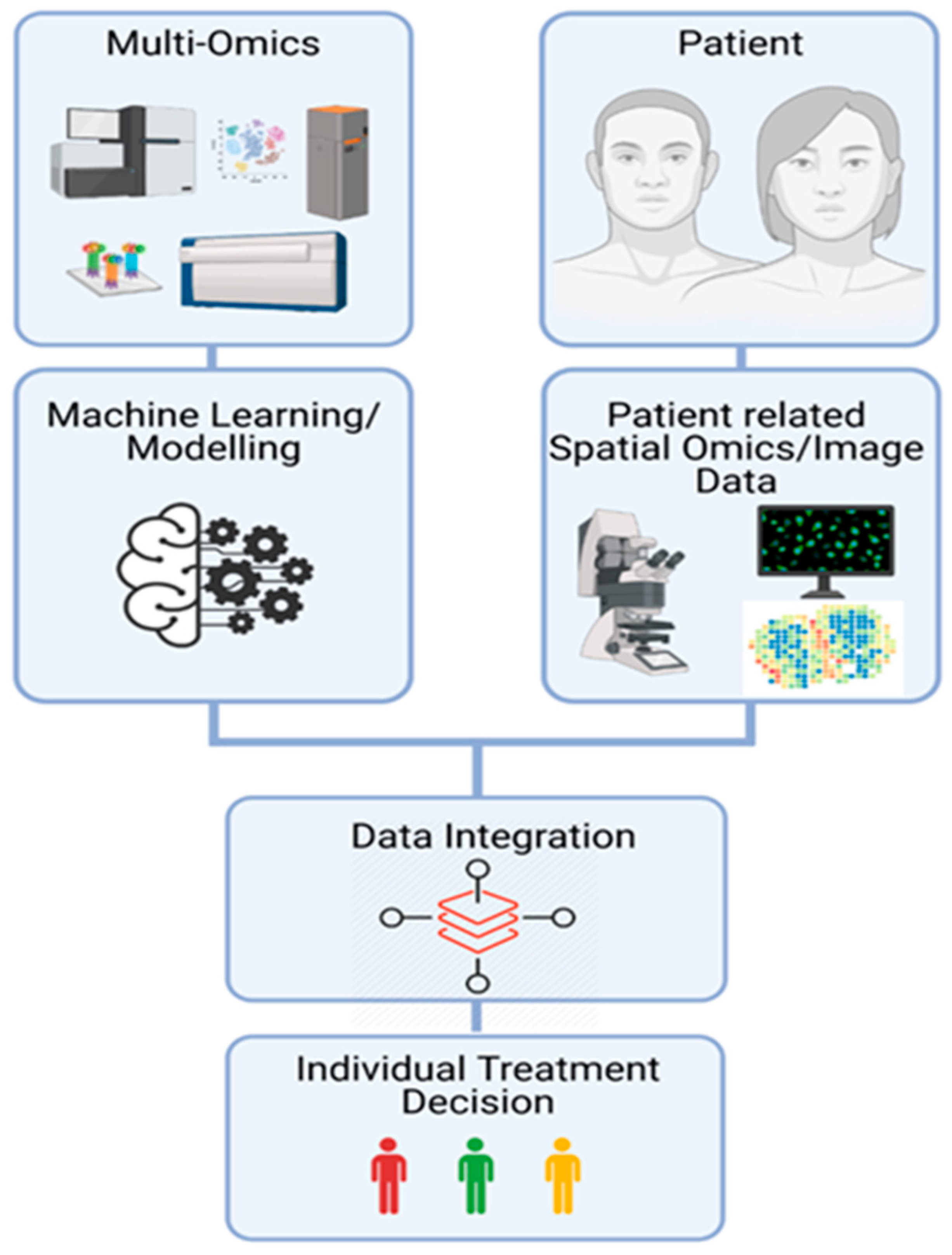
8. Perspectives to Take Advantages of OMICs Technologies in Daily Clinical Practice
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schett, G.; Neurath, M.F. Resolution of chronic inflammatory disease: Universal and tissue-specific concepts. Nat. Commun. 2018, 9, 3261. [Google Scholar] [CrossRef]
- Ramiro, S.; Landewé, R.; van der Heijde, D.; Harrison, D.; Collier, D.; Michaud, K. Discontinuation rates of biologics in patients with rheumatoid arthritis: Are TNF inhibitors different from non-TNF inhibitors? RMD Open 2015, 1, e000155. [Google Scholar] [CrossRef] [Green Version]
- Atreya, R.; Neurath, M.F. Mechanisms of molecular resistance and predictors of response to biological therapy in inflammatory bowel disease. Lancet Gastroenterol. Hepatol. 2018, 3, 790–802. [Google Scholar] [CrossRef]
- Chen, Z.; Bozec, A.; Ramming, A.; Schett, G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat. Rev. Rheumatol. 2019, 15, 9–17. [Google Scholar] [CrossRef]
- Evank, D. Method of the Year 2013. Nat. Methods 2014, 11, 1. [Google Scholar]
- Constantinides, M.G.; Link, V.M.; Tamoutounour, S.; Wong, A.C.; Perez-Chaparro, P.J.; Han, S.J.; Chen, Y.E.; Li, K.; Farhat, S.; Weckel, A.; et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science 2019, 25, 366. [Google Scholar] [CrossRef]
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Satoh-Takayama, N.; Vosshenrich, C.A.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Di Santo, J.P. The expanding family of innate lymphoid cells: Regulators and effectors of immunity and tissue remodeling. Nat. Immunol. 2011, 12, 21–27. [Google Scholar] [CrossRef]
- Klose, C.S.N.; Flach, M.; Möhle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauber, S.; Luber, M.; Weber, S.; Maul, L.; Soare, A.; Wohlfahrt, T.; Lin, N.Y.; Dietel, K.; Bozec, A.; Herrmann, M.; et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat. Med. 2017, 23, 938–944. [Google Scholar] [CrossRef] [Green Version]
- Soare, A.; Weber, S.; Maul, L.; Rauber, S.; Gheorghiu, A.M.; Luber, M.; Houssni, I.; Kleyer, A.; von Pickardt, G.; Gado, M.; et al. Cutting Edge: Homeostasis of Innate Lymphoid Cells Is Imbalanced in Psoriatic Arthritis. J. Immunol. 2018, 200, 1249–1254. [Google Scholar] [CrossRef] [Green Version]
- Guendel, F.; Kofoed-Branzk, M.; Gronke, K.; Tizian, C.; Witkowski, M.; Cheng, H.W.; Heinz, G.A.; Heinrich, F.; Durek, P.; Norris, P.S.; et al. Group 3 innate lymphoid cells program a distinct subset of IL-22BP-producing dendritic cells demarcating solitary intestinal lymphoid tissues. Immunity 2020, 53, 1015–1032. [Google Scholar] [CrossRef] [PubMed]
