
Structural Characterization of EnpA D,L-Endopeptidase from Enterococcus faecalis Prophage Provides Insights into Substrate Specificity of M23 Peptidases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. EnpACD Structure Determination
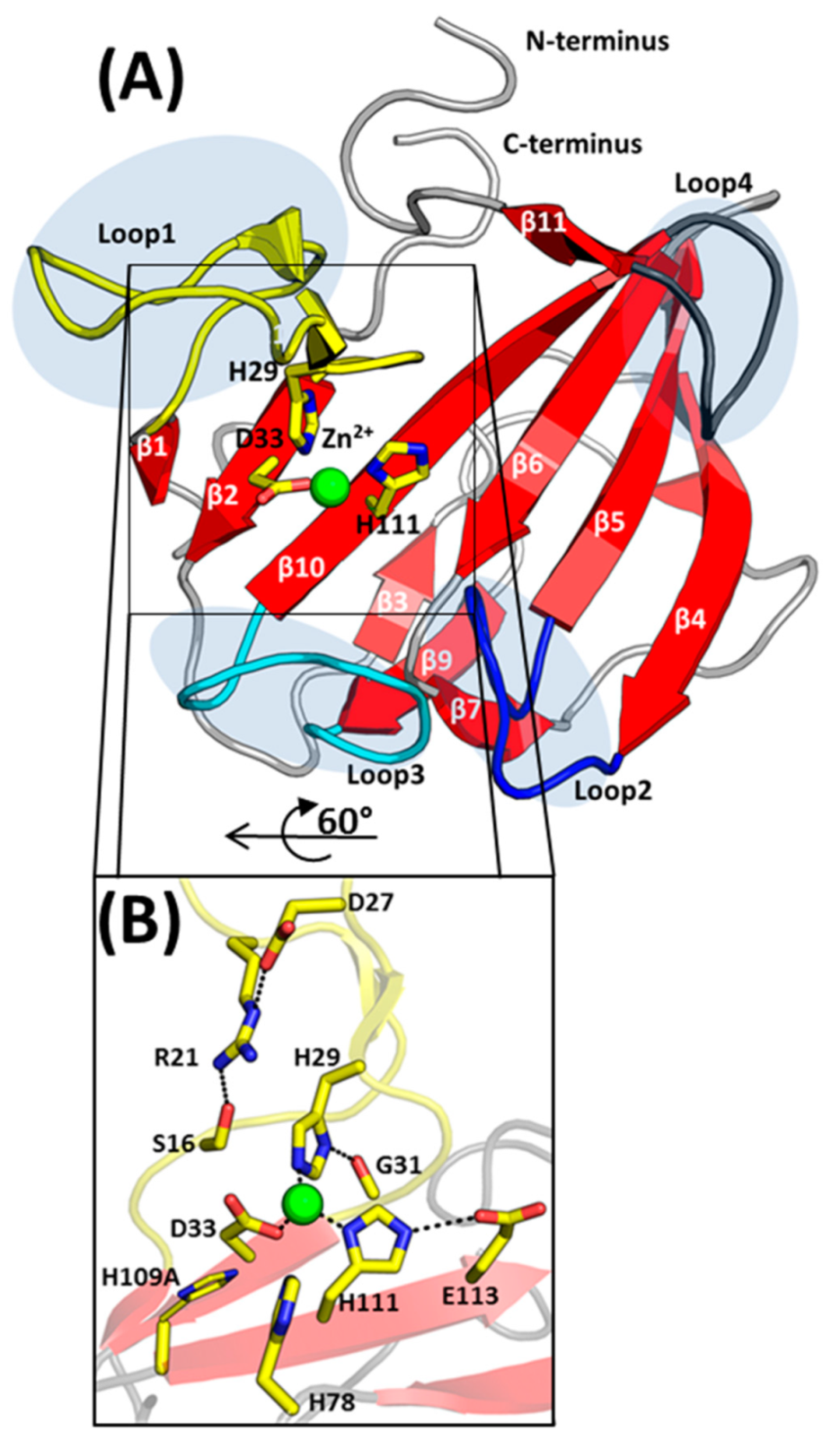
2.2. Overall Structure of EnpACD
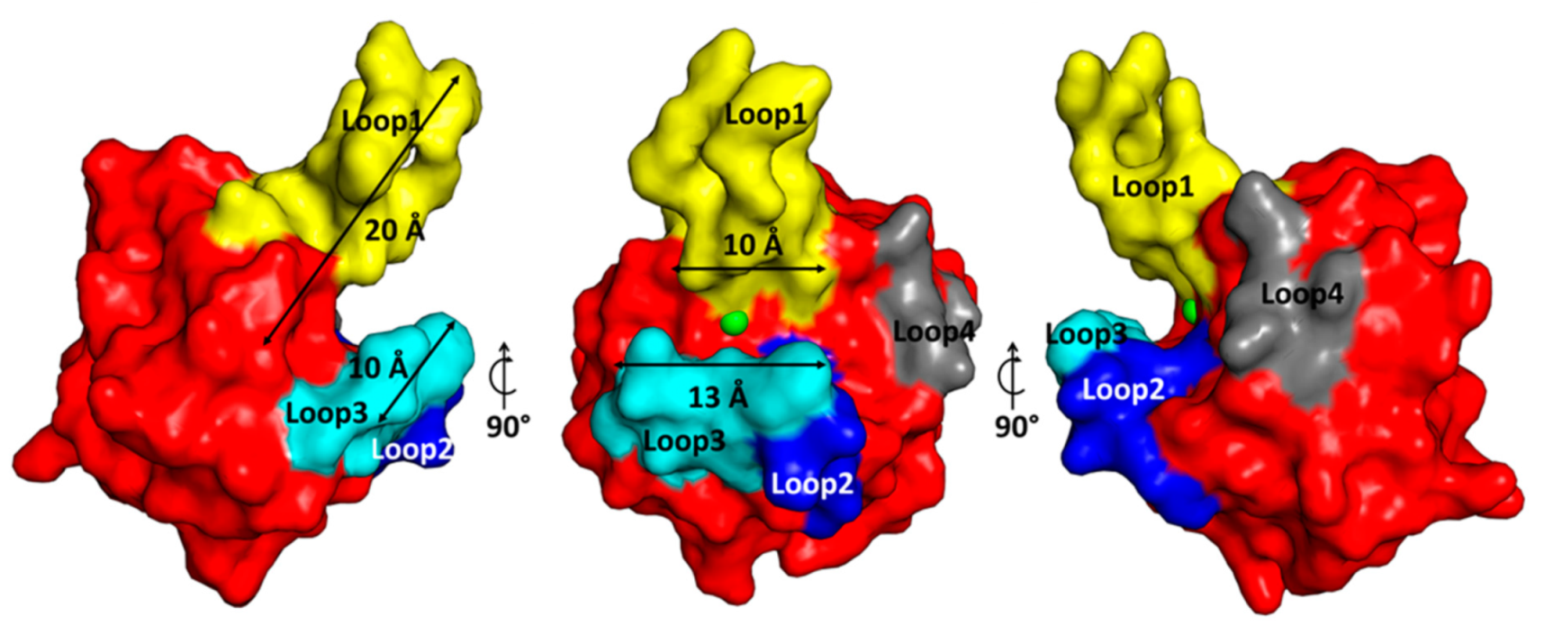
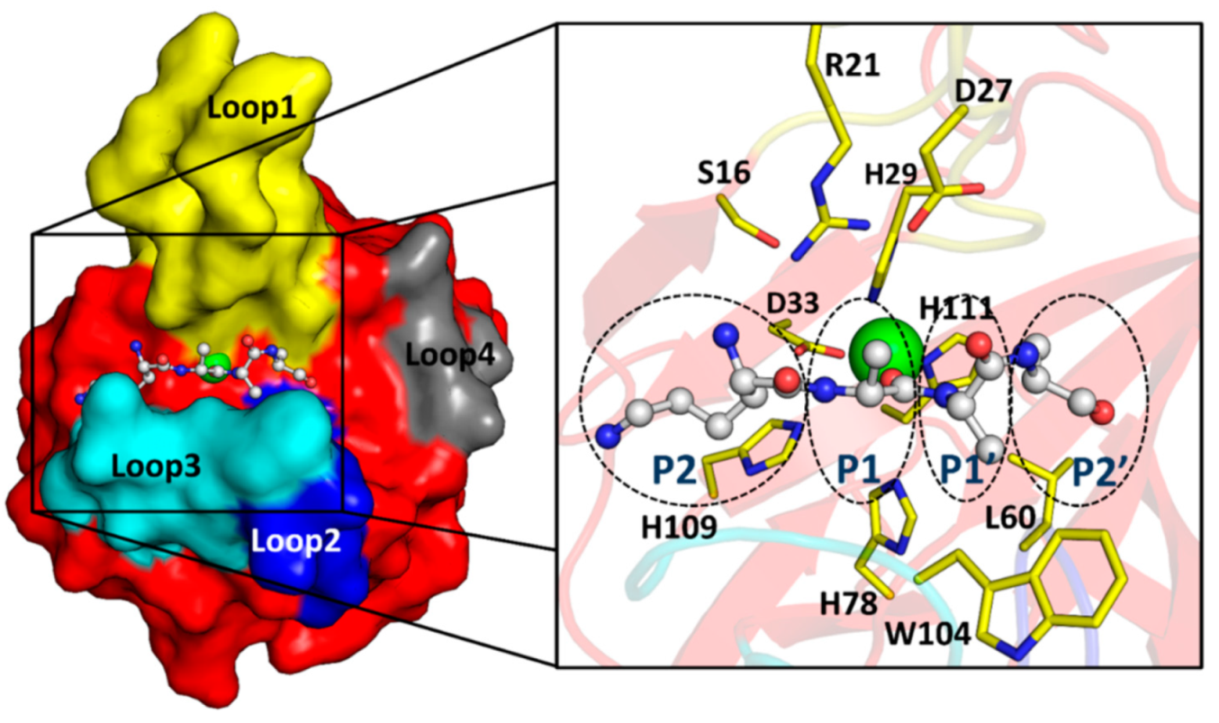
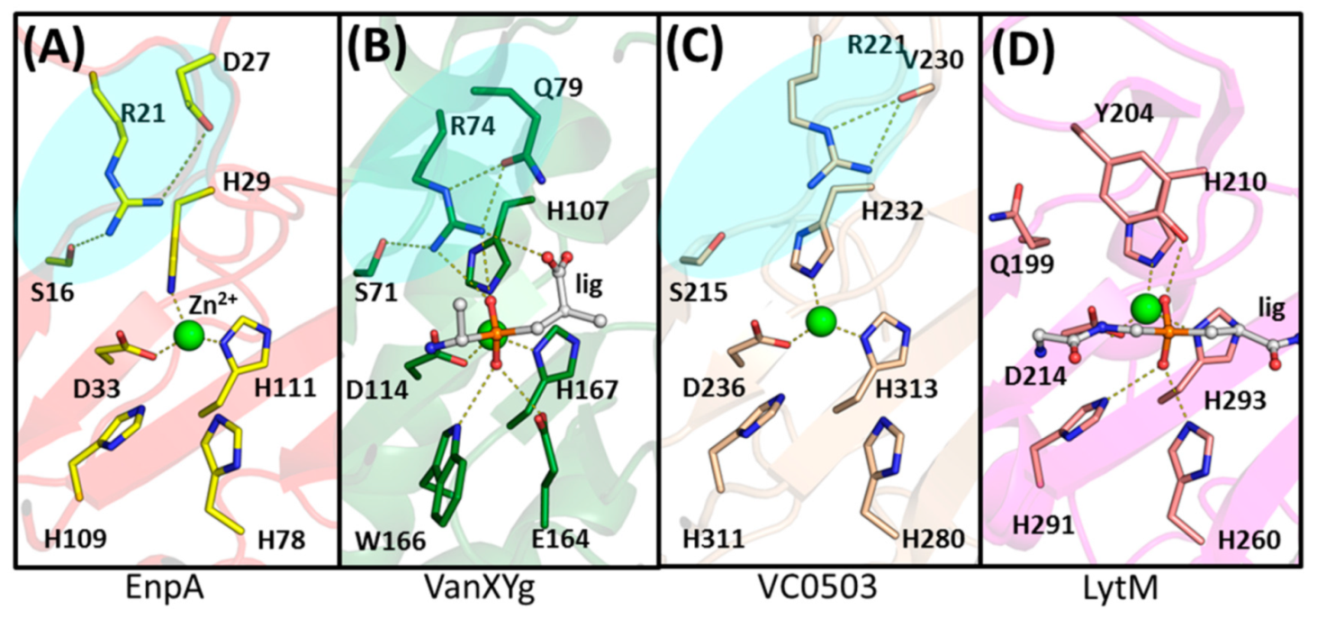
2.3. The Architecture of Active Site and Binding Groove
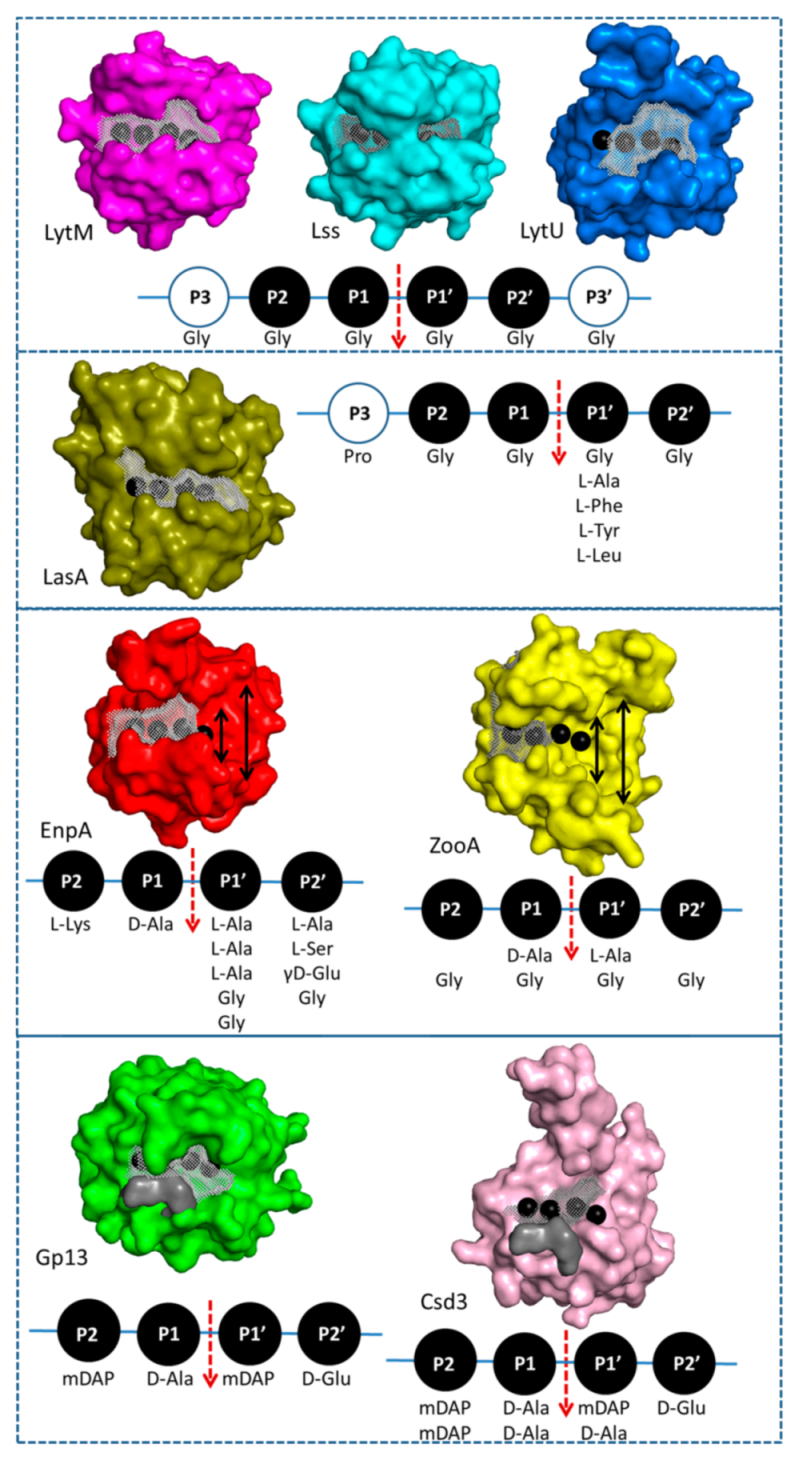
2.4. Structural Comparison with Other Members of M23 Family
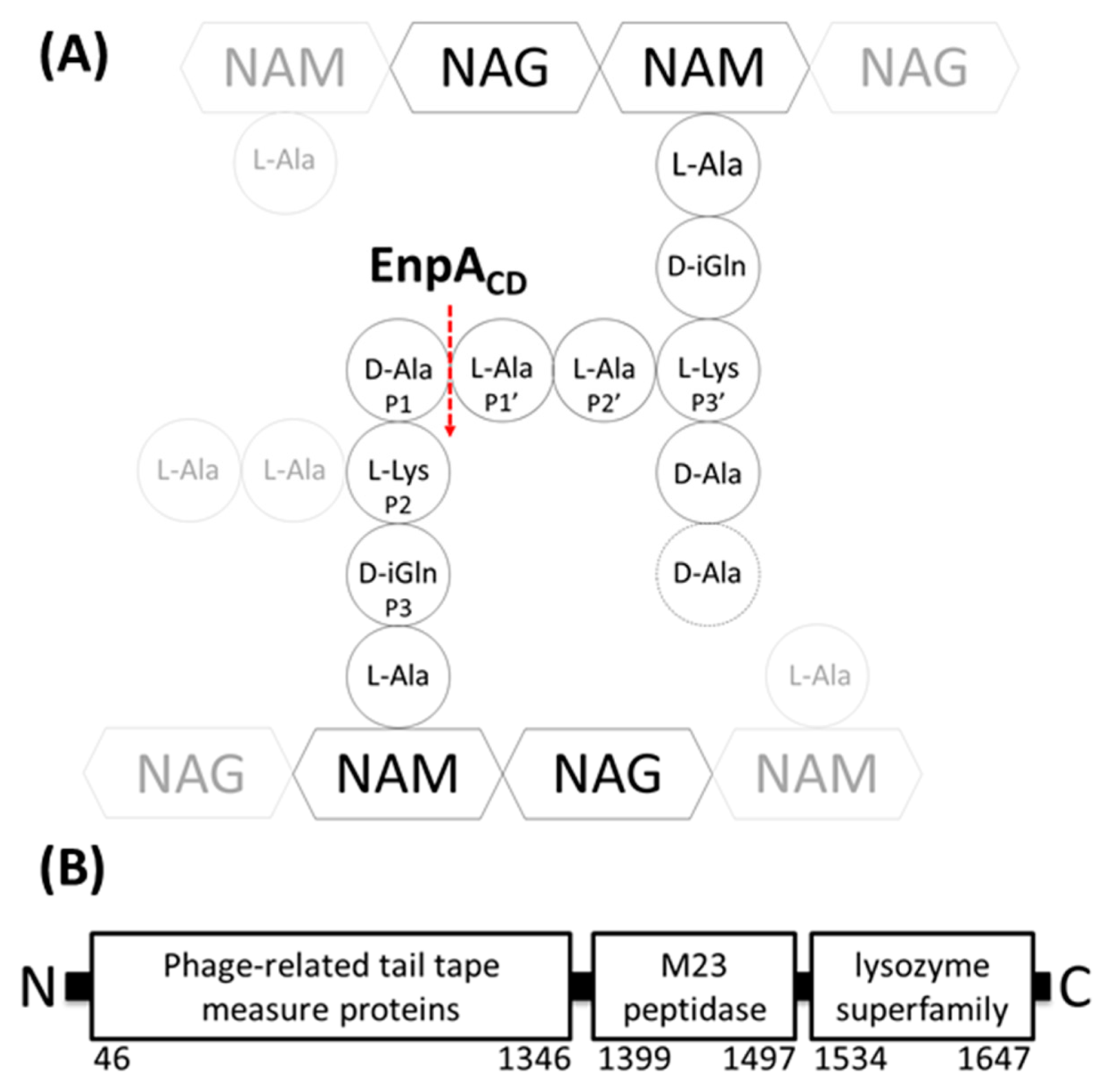
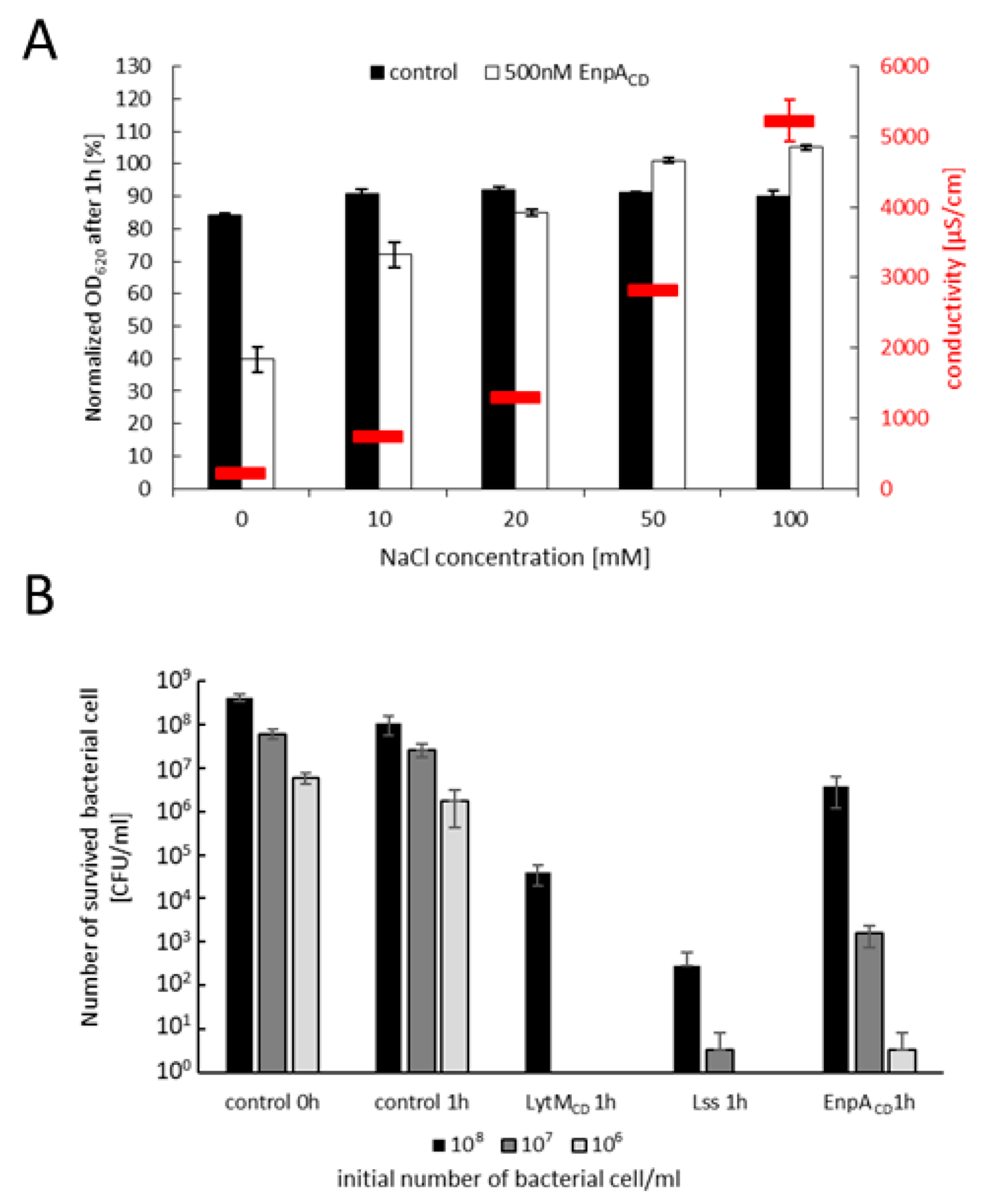
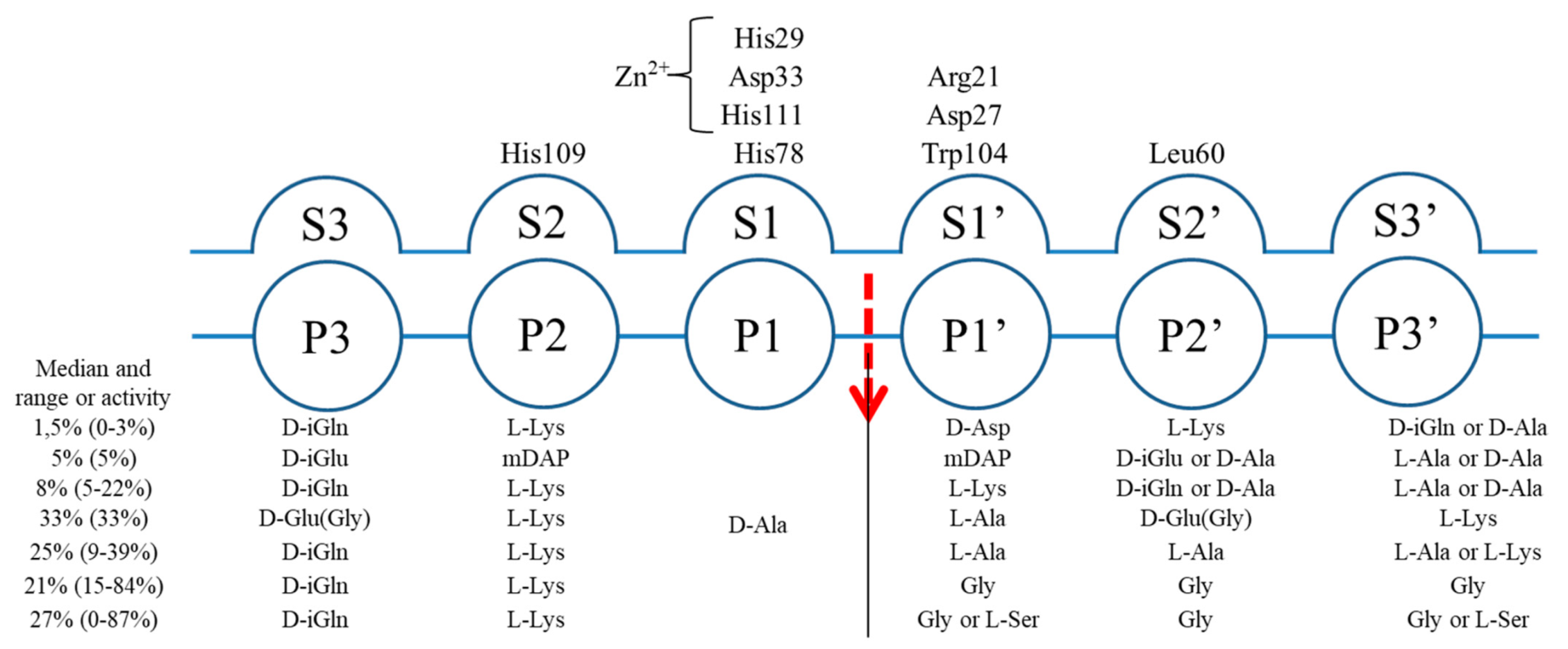
2.5. Specificity and Bacteriolytic Activity of EnpACD
2.6. Substrate Binding Model to EnpACD Reveals Possible Interactions
3. Discussion
3.1. EnpACD Displays a Potent Antibacterial Activity
3.2. General Architecture of the Binding Groove of M23 Reflects Enzyme Specificity
3.3. Arginine 21 Plays a Role in EnpA Activity
4. Materials and Methods
4.1. Gene Cloning
4.2. Mutagenesis
4.3. Protein Purification
4.4. EnpACD H109A Crystallization
4.5. Data Collection, Processing
4.6. Structure Determination and Refinement
4.7. Modelling of the EnpACD Substrate Complex
4.8. Enzymatic Activity Assays
4.9. FTIR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schleifer, K.H.; Kandler, O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 1972, 36, 407–477. [Google Scholar] [CrossRef]
- Vollmer, W.; Joris, B.; Charlier, P.; Foster, S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 2008, 32, 259–286. [Google Scholar] [CrossRef]
- Climo, M.W.; Patron, R.L.; Goldstein, B.P.; Archer, G.L. Lysostaphin treatment of experimental methicillin-resistant Staphylococcus aureus aortic valve endocarditis. Antimicrob. Agents Chemother. 1998, 42, 1355–1360. [Google Scholar] [CrossRef]
