
Effects of Mutations in TSC Genes on Neurodevelopment and Synaptic Transmission


Abstract
1. Introduction
2. Neurodevelopment, Epilepsy, and Autism
3. Mouse Models and Development
4. Morphological Alterations
5. Glutamatergic System
6. GABAergic System
7. Excitation/Inhibition Balance
8. Therapeutical Perspectives
9. Conclusions
Funding
Conflicts of Interest
References
- Hasbani, D.M.; Crino, P.B. Tuberous sclerosis complex. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 148, pp. 813–822. ISBN 978-0-444-64076-5. [Google Scholar]
- Au, K.S.; Williams, A.T.; Roach, E.S.; Batchelor, L.; Sparagana, S.P.; Delgado, M.R.; Wheless, J.W.; Baumgartner, J.E.; Roa, B.B.; Wilson, C.M.; et al. Genotype/Phenotype Correlation in 325 Individuals Referred for a Diagnosis of Tuberous Sclerosis Complex in the United States. Genet. Med. 2007, 9, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. The TSC1–TSC2 Complex: A Molecular Switchboard Controlling Cell Growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Manning, B.D. MTORC1 Signaling and the Metabolic Control of Cell Growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. A Complex Interplay between Akt, TSC2 and the Two MTOR Complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef]
- Chen, C.-J.; Sgritta, M.; Mays, J.; Zhou, H.; Lucero, R.; Park, J.; Wang, I.-C.; Park, J.H.; Kaipparettu, B.A.; Stoica, L.; et al. Therapeutic Inhibition of MTORC2 Rescues the Behavioral and Neurophysiological Abnormalities Associated with Pten-Deficiency. Nat. Med. 2019, 25, 1684–1690. [Google Scholar] [CrossRef]
- Bockaert, J.; Marin, P. MTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef] [PubMed]
- Sato, A. MTOR, a Potential Target to Treat Autism Spectrum Disorder. CNS Neurol. Disord.-Drug Targets (Former. Curr. Drug Targets-CNS Neurol. Disord.) 2016, 15, 533–543. [Google Scholar] [CrossRef]
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular Neurobiology of MTOR. Neuroscience 2017, 341, 112–153. [Google Scholar] [CrossRef]
- de Vries, P.J.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; Dahlin, M.; D’Amato, L.; d’Augères, G.B.; Ferreira, J.C.; et al. TSC-Associated Neuropsychiatric Disorders (TAND): Findings from the TOSCA Natural History Study. Orphanet J. rare Dis. 2018, 13, 157. [Google Scholar] [CrossRef]
- Zöllner, J.P.; Franz, D.N.; Hertzberg, C.; Nabbout, R.; Rosenow, F.; Sauter, M.; Schubert-Bast, S.; Wiemer-Kruel, A.; Strzelczyk, A. A Systematic Review on the Burden of Illness in Individuals with Tuberous Sclerosis Complex (TSC). Orphanet J. Rare Dis. 2020, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Prabowo, A.S.; Anink, J.J.; Lammens, M.; Nellist, M.; van den Ouweland, A.M.W.; Adle-Biassette, H.; Sarnat, H.B.; Flores-Sarnat, L.; Crino, P.B.; Aronica, E. Fetal Brain Lesions in Tuberous Sclerosis Complex: TORC1 Activation and Inflammation: Fetal Brain Lesions and TSC. Brain Pathol. 2013, 23, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.S.; Karas, P.J.; Krueger, D.A.; Weiner, H.L. Central Nervous System Manifestations of Tuberous Sclerosis Complex. Am. J. Med. Genet. 2018, 178, 291–298. [Google Scholar] [CrossRef]
- Cotter, J.A. An Update on the Central Nervous System Manifestations of Tuberous Sclerosis Complex. Acta Neuropathol. 2020, 139, 613–624. [Google Scholar] [CrossRef]
- Marcotte, L.; Aronica, E.; Baybis, M.; Crino, P.B. Cytoarchitectural Alterations Are Widespread in Cerebral Cortex in Tuberous Sclerosis Complex. Acta Neuropathol 2012, 123, 685–693. [Google Scholar] [CrossRef]
- Chiarotti, F.; Venerosi, A. Epidemiology of Autism Spectrum Disorders: A Review of Worldwide Prevalence Estimates Since 2014. Brain Sciences 2020, 10, 274. [Google Scholar] [CrossRef]
- Jeste, S.S.; Varcin, K.J.; Hellemann, G.S.; Gulsrud, A.C.; Bhatt, R.; Kasari, C.; Wu, J.Y.; Sahin, M.; Nelson, C.A. Symptom Profiles of Autism Spectrum Disorder in Tuberous Sclerosis Complex. Neurology 2016, 87, 766–772. [Google Scholar] [CrossRef] [PubMed]
