
The Enigmatic Role of TP53 in Germ Cell Tumours: Are We Missing Something?

 , , , ,
, , , ,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Platinum Resistance and Sensitivity in Germ Cell Tumours
2.1. General Overview
2.2. The Role of TP53 and Its Pathway
3. Germ Cell Tumours and Second Cancers
3.1. General Overview
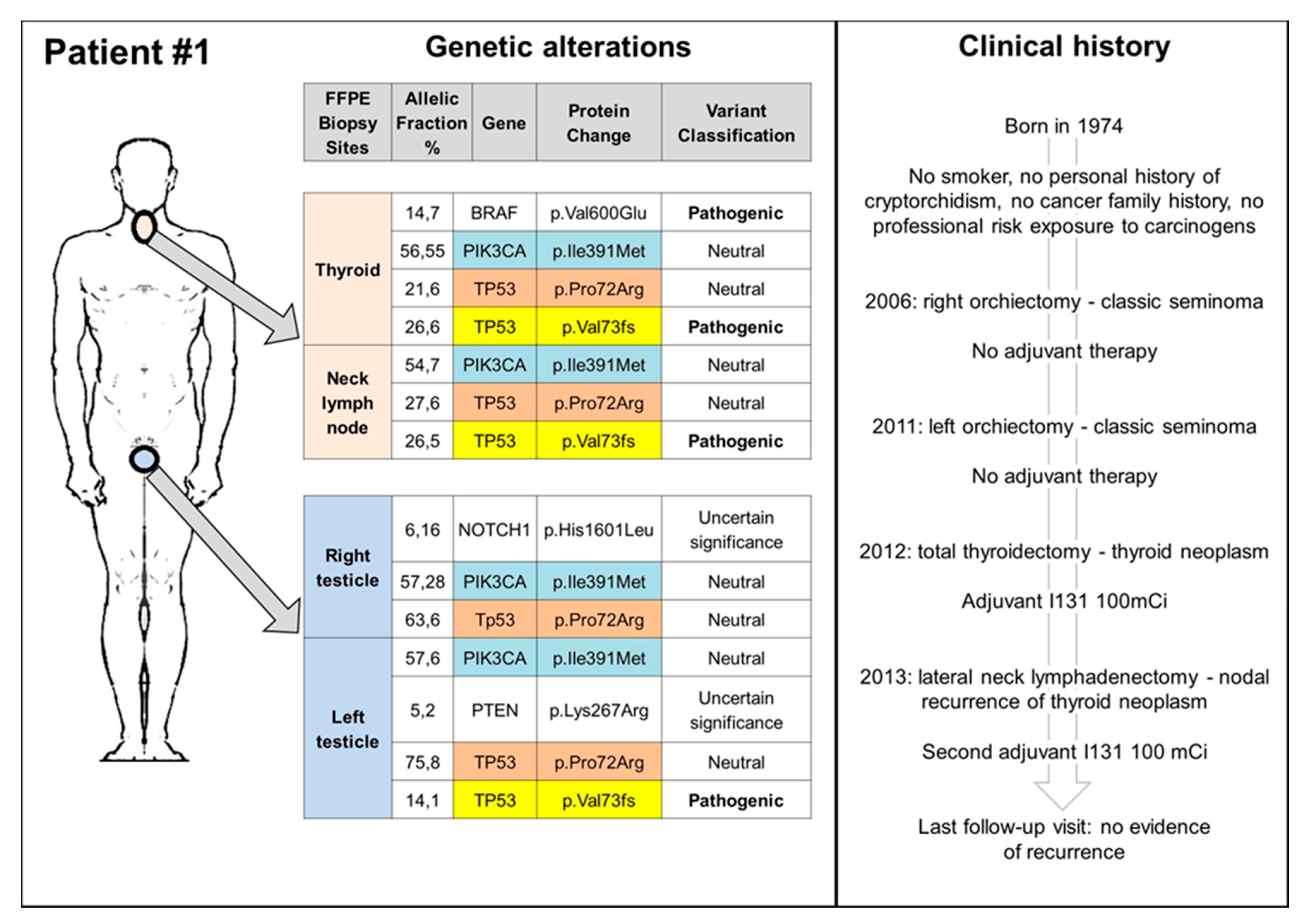
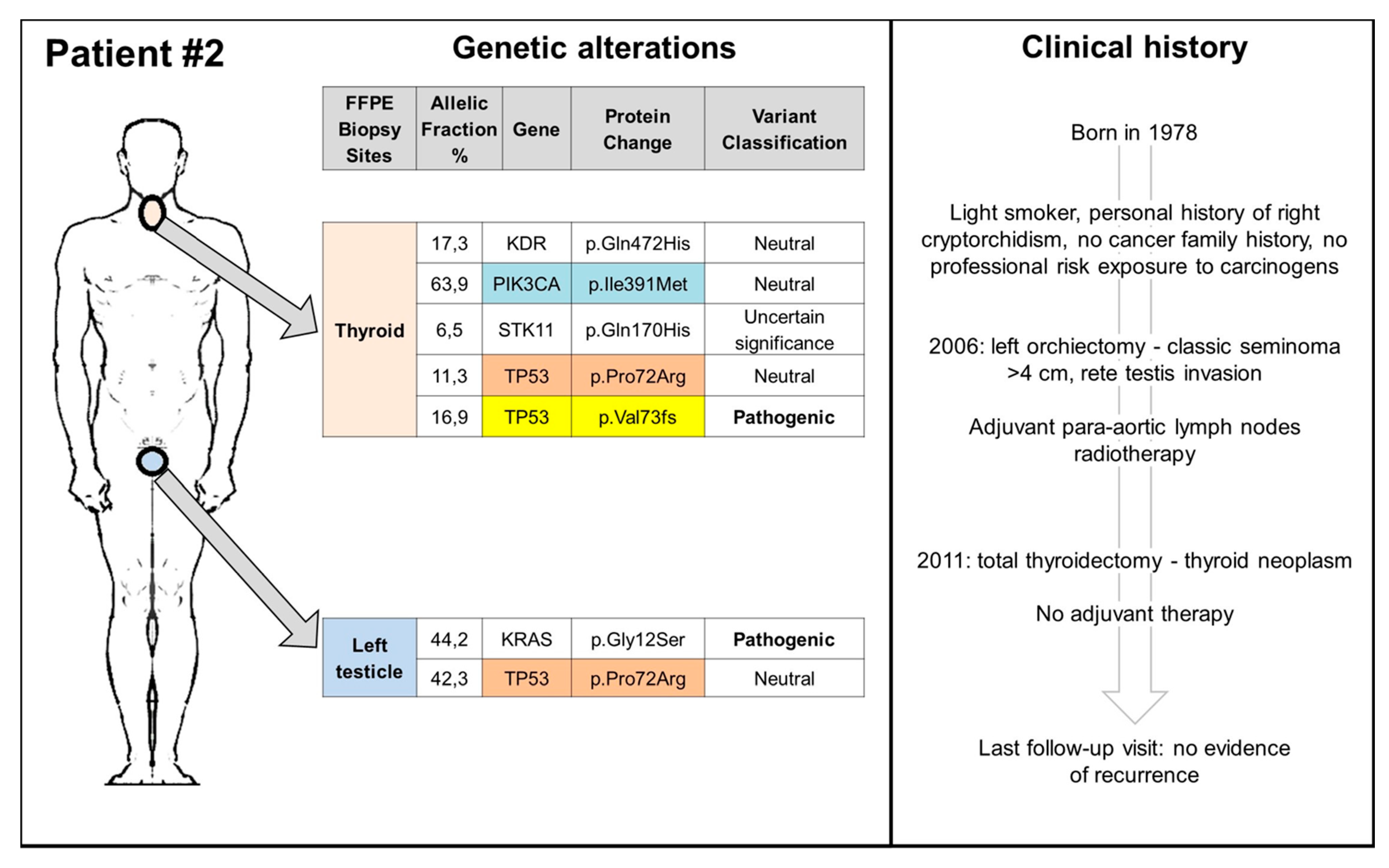
3.2. TGCT, Thyroid Cancers, and TP53: Experience from a Reference Center
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Albers, P.; Albrecht, W.; Algaba, F.; Bokemeyer, C.; Cohn-Cedermark, G.; Fizazi, K.; Horwich, A.; Laguna, M.P.; Nicolai, N.; Oldenburg, J. Guidelines on Testicular Cancer: 2015 Update. Eur. Urol. 2015, 68, 1054–1068. [Google Scholar] [CrossRef] [PubMed]
- Richardson, L.C.; Neri, A.J.; Tai, E.; Glenn, J.D. Testicular cancer: A narrative review of the role of socioeconomic position from risk to survivorship. Urol. Oncol. Semin. Orig. Investig. 2012, 30, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Ylönen, O.; Jyrkkiö, S.; Pukkala, E.; Syvänen, K.; Boström, P.J. Time trends and occupational variation in the incidence of testicular cancer in the Nordic countries. BJU Int. 2018, 122, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Rescigno, P.; Ottaviano, M.; Nappi, L.; Tortora, M.; De Placido, S.; Palmieri, G. Clinical features and psychological aspects of the decision-making process in stage I testicular germ cell tumors. Futur. Oncol. 2018, 14, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, R.; Coebergh, J.; Kiemeney, L.; Koldewijn, E.; Houterman, S. Testicular cancer: Trends in mortality are well explained by changes in treatment and survival in the southern Netherlands since 1970. Eur. J. Cancer 2007, 43, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- Richiardi, L.; Scélo, G.; Boffetta, P.; Hemminki, K.; Pukkala, E.; Olsen, J.H.; Weiderpass, E.; Tracey, E.; Brewster, D.; McBride, M.L.; et al. Second malignancies among survivors of germ-cell testicular cancer: A pooled analysis between 13 cancer registries. Int. J. Cancer 2007, 120, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Murty, V.V.; Chaganti, R.S. A genetic perspective of male germ cell tumors. Semin. Oncol. 1998, 25, 133–144. [Google Scholar]
- Adra, N.; Einhorn, L.H. Testicular cancer update. Clin. Adv. Hematol. Oncol. 2017, 15, 386–396. [Google Scholar]
- Weidner, N. Germ-cell tumors of the mediastinum. Semin. Diagn. Pathol. 1999, 16, 42–50. [Google Scholar]
- Bokemeyer, C.; Nichols, C.R.; Droz, J.-P.; Schmoll, H.-J.; Horwich, A.; Gerl, A.; Fossa, S.D.; Beyer, J.; Pont, J.; Kanz, L.; et al. Extragonadal Germ Cell Tumors of the Mediastinum and Retroperitoneum: Results from an International Analysis. J. Clin. Oncol. 2002, 20, 1864–1873. [Google Scholar] [CrossRef]
- Varmus, H.; Harlow, E. Science funding: Provocative questions in cancer research. Nature 2012, 481, 436–437. [Google Scholar] [CrossRef]