- Kokai-Kun, J.F.; Chanturiya, T.; Mond, J.J. Lysostaphin as a treatment for systemic Staphylococcus aureus infection in a mouse model. J. Antimicrob. Chemother. 2007, 60, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Kokai-Kun, J.F.; Chanturiya, T.; Mond, J.J. Lysostaphin eradicates established Staphylococcus aureus biofilms in jugular vein catheterized mice. J. Antimicrob. Chemother. 2009, 64, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef]
- Schmelcher, M.; Loessner, M.J. Bacteriophage endolysins: Applications for food safety. Curr. Opin. Biotechnol. 2016, 37, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.C.; Schmelcher, M.; Rodriguez-Rubio, L.; Klumpp, J.; Pritchard, D.G.; Dong, S.; Donovan, D.M. Endolysins as antimicrobials. Adv. Virus Res. 2012, 83, 299–365. [Google Scholar]
- Donovan, D.M. Bacteriophage and peptidoglycan degrading enzymes with antimicrobial applications. Recent Pat. Biotechnol. 2007, 1, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: The peptidase database. Nucleic Acids Res. 2008, 36, D320–D325. [Google Scholar] [CrossRef] [PubMed]
- Odintsov, S.G.; Sabala, I.; Marcyjaniak, M.; Bochtler, M. Latent LytM at 1.3A resolution. J. Mol. Biol. 2004, 335, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Sabala, I.; Jagielska, E.; Bardelang, P.T.; Czapinska, H.; Dahms, S.O.; Sharpe, J.A.; James, R.; Than, M.E.; Thomas, N.R.; Bochtler, M. Crystal structure of the antimicrobial peptidase lysostaphin from Staphylococcus simulans. FEBS J. 2014, 281, 4112–4122. [Google Scholar] [CrossRef]
- Firczuk, M.; Bochtler, M. Mutational analysis of peptidoglycan amidase MepA. Biochemistry 2007, 46, 120–128. [Google Scholar] [CrossRef]