- van’t Hof, M.; Tisseur, C.; van Berckelear-Onnes, I.; van Nieuwenhuyzen, A.; Daniels, A.M.; Deen, M.; Hoek, H.W.; Ester, W.A. Age at Autism Spectrum Disorder Diagnosis: A Systematic Review and Meta-Analysis from 2012 to 2019. Autism 2021, 25, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Chawarska, K.; Paul, R.; Klin, A.; Hannigen, S.; Dichtel, L.E.; Volkmar, F. Parental Recognition of Developmental Problems in Toddlers with Autism Spectrum Disorders. J. Autism Dev. Disord. 2007, 37, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Moavero, R.; Benvenuto, A.; Emberti Gialloreti, L.; Siracusano, M.; Kotulska, K.; Weschke, B.; Riney, K.; Jansen, F.; Feucht, M.; Krsek, P.; et al. Early Clinical Predictors of Autism Spectrum Disorder in Infants with Tuberous Sclerosis Complex: Results from the EPISTOP Study. JCM 2019, 8, 788. [Google Scholar] [CrossRef]
- Dickinson, A.; Varcin, K.J.; Sahin, M.; Nelson, C.A.; Jeste, S.S. Early Patterns of Functional Brain Development Associated with Autism Spectrum Disorder in Tuberous Sclerosis Complex. Autism Res. 2019, 12, 1758–1773. [Google Scholar] [CrossRef] [PubMed]
- Prohl, A.K.; Scherrer, B.; Tomas-Fernandez, X.; Davis, P.E.; Filip-Dhima, R.; Prabhu, S.P.; Peters, J.M.; Bebin, E.M.; Krueger, D.A.; Northrup, H.; et al. Early White Matter Development Is Abnormal in Tuberous Sclerosis Complex Patients Who Develop Autism Spectrum Disorder. J. Neurodev. Disord. 2019, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Nabbout, R.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; Dahlin, M.; D’Amato, L.; d’Augères, G.B.; de Vries, P.J.; et al. Epilepsy in Tuberous Sclerosis Complex: Findings from the TOSCA Study. Epilepsia Open 2019, 4, 73–84. [Google Scholar] [CrossRef]
- Curatolo, P.; Moavero, R.; de Vries, P.J. Neurological and Neuropsychiatric Aspects of Tuberous Sclerosis Complex. Lancet Neurol. 2015, 14, 733–745. [Google Scholar] [CrossRef]
- Major, P.; Rakowski, S.; Simon, M.V.; Cheng, M.L.; Eskandar, E.; Baron, J.; Leeman, B.A.; Frosch, M.P.; Thiele, E.A. Are Cortical Tubers Epileptogenic? Evidence from Electrocorticography. Epilepsia 2009, 50, 147–154. [Google Scholar] [CrossRef]
- Neal, A.; Ostrowsky-Coste, K.; Jung, J.; Lagarde, S.; Maillard, L.; Kahane, P.; Touraine, R.; Catenoix, H.; Montavont, A.; Isnard, J.; et al. Epileptogenicity in Tuberous Sclerosis Complex: A Stereoelectroencephalographic Study. Epilepsia 2020, 61, 81–95. [Google Scholar] [CrossRef]
- Specchio, N.; Pietrafusa, N.; Trivisano, M.; Moavero, R.; De Palma, L.; Ferretti, A.; Vigevano, F.; Curatolo, P. Autism and Epilepsy in Patients With Tuberous Sclerosis Complex. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef]
- Tye, C.; Mcewen, F.S.; Liang, H.; Underwood, L.; Woodhouse, E.; Barker, E.D.; Sheerin, F.; Yates, J.R.W.; Bolton, P.F. Long-Term Cognitive Outcomes in Tuberous Sclerosis Complex. Dev. Med. Child. Neurol. 2020, 62, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Moavero, R.; Kotulska, K.; Lagae, L.; Benvenuto, A.; Gialloreti, L.E.; Weschke, B.; Riney, K.; Feucht, M.; Krsek, P.; Nabbout, R.; et al. Is Autism Driven by Epilepsy in Infants with Tuberous Sclerosis Complex? Ann. Clin. Transl. Neurol. 2020, 7, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Hulshof, H.M.; Slot, E.M.H.; Lequin, M.; Breuillard, D.; Boddaert, N.; Jozwiak, S.; Kotulska, K.; Riney, K.; Feucht, M.; Samueli, S.; et al. Fetal Brain Magnetic Resonance Imaging Findings Predict Neurodevelopment in Children with Tuberous Sclerosis Complex. J. Pediatrics 2021, 233, 156–162.e2. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.E.; Kapur, K.; Filip-Dhima, R.; Trowbridge, S.K.; Little, E.; Wilson, A.; Leuchter, A.; Bebin, E.M.; Krueger, D.; Northrup, H.; et al. Increased Electroencephalography Connectivity Precedes Epileptic Spasm Onset in Infants with Tuberous Sclerosis Complex. Epilepsia 2019, 60, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Rennebeck, G.; Kleymenova, E.V.; Anderson, R.; Yeung, R.S.; Artzt, K.; Walker, C.L. Loss of Function of the Tuberous Sclerosis 2 Tumor Suppressor Gene Results in Embryonic Lethality Characterized by Disrupted Neuroepithelial Growth and Development. Proc. Natl. Acad. Sci. USA 1998, 95, 15629–15634. [Google Scholar] [CrossRef]
- Kobayashi, T.; Minowa, O.; Sugitani, Y.; Takai, S.; Mitani, H.; Kobayashi, E.; Noda, T.; Hino, O. A Germ-Line Tsc1 Mutation Causes Tumor Development and Embryonic Lethality That Are Similar, but Not Identical to, Those Caused by Tsc2 Mutation in Mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8762–8767. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Takashima, S.; Yamanouchi, H.; Nakazato, Y.; Mitani, H.; Hino, O. Novel Cerebral Lesions in the Eker Rat Model of Tuberous Sclerosis: Cortical Tuber and Anaplastic Ganglioglioma. J. Neuropathol. Exp. Neurol. 2000, 59, 188–196. [Google Scholar] [CrossRef][Green Version]