- Wohlfahrt, T.; Rauber, S.; Uebe, S.; Luber, M.; Soare, A.; Ekici, A.; Weber, S.; Matei, A.E.; Chen, C.W.; Maier, C.; et al. PU. 1 controls fibroblast polarization and tissue fibrosis. Nature 2019, 566, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019, 570, 246–251. [Google Scholar] [CrossRef]
- Wei, K.; Korsunsky, I.; Marshall, J.L.; Gao, A.; Watts, G.F.M.; Major, T.; Croft, A.P.; Watts, J.; Blazar, P.E.; Lange, J.K.; et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature 2020, 582, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Humby, F.; Durez, P.; Buch, M.H.; Lewis, M.J.; Rizvi, H.; Rivellese, F.; Nerviani, A.; Giorli, G.; Mahto, A.; Montecucco, C.; et al. Rituximab versus tocilizumab in anti-TNF inadequate responder patients with rheumatoid arthritis (R4RA): 16-week outcomes of a stratified, biopsy-driven, multicentre, open-label, phase 4 randomised controlled trial. Lancet 2021, 397, 305–317. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative analysis of single-cell RNA sequencing methods. Mol. Cell. 2007, 65, 631–643.e634. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Cai, W.; Sun, Z. Single-cell sequencing technologies: Current and future. J. Genet. Genom. 2014, 41, 513–528. [Google Scholar] [CrossRef]
- Valihrach, L.; Androvic, P.; Kubista, M. Platforms for single-cell collection and analysis. Int. J. Mol. Sci. 2018, 19, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, T.; Liu, F.; Chen, Y.; Yao, J.; Li, Z.; Huang, Y.; Wang, J. Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol. Cell. 2019, 73, 130–142. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kobayashi, T.; Voisin, B.; Jo, J.H.; Sakamoto, K.; Jin, S.P.; Kelly, M.; Pasieka, H.B.; Naff, J.L.; Meyerle, J.H.; et al. Targeted therapy guided by single-cell transcriptomic analysis in drug-induced hypersensitivity syndrome: A case report. Nat. Med. 2020, 26, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Poddubskaya, E.; Bondarenko, A.; Boroda, A.; Zotova, E.; Glusker, A.; Sletina, S.; Makovskaia, L.; Kopylov, P.; Sekacheva, M.; Moisseev, A.; et al. Transcriptomics-guided personalized prescription of targeted therapeutics for metastatic ALK-positive lung cancer case following recurrence on ALK inhibitors. Front. Oncol. 2019, 9, 1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetterstrand, K.A. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP), National Human Genome Research Institute. 2020. Available online: www.genome.gov/sequencingcostsdata (accessed on 14 February 2021).