- Firczuk, M.; Mucha, A.; Bochtler, M. Crystal structures of active LytM. J. Mol. Biol. 2005, 354, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, M.; Odintsov, S.G.; Marcyjaniak, M.; Sabala, I. Similar active sites in lysostaphins and D-Ala-D-Ala metallopeptidases. Protein Sci. 2004, 13, 854–861. [Google Scholar] [CrossRef]
- Cui, F.; Li, G.; Huang, J.; Zhang, J.; Lu, M.; Lu, W.; Huang, Q. Extension of nasal anti-Staphylococcus aureus efficacy of lysostaphin by its incorporation into a chitosan-o/w cream. Drug Deliv. 2010, 17, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Kokai-Kun, J. Lysostaphin: A Silver Bullet for Staph; Tegos, G., Ed.; CABI: Wallingford, UK, 2012; pp. 147–165. [Google Scholar]
- De Roca, F.R.; Duché, C.; Dong, S.; Rincé, A.; Dubost, L.; Pritchard, D.G.; Baker, J.R.; Arthur, M.; Mesnage, S. Cleavage Specificity of Enterococcus faecalis EnpA (EF1473), a Peptidoglycan Endopeptidase Related to the LytM/Lysostaphin Family of Metallopeptidases. J. Mol. Biol. 2010, 398, 507–517. [Google Scholar] [CrossRef]
- Arias, C.A.; Murray, B.E. The rise of the Enterococcus: Beyond vancomycin resistance. Nat. Rev. Microbiol. 2012, 10, 266–278. [Google Scholar] [CrossRef]
- Jett, B.D.; Huycke, M.M.; Gilmore, M.S. Virulence of enterococci. Clin. Microbiol. Rev. 1994, 7, 462–478. [Google Scholar] [CrossRef]
- Murray, B.E. The life and times of the Enterococcus. Clin. Microbiol. Rev. 1990, 3, 46–65. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Spencer, J.; Murphy, L.M.; Conners, R.; Sessions, R.B.; Gamblin, S.J. Crystal structure of the LasA virulence factor from Pseudomonas aeruginosa: Substrate specificity and mechanism of M23 metallopeptidases. J. Mol. Biol. 2010, 396, 908–923. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Kääriäinen, S.; Wilton, C.; Plewczynski, D. Using Dali for structural comparison of proteins. Curr. Protoc. Bioinform. 2006, 14. [Google Scholar] [CrossRef]