- Takahashi, D.K.; Dinday, M.T.; Barbaro, N.M.; Baraban, S.C. Abnormal Cortical Cells and Astrocytomas in the Eker Rat Model of Tuberous Sclerosis Complex. Epilepsia 2004, 45, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Lozovaya, N.; Gataullina, S.; Tsintsadze, T.; Tsintsadze, V.; Pallesi-Pocachard, E.; Minlebaev, M.; Goriounova, N.A.; Buhler, E.; Watrin, F.; Shityakov, S.; et al. Selective Suppression of Excessive GluN2C Expression Rescues Early Epilepsy in a Tuberous Sclerosis Murine Model. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Gataullina, S.; Lemaire, E.; Wendling, F.; Kaminska, A.; Watrin, F.; Riquet, A.; Ville, D.; Moutard, M.-L.; de Saint Martin, A.; Napuri, S.; et al. Epilepsy in Young Tsc1 +/− Mice Exhibits Age-Dependent Expression That Mimics That of Human Tuberous Sclerosis Complex. Epilepsia 2016, 57, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Kasai, S.; Kobayashi, T.; Takamatsu, Y.; Hino, O.; Ikeda, K.; Mizuguchi, M. Rapamycin Reverses Impaired Social Interaction in Mouse Models of Tuberous Sclerosis Complex. Nat. Commun. 2012, 3, 1292. [Google Scholar] [CrossRef]
- Tang, G.; Gudsnuk, K.; Kuo, S.-H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of MTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Han, S.; Shilyansky, C.; Zhou, Y.; Li, W.; Kwiatkowski, D.J.; Ramesh, V.; Silva, A.J. Reversal of Learning Deficits in a Tsc2+/− Mouse Model of Tuberous Sclerosis. Nat. Med. 2008, 14, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Silva, A.J. Increased Levels of Anxiety-Related Behaviors in a Tsc2 Dominant Negative Transgenic Mouse Model of Tuberous Sclerosis. Behav. Genet. 2011, 41, 357–363. [Google Scholar] [CrossRef][Green Version]
- Goorden, S.M.I.; van Woerden, G.M.; van der Weerd, L.; Cheadle, J.P.; Elgersma, Y. Cognitive Deficits in Tsc1+/−mice in the Absence of Cerebral Lesions and Seizures. Ann. Neurol. 2007, 62, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Haji, N.; Riebe, I.; Aguilar-Valles, A.; Artinian, J.; Laplante, I.; Lacaille, J.-C. Tsc1 Haploinsufficiency in Nkx2.1 Cells Upregulates Hippocampal Interneuron MTORC1 Activity, Impairs Pyramidal Cell Synaptic Inhibition, and Alters Contextual Fear Discrimination and Spatial Working Memory in Mice. Mol. Autism 2020, 11, 29. [Google Scholar] [CrossRef]
- Waltereit, R.; Japs, B.; Schneider, M.; de Vries, P.J.; Bartsch, D. Epilepsy and Tsc2 Haploinsufficiency Lead to Autistic-Like Social Deficit Behaviors in Rats. Behav. Genet. 2011, 41, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Young, D.M.; Schenk, A.K.; Yang, S.-B.; Jan, Y.N.; Jan, L.Y. Altered Ultrasonic Vocalizations in a Tuberous Sclerosis Mouse Model of Autism. Proc. Natl. Acad. Sci. USA 2010, 107, 11074–11079. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Zhang, B.; Gutmann, D.H.; Wong, M. Postnatal Reduction of Tuberous Sclerosis Complex 1 Expression in Astrocytes and Neurons Causes Seizures in an Age-Dependent Manner. Epilepsia 2017, 58, 2053–2063. [Google Scholar] [CrossRef]
- Meikle, L.; Talos, D.M.; Onda, H.; Pollizzi, K.; Rotenberg, A.; Sahin, M.; Jensen, F.E.; Kwiatkowski, D.J. A Mouse Model of Tuberous Sclerosis: Neuronal Loss of Tsc1 Causes Dysplastic and Ectopic Neurons, Reduced Myelination, Seizure Activity, and Limited Survival. J. Neurosci. 2007, 27, 5546–5558. [Google Scholar] [CrossRef]
- Fu, C.; Cawthon, B.; Clinkscales, W.; Bruce, A.; Winzenburger, P.; Ess, K.C. GABAergic Interneuron Development and Function Is Modulated by the Tsc1 Gene. Cereb. Cortex 2012, 22, 2111–2119. [Google Scholar] [CrossRef]
- Goto, J.; Talos, D.M.; Klein, P.; Qin, W.; Chekaluk, Y.I.; Anderl, S.; Malinowska, I.A.; Nardo, A.D.; Bronson, R.T.; Chan, J.A.; et al. Regulable Neural Progenitor-Specific Tsc1 Loss Yields Giant Cells with Organellar Dysfunction in a Model of Tuberous Sclerosis Complex. Proc. Natl. Acad. Sci. USA 2011, 108, E1070–E1079. [Google Scholar] [CrossRef]