- Moore, G.M. Cramming more components onto integrated circuits with unit cost. Electronics 1965, 38, 114. [Google Scholar]
- Culemann, S.; Grüneboom, A.; Nicolás-Ávila, J.; Weidner, D.; Lämmle, K.F.; Rothe, T.; Quintana, J.A.; Kirchner, P.; Krljanac, B.; Eberhardt, M.; et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 2019, 572, 670–675. [Google Scholar] [CrossRef]
- Buckley, C.D.; Ospelt, C.; Gay, S.; Midwood, K.S. Location, location, location: How the tissue microenvironment affects inflammation in RA. Nat. Rev. Rheumatol. 2021, 17, 195–212. [Google Scholar] [CrossRef]
- Eng, C.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 2020, 39, 313–319. [Google Scholar] [CrossRef]
- Cho, C.-S.; Xi, J.; Si, Y.; Park, S.-R.; Hsu, J.-E.; Kim, M.; Jun, G.; Kang, H.M.; Lee, J.H. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 2021, 184, 3559–3572. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. Spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 2020, 183, 1665–1681. [Google Scholar] [CrossRef]
- Carlberg, K.; Korotkova, M.; Larsson, L.; Catrina, A.I.; Ståhl, P.L.; Malmström, V. Exploring inflammatory signatures in arthritic joint biopsies with Spatial Transcriptomics. Sci. Rep. 2019, 9, 18975. [Google Scholar] [CrossRef] [Green Version]
- Rzagalinski, I.; Volmer, D.A. Quantification of low molecular weight compounds by MALDI imaging mass spectrometry—A tutorial review. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 726–739. [Google Scholar] [CrossRef]
- Specht, H.; Emmott, E.; Petelski, A.A.; Huffman, R.G.; Perlman, D.H.; Serra, M.; Kharchenko, P.; Koller, A.; Slavov, N. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021, 22, 50. [Google Scholar] [CrossRef] [PubMed]
- Zollinger, D.R.; Lingle, S.E.; Sorg, K.; Beechem, J.M.; Merritt, C.R. GeoMx™ RNA Assay: High multiplex, digital, spatial analysis of RNA in FFPE tissue. Methods Mol. Biol. 2002, 2148, 331–345. [Google Scholar]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef]
- Penkava, F.; Velasco-Herrera, M.D.C.; Young, M.D.; Yager, N.; Nwosu, L.N.; Pratt, A.G.; Lara, A.L.; Guzzo, C.; Maroof, A.; Mamanova, L.; et al. Single-cell sequencing reveals clonal expansions of pro-inflammatory synovial CD8 T cells expressing tissue-homing receptors in psoriatic arthritis. Nat. Commun. 2020, 11, 4767. [Google Scholar] [CrossRef] [PubMed]
- Koba, W.; Jelicks, L.A.; Fine, E.J. MicroPET/SPECT/CT imaging of small animal models of disease. Am. J. Pathol. 2013, 182, 319–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conchello, J.A.; Lichtman, J.W. Optical sectioning microscopy. Nat. Methods 2005, 2, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell. Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legartová, S.; Suchánková, J.; Krejčí, J.; Kovaříková, A.; Bártová, E. Advanced confocal microscopy techniques to study protein-protein interactions and kinetics at DNA lesions. J. Vis. Exp. 2017, 129, 55999. [Google Scholar] [CrossRef]
- Michel, L.; Grasmuck, C.; Charabati, M.; Lécuyer, M.A.; Zandee, S.; Dhaeze, T.; Alvarez, J.I.; Li, R.; Larouche, S.; Bourbonnière, L.; et al. Activated leukocyte cell adhesion molecule regulates B lymphocyte migration across central nervous system barriers. Sci. Transl. Med. 2019, 11, eaaw0475. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Delon, J.; Brotz, T.M.; Germain, R.N. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science 2002, 296, 1873–1876. [Google Scholar] [CrossRef]