- Raulinaitis, V.; Tossavainen, H.; Aitio, O.; Juuti, J.T.; Hiramatsu, K.; Kontinen, V.; Permi, P. Identification and structural characterization of LytU, a unique peptidoglycan endopeptidase from the lysostaphin family. Sci. Rep. 2017, 7, 6020. [Google Scholar] [CrossRef] [PubMed]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Ehlert, K.; Schroder, W.; Labischinski, H. Specificities of FemA and FemB for different glycine residues: FemB cannot substitute for FemA in Staphylococcal peptidoglycan pentaglycine side chain formation. J. Bacteriol. 1997, 179, 7573–7576. [Google Scholar] [CrossRef] [PubMed]
- Ehlert, K.; Tschierske, M.; Mori, C.; Schröder, W.; Berger-Bächi, B. Site-specific serine incorporation by Lif and Epr into positions 3 and 5 of the staphylococcal peptidoglycan interpeptide bridge. J. Bacteriol. 2000, 182, 2635–2638. [Google Scholar] [CrossRef] [PubMed]
- Ling, B.; Berger-Bachi, B. Increased overall antibiotic susceptibility in Staphylococcus aureus femAB null mutants. Antimicrob. Agents Chemother. 1998, 42, 936–938. [Google Scholar] [CrossRef]
- Brown, S.; Maria, J.P.S., Jr.; Walker, S. Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef]
- Thomas, K.J., 3rd; Rice, C.V. Equilibrium binding behavior of magnesium to wall teichoic acid. Biochim. Biophys. Acta 2015, 1848, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Hancock, I.C.; Wiseman, G.; Baddiley, J. Biosynthesis of the unit that links teichoic acid to the bacterial wall: Inhibition by tunicamycin. FEBS Lett. 1976, 69, 75–80. [Google Scholar] [CrossRef]
- Grabowska, M.; Jagielska, E.; Czapinska, H.; Bochtler, M.; Sabala, I. High resolution structure of an M23 peptidase with a substrate analogue. Sci. Rep. 2015, 5, 14833. [Google Scholar] [CrossRef] [PubMed]