- Magri, L.; Cambiaghi, M.; Cominelli, M.; Alfaro-Cervello, C.; Cursi, M.; Pala, M.; Bulfone, A.; Garcìa-Verdugo, J.M.; Leocani, L.; Minicucci, F.; et al. Sustained Activation of MTOR Pathway in Embryonic Neural Stem Cells Leads to Development of Tuberous Sclerosis Complex-Associated Lesions. Cell Stem Cell 2011, 9, 447–462. [Google Scholar] [CrossRef]
- Tsai, P.T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like Behaviour and Cerebellar Dysfunction in Purkinje Cell Tsc1 Mutant Mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef]
- Normand, E.A.; Crandall, S.R.; Thorn, C.A.; Murphy, E.M.; Voelcker, B.; Browning, C.; Machan, J.T.; Moore, C.I.; Connors, B.W.; Zervas, M. Temporal and Mosaic Tsc1 Deletion in the Developing Thalamus Disrupts Thalamocortical Circuitry, Neural Function, and Behavior. Neuron 2013, 78, 895–909. [Google Scholar] [CrossRef]
- Way, S.W.; McKenna, J.; Mietzsch, U.; Reith, R.M.; Wu, H.C.-J.; Gambello, M.J. Loss of Tsc2 in Radial Glia Models the Brain Pathology of Tuberous Sclerosis Complex in the Mouse. Hum. Mol. Genet. 2009, 18, 1252–1265. [Google Scholar] [CrossRef]
- Fu, C.; Ess, K.C. Conditional and Domain-Specific Inactivation of the Tsc2 Gene in Neural Progenitor Cells. Genesis 2013, 51, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Crowell, B.; Hwa Lee, G.; Nikolaeva, I.; Dal Pozzo, V.; D’Arcangelo, G. Complex Neurological Phenotype in Mutant Mice Lacking Tsc2 in Excitatory Neurons of the Developing Forebrain. Eneuro 2015, 2, ENEURO.0046-15.2015. [Google Scholar] [CrossRef] [PubMed]
- Carson, R.P.; Kelm, N.D.; West, K.L.; Does, M.D.; Fu, C.; Weaver, G.; McBrier, E.; Parker, B.; Grier, M.D.; Ess, K.C. Hypomyelination Following Deletion of Tsc2 in Oligodendrocyte Precursors. Ann. Clin. Transl. Neurol. 2015, 2, 1041–1054. [Google Scholar] [CrossRef]
- Zeng, L.-H.; Rensing, N.R.; Zhang, B.; Gutmann, D.H.; Gambello, M.J.; Wong, M. Tsc2 Gene Inactivation Causes a More Severe Epilepsy Phenotype than Tsc1 Inactivation in a Mouse Model of Tuberous Sclerosis Complex. Hum. Mol. Genet. 2011, 20, 445–454. [Google Scholar] [CrossRef]
- Mietzsch, U.; McKenna, J.; Reith, R.M.; Way, S.W.; Gambello, M.J. Comparative Analysis of Tsc1 and Tsc2 Single and Double Radial Glial Cell Mutants. J. Comp. Neurol. 2013, 521, 3817–3831. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Onda, H.; Lueck, A.; Marks, P.W.; Warren, H.B.; Kwiatkowski, D.J. Tsc2(+/-) Mice Develop Tumors in Multiple Sites That Express Gelsolin and Are Influenced by Genetic Background. J. Clin. Investig. 1999, 104, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.D.; Hockemeyer, D.; Bateup, H.S. Genetically Engineered Human Cortical Spheroid Models of Tuberous Sclerosis. Nat. Med. 2018, 24, 1568–1578. [Google Scholar] [CrossRef]
- Feliciano, D.M.; Su, T.; Lopez, J.; Platel, J.-C.; Bordey, A. Single-Cell Tsc1 Knockout during Corticogenesis Generates Tuber-like Lesions and Reduces Seizure Threshold in Mice. J. Clin. Investig. 2011, 121, 1596–1607. [Google Scholar] [CrossRef]
- Uhlmann, E.J.; Wong, M.; Baldwin, R.L.; Bajenaru, M.L.; Onda, H.; Kwiatkowski, D.J.; Yamada, K.; Gutmann, D.H. Astrocyte-Specific TSC1 Conditional Knockout Mice Exhibit Abnormal Neuronal Organization and Seizures. Ann. Neurol. 2002, 52, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Dooves, S.; van Velthoven, A.J.H.; Suciati, L.G.; Heine, V.M. Neuron–Glia Interactions in Tuberous Sclerosis Complex Affect the Synaptic Balance in 2D and Organoid Cultures. Cells 2021, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-J.; Di Nardo, A.; Kramvis, I.; Meikle, L.; Kwiatkowski, D.J.; Sahin, M.; He, X. Tuberous Sclerosis Complex Proteins Control Axon Formation. Genes Dev. 2008, 22, 2485–2495. [Google Scholar] [CrossRef] [PubMed]
- Nie, D.; Di Nardo, A.; Han, J.M.; Baharanyi, H.; Kramvis, I.; Huynh, T.; Dabora, S.; Codeluppi, S.; Pandolfi, P.P.; Pasquale, E.B.; et al. Tsc2-Rheb Signaling Regulates EphA-Mediated Axon Guidance. Nat. Neurosci. 2010, 13, 163–172. [Google Scholar] [CrossRef]
- Zhang, B.; Zou, J.; Han, L.; Rensing, N.; Wong, M. Microglial Activation during Epileptogenesis in a Mouse Model of Tuberous Sclerosis Complex. Epilepsia 2016, 57, 1317–1325. [Google Scholar] [CrossRef]
- Nadadhur, A.G.; Alsaqati, M.; Gasparotto, L.; Cornelissen-Steijger, P.; van Hugte, E.; Dooves, S.; Harwood, A.J.; Heine, V.M. Neuron-Glia Interactions Increase Neuronal Phenotypes in Tuberous Sclerosis Complex Patient IPSC-Derived Models. Stem Cell Rep. 2019, 12, 42–56. [Google Scholar] [CrossRef]