- Robinson, D.; Moller, H.; Horwich, A. Mortality and incidence of second cancers following treatment for testicular cancer. Br. J. Cancer 2007, 96, 529–533. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Banna, G.L.; Nicolai, N.; Palmieri, G.; Ottaviano, M.; Balzarini, L.; Barone, D.; Basso, U.; Bavila, A.; Bertoni, F.; Calliada, F.; et al. ☆Corrigendum to “Recommendations for surveillance and follow-up of men with testicular germ cell tumors: A multidisciplinary consensus conference by the Italian Germ cell cancer Group and the Associazione Italiana di Oncologia Medica”. Crit. Rev. Oncol. Hematol. 2020, 146, 102865. [Google Scholar] [CrossRef] [PubMed]
- Travis, L.B.; Beard, C.; Allan, J.; Dahl, A.A.; Feldman, D.; Oldenburg, J.; Daugaard, G.; Kelly, J.L.; Dolan, M.E.; Hannigan, R.; et al. Testicular Cancer Survivorship: Research Strategies and Recommendations. J. Natl. Cancer Inst. 2010, 102, 1114–1130. [Google Scholar] [CrossRef]
- Haugnes, H.S.; Bosl, G.; Boer, H.; Gietema, J.; Brydøy, M.; Oldenburg, J.; Dahl, A.A.; Bremnes, R.M.; Fosså, S.D. Long-Term and Late Effects of Germ Cell Testicular Cancer Treatment and Implications for Follow-Up. J. Clin. Oncol. 2012, 30, 3752–3763. [Google Scholar] [CrossRef]
- Curreri, S.A.; Fung, C.; Beard, C.J. Secondary malignant neoplasms in testicular cancer survivors. Urol. Oncol. Semin. Orig. Investig. 2015, 33, 392–398. [Google Scholar] [CrossRef]
- Nichols, C.R.; Nappi, L.; Kollmannsberger, C.; Hamilton, R.; Daneshmand, S. Back to the Future—Moving Forward for Testicular Cancer Survivors. JNCI Cancer Spectr. 2019, 4, pkz082. [Google Scholar] [CrossRef]
- Fung, C.; Fossa, S.D.; Milano, M.T.; Oldenburg, J.; Travis, L.B. Solid Tumors After Chemotherapy or Surgery for Testicular Nonseminoma: A Population-Based Study. J. Clin. Oncol. 2013, 31, 3807–3814. [Google Scholar] [CrossRef] [PubMed]
- Kier, M.G.; Hansen, M.K.; Lauritsen, J.; Mortensen, M.S.; Bandak, M.; Agerbaek, M.; Holm, N.V.; Dalton, S.O.; Andersen, K.K.; Johansen, C.; et al. Second malignant neoplasms and cause of death in patients with germ cell cancer: A Danish Nationwide Cohort Study. JAMA Oncol. 2016, 2, 1624–1627. [Google Scholar] [CrossRef]
- Groot, H.J.; Lubberts, S.; De Wit, R.; Witjes, J.A.; Kerst, J.M.; De Jong, I.J.; Groenewegen, G.; van den Eertwegh, A.J.; Poortmans, P.M.; Klümpen, H.-J.; et al. Risk of Solid Cancer After Treatment of Testicular Germ Cell Cancer in the Platinum Era. J. Clin. Oncol. 2018, 36, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- Hellesnes, R.; Kvammen, Ø.; Myklebust, T.Å.; Bremnes, R.M.; Karlsdottir, Á.; Negaard, H.F.S.; Tandstad, T.; Wilsgaard, T.; Fosså, S.D.; Haugnes, H.S. Continuing increased risk of second cancer in long-term testicular cancer survivors after treatment in the cisplatin era. Int. J. Cancer 2019, 147, 21–32. [Google Scholar] [CrossRef]
- Holzik, M.F.L.; Rapley, E.; Hoekstra, H.; Sleijfer, D.; Nolte, I.; Sijmons, R. Genetic predisposition to testicular germ-cell tumours. Lancet Oncol. 2004, 5, 363–371. [Google Scholar] [CrossRef]