- Tossavainen, H.; Raulinaitis, V.; Kauppinen, L.; Pentikäinen, U.; Maaheimo, H.; Permi, P. Structural and Functional Insights into Lysostaphin–Substrate Interaction. Front. Mol. Biosci. 2018, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Cherif, D.M.; Stogios, P.J.; Evdokimova, E.; Savchenko, A.; Courvalin, P. Structural basis for the evolution of vancomycin resistance D,D-peptidases. Proc. Natl. Acad. Sci. USA 2014, 111, 5872–5877. [Google Scholar] [CrossRef]
- Sabala, I.; Jonsson, I.N.; Tarkowski, A.; Bochtler, M. Anti-staphylococcal activities of lysostaphin and LytM catalytic domain. BMC Microbiol. 2012, 12, 97. [Google Scholar] [CrossRef]
- Kokai-Kun, J.F.; Walsh, S.M.; Chanturiya, T.; Mond, J.J. Lysostaphin cream eradicates Staphylococcus aureus nasal colonization in a cotton rat model. Antimicrob. Agents Chemother. 2003, 47, 1589–1597. [Google Scholar] [CrossRef]
- Nilsen, T.; Nes, I.F.; Holo, H. Enterolysi A, a cell wall-degrading bacteriocin from Enterococcus faecalis LMG 2333. Appl. Environ. Microbiol. 2003, 69, 2975–2984. [Google Scholar] [CrossRef]
- Pastagia, M.; Kleinman, L.C.; de la Cruz, E.G.L.; Jenkins, S.G. Predicting risk for death from MRSA bacteremia. Emerg. Infect. Dis. 2012, 18, 1072–1080. [Google Scholar] [CrossRef]
- Gargis, S.R.; Heath, H.E.; LeBlanc, P.A.; Dekker, L.; Simmonds, R.S.; Sloan, G.L. Inhibition of the Activity of Both Domains of Lysostaphin through Peptidoglycan Modification by the Lysostaphin Immunity Protein. Appl. Environ. Microbiol. 2010, 76, 6944–6946. [Google Scholar] [CrossRef][Green Version]
- Jagielska, E.; Chojnacka, O.; Sabala, I. Lytm Fusion with SH3b-Like Domain Restore Its Activity in Physiological Conditions. Microb. Drug Resist. 2016, 22, 461–469. [Google Scholar] [CrossRef]