- Tavazoie, S.F.; Alvarez, V.A.; Ridenour, D.A.; Kwiatkowski, D.J.; Sabatini, B.L. Regulation of Neuronal Morphology and Function by the Tumor Suppressors Tsc1 and Tsc2. Nat. Neurosci. 2005, 8, 1727–1734. [Google Scholar] [CrossRef]
- Sugiura, H.; Yasuda, S.; Katsurabayashi, S.; Kawano, H.; Endo, K.; Takasaki, K.; Iwasaki, K.; Ichikawa, M.; Kobayashi, T.; Hino, O.; et al. Rheb Activation Disrupts Spine Synapse Formation through Accumulation of Syntenin in Tuberous Sclerosis Complex. Nat. Commun. 2015, 6, 6842. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bartley, C.M.; Gong, X.; Hsieh, L.S.; Lin, T.V.; Feliciano, D.M.; Bordey, A. MEK-ERK1/2-Dependent FLNA Overexpression Promotes Abnormal Dendritic Patterning in Tuberous Sclerosis Independent of MTOR. Neuron 2014, 84, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; de Kok, L.; Willemsen, R.; Elgersma, Y.; Borst, J.G.G. In Vivo Synaptic Transmission and Morphology in Mouse Models of Tuberous Sclerosis, Fragile X Syndrome, Neurofibromatosis Type 1, and Costello Syndrome. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Zhao, J.-P.; Yoshii, A. Hyperexcitability of the Local Cortical Circuit in Mouse Models of Tuberous Sclerosis Complex. Mol. Brain 2019, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Bateup, H.S.; Johnson, C.A.; Denefrio, C.L.; Saulnier, J.L.; Kornacker, K.; Sabatini, B.L. Excitatory/Inhibitory Synaptic Imbalance Leads to Hippocampal Hyperexcitability in Mouse Models of Tuberous Sclerosis. Neuron 2013, 78, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, D.; Lombardi, A.; Kirischuk, S.; Luhmann, H.J. Haploinsufficiency of Tsc2 Leads to Hyperexcitability of Medial Prefrontal Cortex via Weakening of Tonic GABAB Receptor-Mediated Inhibition. Cereb. Cortex 2020. [Google Scholar] [CrossRef]
- Bassetti, D.; Luhmann, H.J.; Kirischuk, S. Presynaptic GABA(B) Receptor-Mediated Network Excitation in the Medial Prefrontal Cortex of Tsc2+/− Mice. Pflugers Arch. 2021, in press. [Google Scholar]
- Antoine, M.W.; Langberg, T.; Schnepel, P.; Feldman, D.E. Increased Excitation-Inhibition Ratio Stabilizes Synapse and Circuit Excitability in Four Autism Mouse Models. Neuron 2019, 101, 648–661.e4. [Google Scholar] [CrossRef]
- von der Brelie, C.; Waltereit, R.; Zhang, L.; Beck, H.; Kirschstein, T. Impaired Synaptic Plasticity in a Rat Model of Tuberous Sclerosis. Eur. J. Neurosci. 2006, 23, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, B.D.; Osterweil, E.K.; Bear, M.F. Mutations Causing Syndromic Autism Define an Axis of Synaptic Pathophysiology. Nature 2011, 480, 63–68. [Google Scholar] [CrossRef]
- Brown, E.A.; Lautz, J.D.; Davis, T.R.; Gniffke, E.P.; VanSchoiack, A.A.W.; Neier, S.C.; Tashbook, N.; Nicolini, C.; Fahnestock, M.; Schrum, A.G.; et al. Clustering the Autisms Using Glutamate Synapse Protein Interaction Networks from Cortical and Hippocampal Tissue of Seven Mouse Models. Mol. Autism 2018, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.D.; Iyer, A.M.; van Scheppingen, J.; Bongaarts, A.; Anink, J.J.; Janssen, B.; Zimmer, T.S.; Spliet, W.G.; van Rijen, P.C.; Jansen, F.E.; et al. Coding and Small Non-Coding Transcriptional Landscape of Tuberous Sclerosis Complex Cortical Tubers: Implications for Pathophysiology and Treatment. Sci. Rep. 2017, 7, 8089. [Google Scholar] [CrossRef]
- Okamoto, S.; Prikhodko, O.; Pina-Crespo, J.; Adame, A.; McKercher, S.R.; Brill, L.M.; Nakanishi, N.; Oh, C.; Nakamura, T.; Masliah, E.; et al. NitroSynapsin for the Treatment of Neurological Manifestations of Tuberous Sclerosis Complex in a Rodent Model. Neurobiol. Dis. 2019, 127, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Schaeffer, S.M.; Dhamne, S.C.; Lipton, J.O.; Lindemann, L.; Honer, M.; Jaeschke, G.; Super, C.E.; Lammers, S.H.; Modi, M.E.; et al. MGluR5 Modulation of Behavioral and Epileptic Phenotypes in a Mouse Model of Tuberous Sclerosis Complex. Neuropsychopharmacology 2018, 43, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Potter, W.B.; Basu, T.; O’Riordan, K.J.; Kirchner, A.; Rutecki, P.; Burger, C.; Roopra, A. Reduced Juvenile Long-Term Depression in Tuberous Sclerosis Complex Is Mitigated in Adults by Compensatory Recruitment of MGluR5 and Erk Signaling. PLoS Biol. 2013, 11, e1001627. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.E.; Ratai, E.-M.; Kanwisher, N. Reduced GABAergic Action in the Autistic Brain. Curr. Biol. 2016, 26, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Halt, A.R.; Stary, J.M.; Kanodia, R.; Schulz, S.C.; Realmuto, G.R. Glutamic Acid Decarboxylase 65 and 67 KDa Proteins Are Reduced in Autistic Parietal and Cerebellar Cortices. Biol. Psychiatry 2002, 52, 805–810. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Reutiman, T.J.; Folsom, T.D.; Thuras, P.D. GABAA Receptor Downregulation in Brains of Subjects with Autism. J. Autism Dev. Disord. 2009, 39, 223–230. [Google Scholar] [CrossRef]
- Talos, D.M.; Sun, H.; Kosaras, B.; Joseph, A.; Folkerth, R.D.; Poduri, A.; Madsen, J.R.; Black, P.M.; Jensen, F.E. Altered Inhibition in Tuberous Sclerosis and Type IIb Cortical Dysplasia. Ann. Neurol. 2012, 71, 539–551. [Google Scholar] [CrossRef]
- Brooks-Kayal, A.R.; Shumate, M.D.; Jin, H.; Rikhter, T.Y.; Coulter, D.A. Selective Changes in Single Cell GABAA Receptor Subunit Expression and Function in Temporal Lobe Epilepsy. Nat. Med. 1998, 4, 1166–1172. [Google Scholar] [CrossRef]
- Raol, Y.H.; Lund, I.V.; Bandyopadhyay, S.; Zhang, G.; Roberts, D.S.; Wolfe, J.H.; Russek, S.J.; Brooks-Kayal, A.R. Enhancing GABAA Receptor 1 Subunit Levels in Hippocampal Dentate Gyrus Inhibits Epilepsy Development in an Animal Model of Temporal Lobe Epilepsy. J. Neurosci. 2006, 26, 11342–11346. [Google Scholar] [CrossRef]
- Cepeda, C.; Levinson, S.; Nariai, H.; Yazon, V.-W.; Tran, C.; Barry, J.; Oikonomou, K.D.; Vinters, H.V.; Fallah, A.; Mathern, G.W.; et al. Pathological High Frequency Oscillations Associate with Increased GABA Synaptic Activity in Pediatric Epilepsy Surgery Patients. Neurobiol. Dis. 2020, 134, 104618. [Google Scholar] [CrossRef]
- Kirischuk, S.; Sinning, A.; Blanquie, O.; Yang, J.-W.; Luhmann, H.J.; Kilb, W. Modulation of Neocortical Development by Early Neuronal Activity: Physiology and Pathophysiology. Front. Cell. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Cherubini, E.; Corradetti, R.; Gaiarsa, J.L. Giant Synaptic Potentials in Immature Rat CA3 Hippocampal Neurones. J. Physiol. 1989, 416, 303–325. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Pai, E.L.-L.; Rubin, A.N.; Stafford, A.M.; Angara, K.; Minasi, P.; Rubenstein, J.L.; Sohal, V.S.; Vogt, D. Tsc1 Represses Parvalbumin Expression and Fast-Spiking Properties in Somatostatin Lineage Cortical Interneurons. Nat. Commun. 2019, 10, 4994. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L.R.; Merzenich, M.M. Model of Autism: Increased Ratio of Excitation/Inhibition in Key Neural Systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef]
- Weston, M.C.; Chen, H.; Swann, J.W. Loss of MTOR Repressors Tsc1 or Pten Has Divergent Effects on Excitatory and Inhibitory Synaptic Transmission in Single Hippocampal Neuron Cultures. Front. Mol. Neurosci. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Bateup, H.S.; Takasaki, K.T.; Saulnier, J.L.; Denefrio, C.L.; Sabatini, B.L. Loss of Tsc1 In Vivo Impairs Hippocampal MGluR-LTD and Increases Excitatory Synaptic Function. J. Neurosci. 2011, 31, 8862–8869. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.L.; Calderon de Anda, F.; Mangoubi, T.; Yoshii, A. Multiple Critical Periods for Rapamycin Treatment to Correct Structural Defects in Tsc-1-Suppressed Brain. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef]
- Magri, L.; Cominelli, M.; Cambiaghi, M.; Cursi, M.; Leocani, L.; Minicucci, F.; Poliani, P.L.; Galli, R. Timing of MTOR Activation Affects Tuberous Sclerosis Complex Neuropathology in Mouse Models. Dis. Models Mech. 2013, 6, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, T.S.; Broekaart, D.W.M.; Gruber, V.-E.; van Vliet, E.A.; Mühlebner, A.; Aronica, E. Tuberous Sclerosis Complex as Disease Model for Investigating MTOR-Related Gliopathy During Epileptogenesis. Front. Neurol. 2020, 11, 1028. [Google Scholar] [CrossRef]
- Bitzenhofer, S.H.; Pöpplau, J.A.; Chini, M.; Marquardt, A.; Hanganu-Opatz, I.L. A Transient Developmental Increase in Prefrontal Activity Alters Network Maturation and Causes Cognitive Dysfunction in Adult Mice. Neuron 2021, 109, 1350–1364.e6. [Google Scholar] [CrossRef]