- Hemminki, K.; Li, X. Familial risk in testicular cancer as a clue to a heritable and environmental aetiology. Br. J. Cancer 2004, 90, 1765–1770. [Google Scholar] [CrossRef]
- Heimdal, K.; Olsson, H.; Tretli, S.; Flodgren, P.; Børresen, A.-L.; Fossa, S.D. Familial testicular cancer in Norway and southern Sweden. Br. J. Cancer 1996, 73, 964–969. [Google Scholar] [CrossRef]
- Rescigno, P.; Ottaviano, M.; Palmieri, G. Platinum drug sensitivity and resistance in testicular germ cell tumors: Two sides of the same coin. Cancer Drug Resist. 2020. [Google Scholar] [CrossRef]
- Facchini, G.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Di Franco, R.; Iovane, G.; Grimaldi, G.; Piscitelli, R.; Muto, P.; Botti, G.; et al. Exploring the molecular aspects associated with testicular germ cell tumors: A review. Oncotarget 2017, 9, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Summersgill, B.; Yost, S.; Sultana, R.; LaBreche, K.; Dudakia, D.; Renwick, A.; Seal, S.; Al-Saadi, R.; Broderick, P.; et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat. Commun. 2015, 6, 5973. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef]
- Cutcutache, I.; Suzuki, Y.; Tan, I.; Ramgopal, S.; Zhang, S.; Ramnarayanan, K.; Gan, A.; Lee, H.H.; Tay, S.T.; Ooi, A.; et al. Exome-wide Sequencing Shows Low Mutation Rates and Identifies Novel Mutated Genes in Seminomas. Eur. Urol. 2015, 68, 77–83. [Google Scholar] [CrossRef]
- Leibeling, D.; Laspe, P.; Emmert, S. Nucleotide excision repair and cancer. J. Mol. Histol. 2006, 37, 225–238. [Google Scholar] [CrossRef]
- Köberle, B.; Masters, J.R.; Hartley, J.A.; Wood, R.D. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr. Biol. 1999, 9, 273–278. [Google Scholar] [CrossRef]
- Welsh, C.; Day, R.; McGurk, C.; Masters, J.R.; Wood, R.D.; Köberle, B. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int. J. Cancer 2004, 110, 352–361. [Google Scholar] [CrossRef]
- Cavallo, F.; Graziani, G.; Antinozzi, C.; Feldman, D.R.; Houldsworth, J.; Bosl, G.J.; Chaganti, R.S.K.; Moynahan, M.E.; Jasin, M.; Barchi, M. Reduced Proficiency in Homologous Recombination Underlies the High Sensitivity of Embryonal Carcinoma Testicular Germ Cell Tumors to Cisplatin and Poly (ADP-Ribose) Polymerase Inhibition. PLoS ONE 2012, 7, e51563. [Google Scholar] [CrossRef]
- Loveday, C.; Litchfield, K.; Proszek, P.Z.; Cornish, A.J.; Santo, F.; Levy, M.; MacIntyre, G.; Holryod, A.; Broderick, P.; Dudakia, D.; et al. Genomic landscape of platinum resistant and sensitive testicular cancers. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef]
- Albany, C.; Alva, A.S.; Aparicio, A.M.; Singal, R.; Yellapragada, S.; Sonpavde, G.; Hahn, N.M. Epigenetics in Prostate Cancer. Prostate Cancer 2011, 2011, 1–12. [Google Scholar] [CrossRef]
- Lind, G.E.; Skotheim, R.I.; Lothe, R.A. The epigenome of testicular germ cell tumors. APMIS 2007, 115, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.H.; Einhorn, L.H. Testicular cancer—Discoveries and updates. N. Engl. J. Med. 2014, 371, 2005–2016. [Google Scholar] [CrossRef]