- Shin, J.U.; Abaci, H.E.; Herron, L.; Guo, Z.; Sallee, B.; Pappalardo, A.; Jackow, J.; Wang, E.H.C.; Doucet, Y.; Christiano, A.M. Recapitulating T cell infiltration in 3D psoriatic skin models for patient-specific drug testing. Sci. Rep. 2020, 10, 4123. [Google Scholar] [CrossRef] [PubMed]
- Mohan, J.F.; Kohler, R.H.; Hill, J.A.; Weissleder, R.; Mathis, D.; Benoist, C. Imaging the emergence and natural progression of spontaneous autoimmune diabetes. Proc. Natl. Acad. Sci. USA 2017, 114, E7776–E7785. [Google Scholar] [CrossRef] [Green Version]
- Cahalan, M.D.; Parker, I.; Wei, S.H.; Miller, M.J. Two-photon tissue imaging: Seeing the immune system in a fresh light. Nat. Rev. Immunol. 2002, 2, 872–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, Y.M.; Choe, Y.H.; Park, S.A.; Kim, M. LFA-1 (CD11a/CD18) and Mac-1 (CD11b/CD18) distinctly regulate neutrophil extravasation through hotspots I and II. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, R.; Linington, C.; Lassmann, H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006, 129, 1953–1971. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, F.; Murooka, T.T.; Manzo, T.; Kim, E.Y.; Carrizosa, E.; Elpek, N.M.; Mempel, T.R. The transcription factor NFAT exhibits signal memory during serial T cell interactions with antigen-presenting cells. Immunity 2013, 38, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Bartholomäus, I.; Kawakami, N.; Odoardi, F.; Schläger, C.; Miljkovic, D.; Ellwart, J.W.; Klinkert, W.E.; Flügel-Koch, C.; Issekutz, T.B.; Wekerle, H.; et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature 2009, 462, 94–98. [Google Scholar] [CrossRef]
- Horton, N.G.; Wang, K.; Kobat, D.; Clark, C.G.; Wise, F.W.; Schaffer, C.B.; Xu, C. In vivo three-photon microscopy of subcortical structures within an intact mouse brain. Nat. Photonics 2013, 7, 205–209. [Google Scholar] [CrossRef]
- Klioutchnikov, A.; Wallace, D.J.; Frosz, M.H.; Zeltner, R.; Sawinski, J.; Pawlak, V.; Voit, K.M.; Russell, P.S.J.; Kerr, J.N.D. Three-photon head-mounted microscope for imaging deep cortical layers in freely moving rats. Nat. Methods 2020, 17, 509–513. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K.; von Andrian, U.H.; Harms, G. Biological second and third harmonic generation microscopy. Curr. Protoc. Cell. Biol. 2007, 34, 4–15. [Google Scholar] [CrossRef]
- Weigelin, B.; Bakker, G.J.; Friedl, P. Intravital third harmonic generation microscopy of collective melanoma cell invasion: Principles of interface guidance and microvesicle dynamics. Intravital 2012, 1, 32–43. [Google Scholar] [CrossRef]
- Sorokin, L. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Siedentopf, H.; Zsigmondy, R. Uber Sichtbarmachung und Größenbestimmung ultramikoskopischer Teilchen, mit besonderer Anwendung auf Goldrubingläser. Annalen Physik. 1902, 315, 1–39. [Google Scholar] [CrossRef] [Green Version]
- Huisken, J.; Stainier, D.Y. Selective plane illumination microscopy techniques in developmental biology. Development 2009, 136, 1963–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomer, R.; Ye, L.; Hsueh, B.; Deisseroth, K. Advanced CLARITY for rapid and high-resolution imaging of intact tissues. Nat. Protoc. 2014, 9, 1682–1697. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.R.; Dodt, H.U.; Osten, P.; Economo, M.N.; Chandrashekar, J.; Keller, P.J. Whole-brain profiling of cells and circuits in mammals by tissue clearing and light-sheet microscopy. Neuron 2002, 106, 369–387. [Google Scholar] [CrossRef] [PubMed]
