
Overlap Arrhythmia Syndromes Resulting from Multiple Genetic Variations Studied in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes



Abstract
:1. Introduction
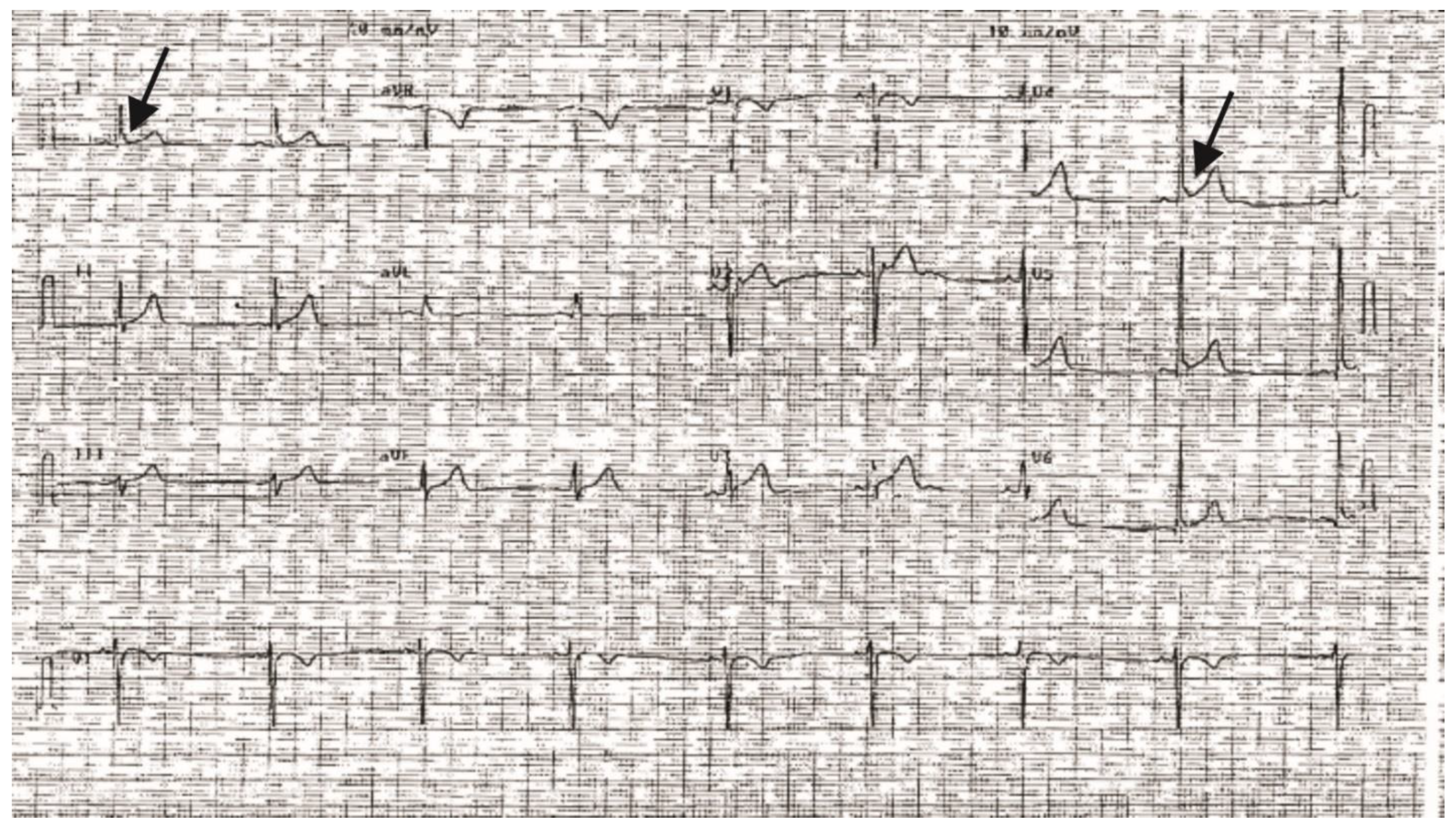