- Cheung, H.H.; Lee, T.-L.; Davis, A.J.; Taft, D.H.; Rennert, O.M.; Chan, W.Y. Genome-wide DNA methylation profiling reveals novel epigenetically regulated genes and non-coding RNAs in human testicular cancer. Br. J. Cancer 2010, 102, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W.; et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 39, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Brait, M.; Maldonado, L.; Begum, S.; Loyo, M.; Wehle, D.; Tavora, F.; Looijenga, L.H.J.; Kowalski, J.; Zhang, Z.; Rosenbaum, E.; et al. DNA methylation profiles delineate epigenetic heterogeneity in seminoma and non-seminoma. Br. J. Cancer 2011, 106, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Haruta, M.; Arai, Y.; Honda, S.; Ohshima, J.; Sugawara, W.; Kageyama, Y.; Higashi, Y.; Nishida, K.; Tsunematsu, Y.; et al. Yolk sac tumor but not seminoma or teratoma is associated with abnormal epigenetic reprogramming pathway and shows frequent hypermethylation of various tumor suppressor genes. Cancer Sci. 2009, 100, 698–708. [Google Scholar] [CrossRef]
- Houldsworth, J.; Xiao, H.; Murty, V.; Chen, W.; Ray, B.; Reuter, V.E.; Bosl, G.J.; Chaganti, R. Human male germ cell tumor resistance to cisplatin is linked to TP53 gene mutation. Oncogene 1998, 16, 2345–2349. [Google Scholar] [CrossRef]
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients with Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Romano, F.J.; Rossetti, S.; Conteduca, V.; Schepisi, G.; Cavaliere, C.; Di Franco, R.; La Mantia, E.; Castaldo, L.; Nocerino, F.; Ametrano, G.; et al. Role of DNA repair machinery and p53 in the testicular germ cell cancer: A review. Oncotarget 2016, 7, 85641–85649. [Google Scholar] [CrossRef]
- Lafin, J.; Bagrodia, A.; Woldu, S.; Amatruda, J.F. New insights into germ cell tumor genomics. Andrology 2019, 7, 507–515. [Google Scholar] [CrossRef] [PubMed]
- International Germ Cell Cancer Collaborative Group. International Germ Cell Consensus Classification: A prognostic fac-tor-based staging system for metastatic germ cell cancers. J. Clin. Oncol. 1997, 15, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Koster, R.; Timmer-Bosscha, H.; Bischoff, R.; Gietema, J.; De Jong, S. Disruption of the MDM2–p53 interaction strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell Death Dis. 2011, 2, e148. [Google Scholar] [CrossRef]
- Zhu, J.; Dou, Z.; Sammons, M.A.; Levine, A.J.; Berger, S.L. Lysine methylation represses p53 activity in teratocarcinoma cancer cells. Proc. Natl. Acad. Sci. USA 2016, 113, 9822–9827. [Google Scholar] [CrossRef] [PubMed]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.-P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A Genetic Screen Implicates miRNA-372 and miRNA-373 As Oncogenes in Testicular Germ Cell Tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Duale, N.; Lindeman, B.; Komada, M.; Olsen, A.-K.; Andreassen, A.; Soderlund, E.J.; Brunborg, G. Molecular portrait of cis-platin induced response in human testis cancer cell lines based on gene expression profiles. Mol. Cancer 2007, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Port, M.; Glaesener, S.; Ruf, C.; Riecke, A.; Bokemeyer, C.; Meineke, V.; Honecker, F.; Abend, M. Micro-RNA expression in cisplatin resistant germ cell tumor cell lines. Mol. Cancer 2011, 10, 52. [Google Scholar] [CrossRef]