- Grüneboom, A.; Hawwari, I.; Weidner, D.; Culemann, S.; Müller, S.; Henneberg, S.; Brenzel, A.; Merz, S.; Bornemann, L.; Zec, K.; et al. A network of trans-cortical capillaries as mainstay for blood circulation in long bones. Nat. Metab. 2019, 1, 236–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, R.; Pan, C.; Ghasemigharagoz, A.; Todorov, M.I.; Förstera, B.; Zhao, S.; Bhatia, H.S.; Parra-Damas, A.; Mrowka, L.; Theodorou, D.; et al. Panoptic imaging of transparent mice reveals whole-body neuronal projections and skull-meninges connections. Nat. Neurosci. 2019, 22, 317–327. [Google Scholar] [CrossRef]
- Zhao, S.; Todorov, M.I.; Cai, R.; Maskari, R.A.; Steinke, H.; Kemter, E.; Mai, H.; Rong, Z.; Warmer, M.; Stanic, K.; et al. Cellular and molecular probing of intact human organs. Cell 2020, 180, 796–812. [Google Scholar] [CrossRef]
- Belle, M.; Godefroy, D.; Couly, G.; Malone, S.A.; Collier, F.; Giacobini, P.; Chédotal, A. Tridimensional visualization and analysis of early human development. Cell 2017, 169, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uderhardt, S.; Martins, A.J.; Tsang, J.S.; Lämmermann, T.; Germain, R.N. Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell 2019, 177, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Klar, T.A.; Hell, S.W. Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett. 1999, 24, 954–956. [Google Scholar] [CrossRef]
- Heintzmann, R.; Huser, T. Super-Resolution Structured Illumination Microscopy. Chem. Rev. 2017, 117, 13890–13908. [Google Scholar] [CrossRef]
- Hell, S.W. Far-field optical nanoscopy. Science 2017, 316, 1153–1158. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, M.G. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA 2005, 102, 13081–13086. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [Green Version]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [Green Version]
- Irvine, D.J.; Purbhoo, M.A.; Krogsgaard, M.; Davis, M.M. Direct observation of ligand recognition by T cells. Nature 2002, 419, 845–849. [Google Scholar] [CrossRef]
- Hu, Y.S.; Cang, H.; Lillemeier, B.F. Superresolution imaging reveals nanometer- and micrometer-scale spatial distributions of T-cell receptors in lymph nodes. Proc. Natl. Acad. Sci. USA 2016, 113, 7201–7206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, M.P.; Gemeinhardt, A.; König, K.; Piliarik, M.; Schaffer, S.; Völkl, S.; Aigner, M.; Mackensen, A.; Sandoghdar, V. Visualizing single-cell secretion dynamics with single-protein sensitivity. Nano. Lett. 2018, 18, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Prevedel, R.; Diz-Muñoz, A.; Ruocco, G.; Antonacci, G. Brillouin microscopy: An emerging tool for mechanobiology. Nat. Methods 2019, 16, 969–977. [Google Scholar] [CrossRef]
- Gargis, A.S.; Kalman, L.; Berry, M.W.; Bick, D.P.; Dimmock, D.P.; Hambuch, T.; Lu, F.; Lyon, E.; Voelkerding, K.V.; Zehnbauer, B.A.; et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol. 2012, 30, 1033–1036. [Google Scholar] [CrossRef]
- Hoen, P.A.; Friedländer, M.R.; Almlöf, J.; Sammeth, M.; Pulyakhina, I.; Anvar, S.Y.; Laros, J.F.; Buermans, H.P.; Karlberg, O.; Brännvall, M.; et al. Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat. Biotechnol. 2013, 31, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Brandão, M.; Pondé, N.; Piccart-Gebhart, M. Mammaprint™: A comprehensive review. Future Oncol. 2019, 15, 207–224. [Google Scholar] [CrossRef]
- Kirschner, M.W. The meaning of systems biology. Cell 2005, 121, 503–504. [Google Scholar] [CrossRef] [Green Version]
- Kitano, H. Systems biology: A brief overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef] [Green Version]