- Osipovitch, D.C.; Griswold, K.E. Fusion with a cell wall binding domain renders autolysin LytM a potent anti-Staphylococcus aureus agent. FEMS Microbiol. Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Oda, A.; Nakayoshi, T.; Kato, K.; Fukuyoshi, S.; Kurimoto, E. Three dimensional structures of putative, primitive proteins to investigate the origin of homochirality. Sci. Rep. 2019, 9, 11594. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Simmonds, R.S.; Timkovich, R. Solution structure of the Cys74 to Ala74 mutant of the recombinant catalytic domain of Zoocin A. Proteins 2017, 85, 177–181. [Google Scholar] [CrossRef]
- Chen, Y.; Simmonds, R.S.; Sloan, G.L.; Timkovich, R. The metal binding site of zoocin A. J. Biol. Inorg. Chem. 2008, 13, 855–860. [Google Scholar] [CrossRef]
- Grundling, A.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus mutants with increased lysostaphin resistance. J. Bacteriol. 2006, 188, 6286–6297. [Google Scholar] [CrossRef]
- Bardelang, P.; Vankemmelbeke, M.; Zhang, Y.; Jarvis, H.; Antoniadou, E.; Rochette, S.; Thomas, N.R.; Penfold, C.N.; James, R. Design of a polypeptide FRET substrate that facilitates study of the antimicrobial protease lysostaphin. Biochem. J. 2009, 418, 615–624. [Google Scholar] [CrossRef]
- Gargis, S.R.; Gargis, A.S.; Heath, H.E.; Heath, L.S.; LeBlanc, P.A.; Senn, M.M.; Berger-Bächi, B.; Simmonds, R.S.; Sloan, G.L. Zif, the zoocin A immunity factor, is a FemABX-like immunity protein with a novel mode of action. Appl. Environ. Microbiol. 2009, 75, 6205–6210. [Google Scholar] [CrossRef]
- Vessillier, S.; Delolme, F.; Bernillon, J.; Saulnier, J.; Wallach, J. Hydrolysis of glycine-containing elastin pentapeptides by LasA, a metalloelastase from Pseudomonas aeruginosa. Eur. J. Biochem. 2001, 268, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.N.; Sham, Y.Y.; Haugstad, G.D.; Xiang, Y.; Rossmann, M.G.; Anderson, D.L.; Popham, D.L. Shared catalysis in virus entry and bacterial cell wall depolymerization. J. Mol. Biol. 2009, 387, 607–618. [Google Scholar] [CrossRef]