- Yi, J.; Luo, J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 1684–1689. [Google Scholar] [CrossRef]
- Spierings, D.; De Vries, E.; Vellenga, E.; De Jong, S. Loss of drug-induced activation of the CD95 apoptotic pathway in a cispla-tin-resistant testicular germ cell. Cell Death Differ. 2003, 10, 808–822. [Google Scholar]
- Mueller, T.; Voigt, W.; Simon, H.; Fruehauf, A.; Bulankin, A.; Grothey, A.; Schmoll, H.-J. Failure of activation of caspase-9 induces a higher threshold for apoptosis and cisplatin resistance in testicular cancer. Cancer Res. 2003, 63, 513–521. [Google Scholar]
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; van der Kuip, H. p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cis-platin. PLoS ONE 2011, 6, e19198. [Google Scholar] [CrossRef]
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; Van Der Kuip, H. Cisplatin Hypersensitivity of Testicular Germ Cell Tumors Is Determined by High Constitutive Noxa Levels Mediated by Oct-4. Cancer Res. 2013, 73, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Grande, L.; Bretones, G.; Rosa-Garrido, M.; Martin, E.M.G.; Hernandez, T.; Fraile, S.; Botella, L.; de Alava, E.; Vidal, A.; del Muro, X.G.; et al. Transcription Factors Sp1 and p73 Control the Expression of the Proapoptotic Protein NOXA in the Response of Testicular Embryonal Carcinoma Cells to Cisplatin. J. Biol. Chem. 2012, 287, 26495–26505. [Google Scholar] [CrossRef]
- Taylor-Weiner, A.; Zack, A.T.-W.T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.H.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nat. Cell Biol. 2016, 540, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, M.; Maddalena, C.; D’Armiento, M.; Lauria, R.; D’Alessandro, V.; Tortora, M.; Matano, E.; Di Lauro, V.; Mucci, B.; Ferraro, G.; et al. Systemic treatment of malignant gastrointestinal neuroectodermal tumour after childhood neuroblastoma: Chemotherapy in malignant gastrointestinal neuroectodermal tumour. Anti-Cancer Drugs 2019, 30, 959–963. [Google Scholar] [CrossRef]
- Belt-Dusebout, A.W.V.D.; De Wit, R.; Gietema, J.; Horenblas, S.; Louwman, M.W.J.; Ribot, J.G.; Hoekstra, H.J.; Ouwens, G.M.; Aleman, B.M.P.; Van Leeuwen, F.E. Treatment-Specific Risks of Second Malignancies and Cardiovascular Disease in 5-Year Survivors of Testicular Cancer. J. Clin. Oncol. 2007, 25, 4370–4378. [Google Scholar] [CrossRef]
- Kier, M.G.; Lauritsen, J.; Mortensen, M.S.; Bandak, M.; Andersen, K.K.; Hansen, M.K.; Agerbaek, M.; Holm, N.V.; Dalton, S.O.; Johansen, C.; et al. Prognostic Factors and Treatment Results after Bleomycin, Etoposide, and Cisplatin in Germ Cell Cancer: A Population-based Study. Eur. Urol. 2017, 71, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Zagars, G.K.; Babaian, R. Stage I testicular seminoma: Rationale for postorchiectomy radiation therapy. Int. J. Radiat. Oncol. 1987, 13, 155–162. [Google Scholar] [CrossRef]