- Amin, S.; Lux, A.; Calder, N.; Laugharne, M.; Osborne, J.; O’callaghan, F. Causes of Mortality in Individuals with Tuberous Sclerosis Complex. Dev. Med. Child. Neurology 2017, 59, 612–617. [Google Scholar] [CrossRef]
- Canevini, M.P.; Kotulska-Jozwiak, K.; Curatolo, P.; La Briola, F.; Peron, A.; Słowińska, M.; Strzelecka, J.; Vignoli, A.; Jóźwiak, S. Current Concepts on Epilepsy Management in Tuberous Sclerosis Complex. Am. J. Med. Genet. 2018, 178, 299–308. [Google Scholar] [CrossRef]
- Jozwiak, S.; Kotulska, K.; Wong, M.; Bebin, M. Modifying Genetic Epilepsies - Results from Studies on Tuberous Sclerosis Complex. Neuropharmacology 2020, 166, 107908. [Google Scholar] [CrossRef]
- Strzelczyk, A.; Grau, J.; Bast, T.; Bertsche, A.; Bettendorf, U.; Hahn, A.; Hartmann, H.; Hertzberg, C.; Hornemann, F.; Immisch, I.; et al. Prescription Patterns of Antiseizure Drugs in Tuberous Sclerosis Complex (TSC)-Associated Epilepsy: A Multicenter Cohort Study from Germany and Review of the Literature. Expert Rev. Clin. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Jozwiak, S.; Słowińska, M.; Borkowska, J.; Sadowski, K.; Łojszczyk, B.; Domańska-Pakieła, D.; Chmielewski, D.; Kaczorowska-Frontczak, M.; Głowacka, J.; Sijko, K.; et al. Preventive Antiepileptic Treatment in Tuberous Sclerosis Complex: A Long-Term, Prospective Trial. Pediatric Neurol. 2019, 101, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; McDaniel, S.S.; Rensing, N.R.; Wong, M. Vigabatrin Inhibits Seizures and MTOR Pathway Activation in a Mouse Model of Tuberous Sclerosis Complex. PLoS ONE 2013, 8, e57445. [Google Scholar] [CrossRef]
- Kotulska, K.; Kwiatkowski, D.J.; Curatolo, P.; Weschke, B.; Riney, K.; Jansen, F.; Feucht, M.; Krsek, P.; Nabbout, R.; Jansen, A.C.; et al. Prevention of Epilepsy in Infants with Tuberous Sclerosis Complex in the EPISTOP Trial. Ann. Neurol. 2021, 89, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 2017, 40, 536–554. [Google Scholar] [CrossRef]
- Fernell, E.; Gustafsson, P.; Gillberg, C. Bumetanide for Autism: Open-Label Trial in Six Children. Acta Paediatr. 2021, 110, 1548–1553. [Google Scholar] [CrossRef]
- van Andel, D.M.; Sprengers, J.J.; Oranje, B.; Scheepers, F.E.; Jansen, F.E.; Bruining, H. Effects of Bumetanide on Neurodevelopmental Impairments in Patients with Tuberous Sclerosis Complex: An Open-Label Pilot Study. Mol. Autism 2020, 11, 30. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Delpire, E. Phenobarbital, Midazolam, Bumetanide, and Neonatal Seizures: The Devil Is in the Details. Epilepsia 2021, 62, 935–940. [Google Scholar] [CrossRef]
- Moavero, R.; Mühlebner, A.; Luinenburg, M.J.; Craiu, D.; Aronica, E.; Curatolo, P. Genetic Pathogenesis of the Epileptogenic Lesions in Tuberous Sclerosis Complex: Therapeutic Targeting of the MTOR Pathway. Epilepsy Behav. 2021, 107713. [Google Scholar] [CrossRef]
- Zeng, L.-H.; Xu, L.; Gutmann, D.H.; Wong, M. Rapamycin Prevents Epilepsy in a Mouse Model of Tuberous Sclerosis Complex. Ann. Neurol. 2008, 63, 444–453. [Google Scholar] [CrossRef]
- French, J.A.; Lawson, J.A.; Yapici, Z.; Ikeda, H.; Polster, T.; Nabbout, R.; Curatolo, P.; de Vries, P.J.; Dlugos, D.J.; Berkowitz, N.; et al. Adjunctive Everolimus Therapy for Treatment-Resistant Focal-Onset Seizures Associated with Tuberous Sclerosis (EXIST-3): A Phase 3, Randomised, Double-Blind, Placebo-Controlled Study. Lancet 2016, 388, 2153–2163. [Google Scholar] [CrossRef]
- Stockinger, J.; Strzelczyk, A.; Nemecek, A.; Cicanic, M.; Bösebeck, F.; Brandt, C.; Hamer, H.; Intravooth, T.; Steinhoff, B.J. Everolimus in Adult Tuberous Sclerosis Complex Patients with Epilepsy: Too Late for Success? A Retrospective Study. Epilepsia 2021, 62, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Saffari, A.; Brösse, I.; Wiemer-Kruel, A.; Wilken, B.; Kreuzaler, P.; Hahn, A.; Bernhard, M.K.; van Tilburg, C.M.; Hoffmann, G.F.; Gorenflo, M.; et al. Safety and Efficacy of MTOR Inhibitor Treatment in Patients with Tuberous Sclerosis Complex under 2 Years of Age – a Multicenter Retrospective Study. Orphanet J. Rare Dis. 2019, 14, 96. [Google Scholar] [CrossRef]
- Overwater, I.E.; Rietman, A.B.; Mous, S.E.; Bindels-de Heus, K.; Rizopoulos, D.; ten Hoopen, L.W.; van der Vaart, T.; Jansen, F.E.; Elgersma, Y.; Moll, H.A.; et al. A Randomized Controlled Trial with Everolimus for IQ and Autism in Tuberous Sclerosis Complex. Neurology 2019, 93, e200–e209. [Google Scholar] [CrossRef] [PubMed]