- Hood, L.; Heath, J.R.; Phelps, M.E.; Lin, B. Systems biology and new technologies enable predictive and preventative medicine. Science 2004, 306, 640–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.S.; Maron, B.A.; Loscalzo, J. Systems medicine: Evolution of systems biology from bench to bedside. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 141–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleidgen, S.; Fernau, S.; Fleischer, H.; Schickhardt, C.; Oßa, A.K.; Winkler, E.C. Applying systems biology to biomedical research and health care: A précising definition of systems medicine. BMC Health Serv. Res. 2017, 17, 761. [Google Scholar] [CrossRef]
- Kreeger, P.K.; Lauffenburger, D.A. Cancer systems biology: A network modeling perspective. Carcinogenesis 2010, 31, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Germain, R.N.; Meier-Schellersheim, M.; Nita-Lazar, A.; Fraser, I.D. Systems biology in immunology: A computational modeling perspective. Annu. Rev. Immunol. 2011, 29, 527–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.M.; Tato, C.M.; Furman, D. Systems immunology: Just getting started. Nat. Immunol. 2017, 18, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States department of energy systems biology knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Szostak, J.; Ansari, S.; Madan, S.; Fluck, J.; Talikka, M.; Iskandar, A.; De Leon, H.; Hofmann-Apitius, M.; Peitsch, M.C.; Hoeng, J. Construction of biological networks from unstructured information based on a semi-automated curation workflow. Database 2015, 2015, bav057. [Google Scholar] [CrossRef] [Green Version]
- Gehlenborg, N.; O’Donoghue, S.I.; Baliga, N.S.; Goesmann, A.; Hibbs, M.A.; Kitano, H.; Kohlbacher, O.; Neuweger, H.; Schneider, R.; Tenenbaum, D.; et al. Visualization of omics data for systems biology. Nat. Methods 2010, 7, S56–S68. [Google Scholar] [CrossRef]
- Costanzo, M.; Baryshnikova, A.; Bellay, J.; Kim, Y.; Spear, E.D.; Sevier, C.S.; Ding, H.; Koh, J.L.; Toufighi, K.; Mostafavi, S.; et al. The genetic landscape of a cell. Science 2010, 327, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Livigni, A.; O’Hara, L.; Polak, M.E.; Angus, T.; Wright, D.W.; Smith, L.B.; Freeman, T.C. A graphical and computational modeling platform for biological pathways. Nat. Protoc. 2018, 13, 705–722. [Google Scholar] [CrossRef] [Green Version]
- Tyson, J.J.; Chen, K.; Novak, B. Network dynamics and cell physiology. Nat. Rev. Mol. Cell. Biol. 2001, 2, 908–916. [Google Scholar] [CrossRef]
- Wentker, P.; Eberhardt, M.; Dreyer, F.S.; Bertrams, W.; Cantone, M.; Griss, K.; Schmeck, B.; Vera, J. An interactive macrophage signal transduction map facilitates comparative analyses of high-throughput data. J. Immunol. 2017, 198, 2191–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuller, T.; Atar, S.; Ruppin, E.; Gurevich, M.; Achiron, A. Common and specific signatures of gene expression and protein-protein interactions in autoimmune diseases. Genes Immun. 2013, 14, 67–82. [Google Scholar] [CrossRef]
- Singh, V.; Kalliolias, G.D.; Ostaszewski, M.; Veyssiere, M.; Pilalis, E.; Gawron, P.; Mazein, A.; Bonnet, E.; Petit-Teixeira, E.; Niarakis, A. RA-map: Building a state-of-the-art interactive knowledge base for rheumatoid arthritis. Database 2020, 2020, baaa017. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Moreau, Y.; Tranchevent, L.C. Computational tools for prioritizing candidate genes: Boosting disease gene discovery. Nat. Rev. Genet. 2012, 13, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Cimermančič, P.; Szpiech, Z.A.; Sali, A.; Hernandez, R.D.; Krogan, N.J. High-resolution network biology: Connecting sequence with function. Nat. Rev. Genet. 2013, 14, 865–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, K.; Carvunis, A.R.; Ramesh, S.K.; Ideker, T. Integrative approaches for finding modular structure in biological networks. Nat. Rev. Genet. 2013, 14, 719–732. [Google Scholar] [CrossRef]