- Horwich, A.; Fossa, S.D.; Huddart, R.; Dearnaley, D.P.; Stenning, S.; Aresu, M.; Bliss, J.; Hall, E. Second cancer risk and mortality in men treated with radiotherapy for stage I seminoma. Br. J. Cancer 2013, 110, 256–263. [Google Scholar] [CrossRef]
- Hemminki, K.; Liu, H.; Sundquist, J. Second cancers after testicular cancer diagnosed after 1980 in Sweden. Ann. Oncol. 2009, 21, 1546–1551. [Google Scholar] [CrossRef]
- Hauptmann, M.; Johannesen, T.B.; Gilbert, E.S.; Stovall, M.; Van Leeuwen, F.E.; Rajaraman, P.; Smith, S.A.; Weathers, R.E.; Aleman, B.M.P.; Andersson, M.; et al. Increased pancreatic cancer risk following radiotherapy for testicular cancer. Br. J. Cancer 2016, 115, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Dinh, P.; Fossa, S.D.; Travis, L.B. Testicular Cancer Survivorship. J. Natl. Compr. Cancer Netw. 2019, 17, 1557–1568. [Google Scholar] [CrossRef] [PubMed]
- Travis, L.B.; Andersson, M.; Gospodarowicz, M.; Van Leeuwen, F.E.; Bergfeldt, K.; Lynch, C.F.; Curtis, R.E.; Kohler, B.A.; Wiklund, T.; Storm, H.; et al. Treatment-associated leukemia following testicular cancer. J. Natl. Cancer Inst. 2000, 92, 1165–1171. [Google Scholar] [CrossRef]
- Kollmannsberger, C.; Kanz, L.; Bokemeyer, C. Therapy-related malignancies following treatment of germ cell cancer. Int. J. Cancer 1999, 83, 860–863. [Google Scholar] [CrossRef]
- Fung, C.; Bhatia, S.; Allan, J.M. Second cancers. In DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of On-cology, 11th ed.; DeVita, V.T., Rosenberg, S.A., Lawrence, T.S., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2017; pp. 2155–2173. [Google Scholar]
- Travis, L.B.; Fosså, S.D.; Schonfeld, S.J.; McMaster, M.L.; Lynch, C.F.; Storm, H.; Hall, P.; Holowaty, E.J.; Andersen, A.; Pukkala, E.; et al. Second Cancers among 40 576 Testicular Cancer Patients: Focus on Long-term Survivors. J. Natl. Cancer Inst. 2005, 97, 1354–1365. [Google Scholar] [CrossRef]
- Hauptmann, M.; Fossa, S.D.; Stovall, M.; E Van Leeuwen, F.; Johannesen, T.B.; Rajaraman, P.; Gilbert, E.S.; A Smith, S.; E Weathers, R.; Aleman, B.M.P.; et al. Increased stomach cancer risk following radiotherapy for testicular cancer. Br. J. Cancer 2014, 112, 44–51. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Schmoll, H.J. Secondary neoplasms following treatment of malignant germ cell tumors. J. Clin. Oncol. 1993, 11, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Wanderås, E.; Fosså, S.; Tretli, S. Risk of subsequent non-germ cell cancer after treatment of germ cell cancer in 2006 Norwegian male patients. Eur. J. Cancer 1997, 33, 253–262. [Google Scholar] [CrossRef]