- Fohlen, M.; Taussig, D.; Ferrand-Sorbets, S.; Chipaux, M.; Dorison, N.; Delalande, O.; Dorfmüller, G. Refractory Epilepsy in Preschool Children with Tuberous Sclerosis Complex: Early Surgical Treatment and Outcome. Seizure 2018, 60, 71–79. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
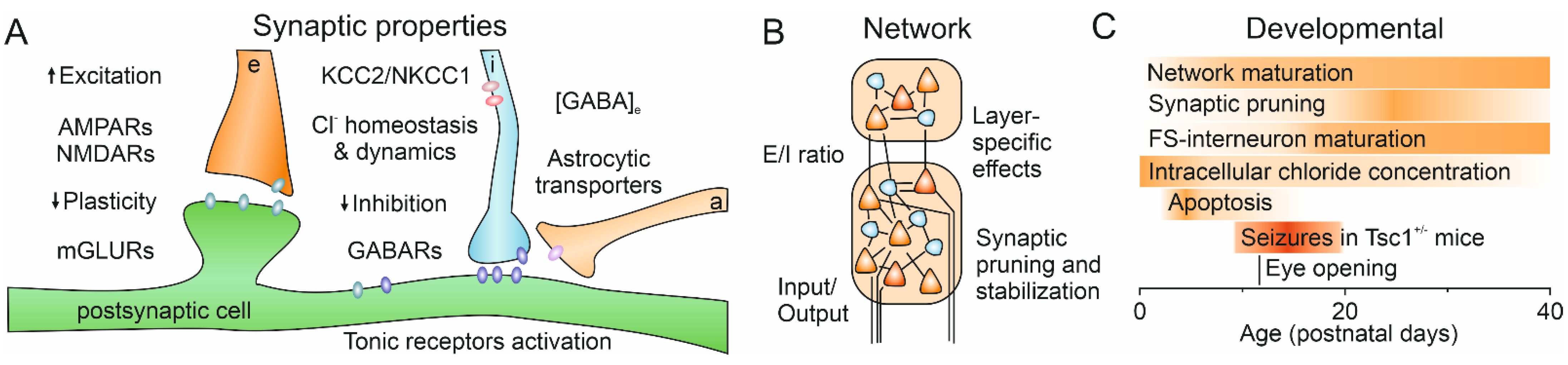{kind=link}
| Model/Specificity | Morphology and Behavior | Synaptic Transmission | Lethality |
|---|---|---|---|
| Tsc1+/− | No major defects Cognitive/behavioral deficits | Transient spontaneous seizures (P9–P18) Altered synaptic transmission | |
| Tsc1c/−; SynI-CKO Neurons [48] | Morphological alterations Myelination deficits | Seizures | Early death (median survival 35 days) |
| Tsc1c/c; Dlx5/6-CKO Interneurons [49] | Morphological alterations Cellular clusters in superficial layers | Reduced seizure threshold | Early death (40% by P30, 60% by P130) |
| Tsc1c/+; Nkx2.1-CKO MGE-derived interneurons [44] | Cognitive deficits | Decreased inhibition on hippocampal pyramidal cells | |
| Tsc1c/c; Nestin-CKO (rtTA+ TetOp-Cre+) Neural progenitors [50] | Increased brain size Giant cells in hippocampus and cortex | Spontaneous seizures (around 3rd week) | Early death |
| Tsc1c/c; Emx1-CKO Dorsal neural progenitors [51] | Increased brain size and cortical thickness Lamination deficits Subependymal nodules Astrocytosis Myelination deficits | Spontaneous seizures (100% of mutants at P13) | Early death (median survival 18 days) |
| Tsc1c/+ and Tsc1c/c; L7 CKO Purkinje cells (PC) [52] | Decreased PC number Behavioral deficits | Increased spine density and decreased excitability in PC | |
| Tsc1c/c; Gbx2 CKO (CreER) Thalamus Tamoxifen at E12 or E18 [53] | Morphological alterations (stronger for earlier deletions), Behavioral deficits | Spontaneous seizures Altered active properties of thalamic neurons for early deletions | |
| Tsc2+/− | No major morphological defects Cognitive/behavioral deficits | Altered synaptic transmission and plasticity | |
| Tsc2c/−; HGFAP CKO Radial glia progenitors [54] | Increased brain size and cortical thickness Lamination deficits | Possible seizures | Early death (by 4th week) |
| Tsc2c/c; Emx1 CKO Dorsal neural progenitors [55] | Spontaneous seizures | Early death (by 3rd week) | |
| Tsc2c/c; Nex CKO Postmitotic excitatory forebrain neurons [56] | Alterations in neurons and glia Minor lamination defects in hippocampus and deep layers Astrogliosis | Early death (0% survival P22) | |
| Tsc2c/c; Olig2 CKO Oligodendrocyte precursor cells [57] | Hypomyelination Astrogliosis | No seizures | |
| Tsc1 GFAP1 CKO and Tsc2 GFAP1 CKO GFAP-positive cells [58] | Increased brain size Astrocytic proliferation Structural alterations of the hippocampus | Spontaneous seizures (onset: 4 wks for Tsc1 and 3 wks for Tsc2) Decreased glutamate transporter expression | Early death (Tsc1: 50% survival at 9 wks, 0% at 18 wks; Tsc2: 50% survival at 7 wks, 0% at 10 wks) |
| Tsc1fl/−, Tsc2fl/− and Tsc1fl/−;Tsc2fl/−; FVB-Tg(GFAP-cre)25Mes/J CKO Radial glia [59] | Macrocephaly Lamination deficits Astrogliosis Myelination and oligodendrocyte alterations | Possible seizures | Early death (50% death at around P20–23) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bassetti, D.; Luhmann, H.J.; Kirischuk, S. Effects of Mutations in TSC Genes on Neurodevelopment and Synaptic Transmission. Int. J. Mol. Sci. 2021, 22, 7273. https://doi.org/10.3390/ijms22147273
Bassetti D, Luhmann HJ, Kirischuk S. Effects of Mutations in TSC Genes on Neurodevelopment and Synaptic Transmission. International Journal of Molecular Sciences. 2021; 22(14):7273. https://doi.org/10.3390/ijms22147273
Chicago/Turabian StyleBassetti, Davide, Heiko J. Luhmann, and Sergei Kirischuk. 2021. "Effects of Mutations in TSC Genes on Neurodevelopment and Synaptic Transmission" International Journal of Molecular Sciences 22, no. 14: 7273. https://doi.org/10.3390/ijms22147273
APA StyleBassetti, D., Luhmann, H. J., & Kirischuk, S. (2021). Effects of Mutations in TSC Genes on Neurodevelopment and Synaptic Transmission. International Journal of Molecular Sciences, 22(14), 7273. https://doi.org/10.3390/ijms22147273