- Cowen, L.; Ideker, T.; Raphael, B.J.; Sharan, R. Network propagation: A universal amplifier of genetic associations. Nat. Rev. Genet. 2017, 18, 551–562. [Google Scholar] [CrossRef]
- Fromer, M.; Roussos, P.; Sieberts, S.K.; Johnson, J.S.; Kavanagh, D.H.; Perumal, T.M.; Ruderfer, D.M.; Oh, E.C.; Topol, A.; Shah, H.R.; et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016, 19, 1442–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, D.W.; O’Shaughnessy, J.A.; Kiefer, J.A.; Aldrich, J.; Sinariet, S. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol. Cancer Ther. 2013, 12, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Borad, M.J.; Champion, M.D.; Egan, J.B.; Liang, W.S.; Fonseca, R.; Bryce, A.H.; McCullough, A.E.; Barrett, M.T.; Hunt, K.; Patel, M.D.; et al. WIntegrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 2014, 10, e1004135. [Google Scholar] [CrossRef]
- Maier, A.K.; Syben, C.; Stimpel, B.; Würfl, T.; Hoffmann, M.; Schebesch, F.; Fu, W.; Mill, L.; Kling, L.; Christiansen, S. Learning with known operators reduces maximum training error bounds. Nat. Mach. Intell. 2019, 1, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Bengio, Y.; Courville, A.; Vincent, P. Representation learning: A review and new perspectives. IEEE Trans. Pattern Anal. Mach. Intell. 2013, 35, 1798–1828. [Google Scholar] [CrossRef] [PubMed]
- Ranard, B.L.; Ha, Y.P.; Meisel, Z.F.; Asch, D.A.; Hill, S.S.; Becker, L.B.; Seymour, A.K.; Merchant, R.M. Crowdsourcing--harnessing the masses to advance health and medicine, a systematic review. J Gen. Intern. Med. 2014, 29, 187–203. [Google Scholar] [CrossRef]
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. The human cell atlas. eLife 2017, 6, e27041. [Google Scholar] [CrossRef]
- Rood, J.E.; Stuart, T.; Ghazanfar, S.; Biancalani, T.; Fisher, E.; Butler, A.; Hupalowska, A.; Gaffney, L.; Mauck, W.; Eraslan, G.; et al. Toward a common coordinate framework for the human body. Cell 2019, 179, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anchang, C.G.; Xu, C.; Raimondo, M.G.; Atreya, R.; Maier, A.; Schett, G.; Zaburdaev, V.; Rauber, S.; Ramming, A. The Potential of OMICs Technologies for the Treatment of Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2021, 22, 7506. https://doi.org/10.3390/ijms22147506
Anchang CG, Xu C, Raimondo MG, Atreya R, Maier A, Schett G, Zaburdaev V, Rauber S, Ramming A. The Potential of OMICs Technologies for the Treatment of Immune-Mediated Inflammatory Diseases. International Journal of Molecular Sciences. 2021; 22(14):7506. https://doi.org/10.3390/ijms22147506
Chicago/Turabian StyleAnchang, Charles Gwellem, Cong Xu, Maria Gabriella Raimondo, Raja Atreya, Andreas Maier, Georg Schett, Vasily Zaburdaev, Simon Rauber, and Andreas Ramming. 2021. "The Potential of OMICs Technologies for the Treatment of Immune-Mediated Inflammatory Diseases" International Journal of Molecular Sciences 22, no. 14: 7506. https://doi.org/10.3390/ijms22147506
APA StyleAnchang, C. G., Xu, C., Raimondo, M. G., Atreya, R., Maier, A., Schett, G., Zaburdaev, V., Rauber, S., & Ramming, A. (2021). The Potential of OMICs Technologies for the Treatment of Immune-Mediated Inflammatory Diseases. International Journal of Molecular Sciences, 22(14), 7506. https://doi.org/10.3390/ijms22147506