- Spiliopoulou, P.; Bowers, S.P.; Gibson, S.; White, J.; Reed, N. Three cases of thyroid cancer following the diagnosis of testicular cancer: Treatment-related complication or genetics? Scott. Med. J. 2016, 61, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Brey, F.; Ferlay, J. Global cancer statistics 2018. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Sipos, J.; Mazzaferri, E. Thyroid Cancer Epidemiology and Prognostic Variables. Clin. Oncol. 2010, 22, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Cossu, A.; Budroni, M.; Paliogiannis, P.; Palmieri, G.; Scognamillo, F.; Cesaraccio, R.; Attene, F.; Trignano, M.; Tanda, F. Epidemiology of Thyroid Cancer in an Area of Epidemic Thyroid Goiter. J. Cancer Epidemiol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Wartofsky, L. Increasing world incidence of thyroid cancer: Increased detection or higher radiation exposure? Hormones 2010, 9, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.P. Thyroid carcinoma: Epidemiology, histology, and diagnosis. Clin. Adv. Hematol. Oncol. H&O 2015, 13, 3–6. [Google Scholar]
- Nosé, V. Familial thyroid cancer: A review. Mod. Pathol. 2011, 24, S19–S33. [Google Scholar] [CrossRef] [PubMed]
- Hińcza, K.; Kowalik, A.; Kowalska, A. Current Knowledge of Germline Genetic Risk Factors for the Development of Non-Medullary Thyroid Cancer. Genes 2019, 10, 482. [Google Scholar] [CrossRef]
- Guilmette, J.; Nosé, V. Hereditary and familial thyroid tumours. Histopathology 2017, 72, 70–81. [Google Scholar] [CrossRef]
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.-M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugières, L.; et al. Revisiting Li-Fraumeni Syndrome from TP53 Mutation Carriers. J. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef]
- Formiga, M.N.D.C.; De Andrade, K.C.; Kowalski, L.P.; Achatz, M.I. Frequency of Thyroid Carcinoma in Brazilian TP53 p.R337H Carriers with Li Fraumeni Syndrome. JAMA Oncol. 2017, 3, 1400–1402. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Wen-Fong, C.Y.; Nguyen, V.Q.; Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li Fraumeni Syndrome: Clinical Characteristics of Families With p53 Germline Mutations. J. Clin. Oncol. 2009, 27, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Kamihara, J.; Rana, H.Q.; Garber, J.E. GermlineTP53Mutations and the Changing Landscape of Li-Fraumeni Syndrome. Hum. Mutat. 2014, 35, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Li, F.P.; Fraumeni, J.F., Jr.; Mulvihill, J.J.; Blattner, W.A.; Dreyfus, M.G.; Tucker, M.A.; Miller, R.W. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988, 48, 5358–5362. [Google Scholar] [PubMed]
- Li, F.P.; Fraumeni, J.F., Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann. Intern. Med. 1969, 71, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.; Nelson, E.C.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- Srivastava, S.; Zou, Z.; Pirollo, K.F.; A Blattner, W.; Chang, E.H. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li–Fraumeni syndrome. Nat. Cell Biol. 1990, 348, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Tinat, J.; Bougeard, G.; Baert-Desurmont, S.; Vasseur, S.; Martin, C.; Bouvignies, E.; Caron, O.; Paillerets, B.B.-D.; Berthet, P.; Dugast, C.; et al. 2009 Version of the Chompret Criteria for Li Fraumeni Syndrome. J. Clin. Oncol. 2009, 27, e108–e109. [Google Scholar] [CrossRef]