- Xiang, Y.; Morais, M.C.; Cohen, D.N.; Bowman, V.D.; Anderson, D.L.; Rossmann, M.G. Crystal and cryoEM structural studies of a cell wall degrading enzyme in the bacteriophage phi29 tail. Proc. Natl. Acad. Sci. USA 2008, 105, 9552–9557. [Google Scholar] [CrossRef] [PubMed]
- An, D.R.; Im, H.N.; Jang, J.Y.; Kim, H.S.; Kim, J.; Yoon, H.J.; Hesek, D.; Lee, M.; Mobashery, S.; Kim, S.O.; et al. Structural Basis of the Heterodimer Formation between Cell Shape-Determining Proteins Csd1 and Csd2 from Helicobacter pylori. PLoS ONE 2016, 11, e0164243. [Google Scholar] [CrossRef]
- Bussiere, D.E.; Pratt, S.D.; Katz, L.; Severin, J.M.; Holzman, T.; Park, C.H. The structure of VanX reveals a novel amino-dipeptidase involved in mediating transposon-based vancomycin resistance. Mol. Cell. 1998, 2, 75–84. [Google Scholar] [CrossRef]
- Keary, R.; Gaitero, M.S.; van Raaij, M.J.; O’Mahony, J.; Fenton, M.; McAuliffe, O.; Hill, C.; Ross, R.P.; Coffey, A. Characterization of a Bacteriophage-Derived Murein Peptidase for Elimination of Antibiotic-Resistant Staphylococcus aureus. Curr. Protein Pept. Sci. 2016, 17, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed]
- Charlier, P.; Wery, J.P.; Dideberg, O.; Frère, J.M. Streptomyces Albus GD-Ala-D-Ala Carboxypeptidase. In Handbook of Metalloproteins; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Phillips, M.A.; Fletterick, R.; Rutter, W.J. Arginine 127 stabilizes the transition state in carboxypeptidase. J. Biol. Chem. 1990, 265, 20692–20698. [Google Scholar] [CrossRef]
- Cianci, M.; Bourenkov, G.; Pompidor, G.; Karpics, I.; Kallio, J.; Bento, I.; Roessle, M.; Cipriani, F.; Fiedler, S.; Schneider, T.R. P13, the EMBL macromolecular crystallography beamline at the low-emittance PETRA III ring for high- and low-energy phasing with variable beam focusing. J. Synchrotron. Radiat. 