- Gonzalez, K.D.; Buzin, C.H.; A Noltner, K.; Gu, D.; Li, W.; Malkin, D.; Sommer, S.S. High frequency of de novo mutations in Li-Fraumeni syndrome. J. Med. Genet. 2009, 46, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Zerdoumi, Y.; Lanos, R.; Raad, S.; Flaman, J.-M.; Bougeard, G.; Frebourg, T.; Tournier, I. Germline TP53 mutations result into a constitutive defect of p53 DNA binding and transcriptional response to DNA damage. Hum. Mol. Genet. 2017, 26, 2591–2602. [Google Scholar] [CrossRef] [PubMed]
- Travis, L.B.; Curtis, R.E.; Fraumeni, J.F.; Boice, J.D.; Storm, H.; Andersson, M.; Hall, P.; Bergfeldt, K.; Holowaty, E.; Clarke, E.A.; et al. Risk of Second Malignant Neoplasms Among Long-term Survivors of Testicular Cancer. J. Natl. Cancer Inst. 1997, 89, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Veiga, L.H.S.; Bhatti, P.; Ronckers, C.M.; Sigurdson, A.J.; Stovall, M.; Smith, S.A.; Weathers, R.; Leisenring, W.; Mertens, A.C.; Hammond, S.; et al. Chemotherapy and Thyroid Cancer Risk: A Report from the Childhood Cancer Survivor Study. Cancer Epidemiol. Biomark. Prev. 2012, 21, 92–101. [Google Scholar] [CrossRef]
- Toguchida, J.; Yamaguchi, T.; Dayton, S.H.; Beaughamp, R.L.; Herrera, G.E.; Ishizaki, K.; Yamamuro, T.; Meyers, P.A.; Little, J.B.; Sasaki, M.S.; et al. Prevalence and Spectrum of Germline Mutations of the p53 Gene among Patients with Sarcoma. N. Engl. J. Med. 1992, 326, 1301–1308. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottaviano, M.; Giunta, E.F.; Rescigno, P.; Pereira Mestre, R.; Marandino, L.; Tortora, M.; Riccio, V.; Parola, S.; Casula, M.; Paliogiannis, P.; et al. The Enigmatic Role of TP53 in Germ Cell Tumours: Are We Missing Something? Int. J. Mol. Sci. 2021, 22, 7160. https://doi.org/10.3390/ijms22137160
Ottaviano M, Giunta EF, Rescigno P, Pereira Mestre R, Marandino L, Tortora M, Riccio V, Parola S, Casula M, Paliogiannis P, et al. The Enigmatic Role of TP53 in Germ Cell Tumours: Are We Missing Something? International Journal of Molecular Sciences. 2021; 22(13):7160. https://doi.org/10.3390/ijms22137160
Chicago/Turabian StyleOttaviano, Margaret, Emilio Francesco Giunta, Pasquale Rescigno, Ricardo Pereira Mestre, Laura Marandino, Marianna Tortora, Vittorio Riccio, Sara Parola, Milena Casula, Panagiotis Paliogiannis, and et al. 2021. "The Enigmatic Role of TP53 in Germ Cell Tumours: Are We Missing Something?" International Journal of Molecular Sciences 22, no. 13: 7160. https://doi.org/10.3390/ijms22137160
APA StyleOttaviano, M., Giunta, E. F., Rescigno, P., Pereira Mestre, R., Marandino, L., Tortora, M., Riccio, V., Parola, S., Casula, M., Paliogiannis, P., Cossu, A., Vogl, U. M., Bosso, D., Rosanova, M., Mazzola, B., Daniele, B., Palmieri, G., & Palmieri, G. (2021). The Enigmatic Role of TP53 in Germ Cell Tumours: Are We Missing Something? International Journal of Molecular Sciences, 22(13), 7160. https://doi.org/10.3390/ijms22137160