2017, 24, 323–332. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Wang, X.; Yang, X.; Yang, C.; Wu, Z.; Xu, H.; Shen, Y. Crystal structure of outer membrane protein NMB0315 from Neisseria meningitidis. PLoS ONE 2011, 6, e26845. [Google Scholar] [CrossRef]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef]
- McCoy, A.J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 32–41. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Małecki, P.H.; Mitkowski, P.; Jagielska, E.; Trochimiak, K.; Mesnage, S.; Sabała, I. Structural Characterization of EnpA D,L-Endopeptidase from Enterococcus faecalis Prophage Provides Insights into Substrate Specificity of M23 Peptidases. Int. J. Mol. Sci. 2021, 22, 7136. https://doi.org/10.3390/ijms22137136
Małecki PH, Mitkowski P, Jagielska E, Trochimiak K, Mesnage S, Sabała I. Structural Characterization of EnpA D,L-Endopeptidase from Enterococcus faecalis Prophage Provides Insights into Substrate Specificity of M23 Peptidases. International Journal of Molecular Sciences. 2021; 22(13):7136. https://doi.org/10.3390/ijms22137136
Chicago/Turabian StyleMałecki, Piotr Henryk, Paweł Mitkowski, Elżbieta Jagielska, Karolina Trochimiak, Stéphane Mesnage, and Izabela Sabała. 2021. "Structural Characterization of EnpA D,L-Endopeptidase from Enterococcus faecalis Prophage Provides Insights into Substrate Specificity of M23 Peptidases" International Journal of Molecular Sciences 22, no. 13: 7136. https://doi.org/10.3390/ijms22137136
APA StyleMałecki, P. H., Mitkowski, P., Jagielska, E., Trochimiak, K., Mesnage, S., & Sabała, I. (2021). Structural Characterization of EnpA D,L-Endopeptidase from Enterococcus faecalis Prophage Provides Insights into Substrate Specificity of M23 Peptidases. International Journal of Molecular Sciences, 22(13), 7136. https://doi.org/10.3390/ijms22137136