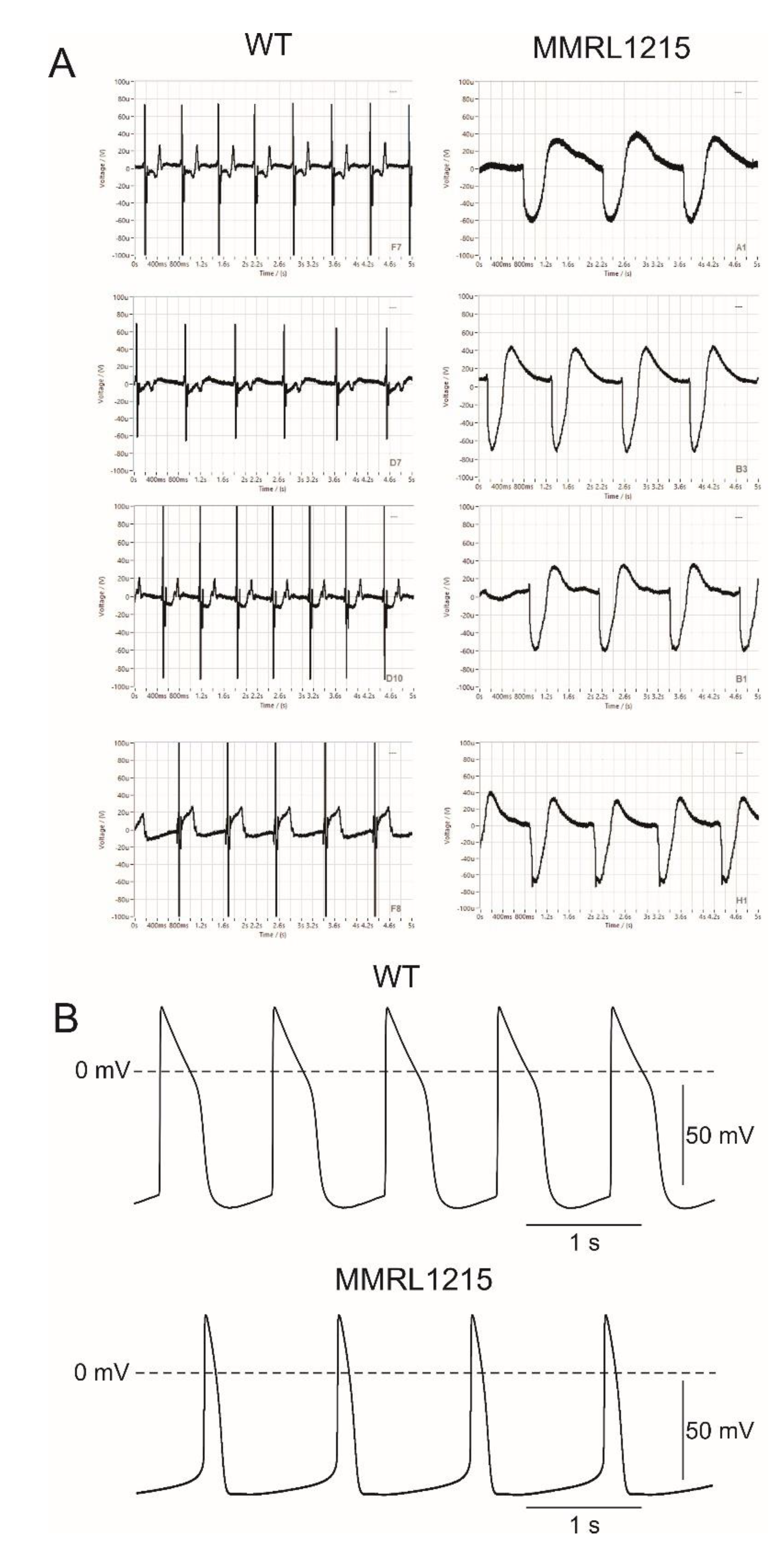
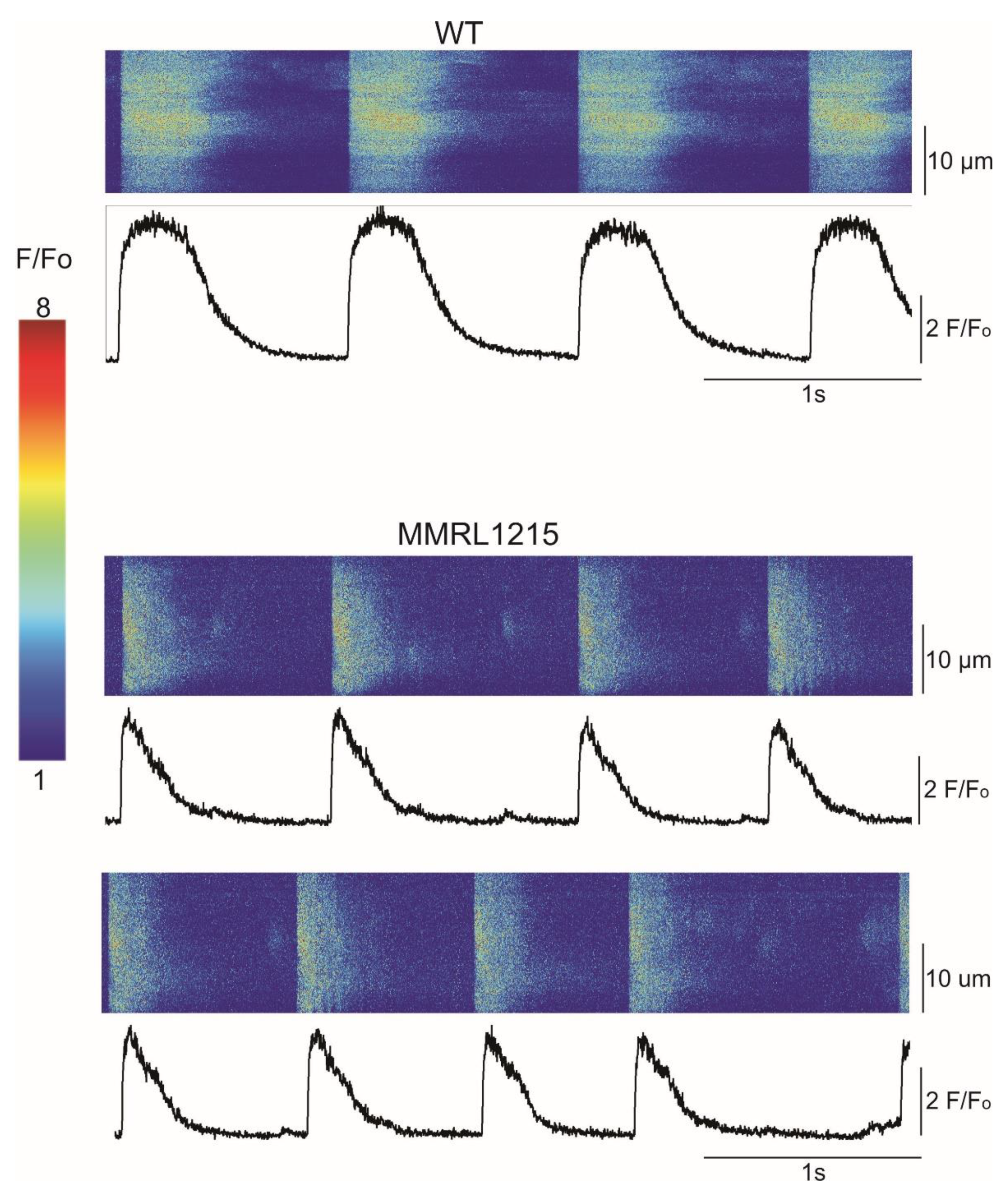
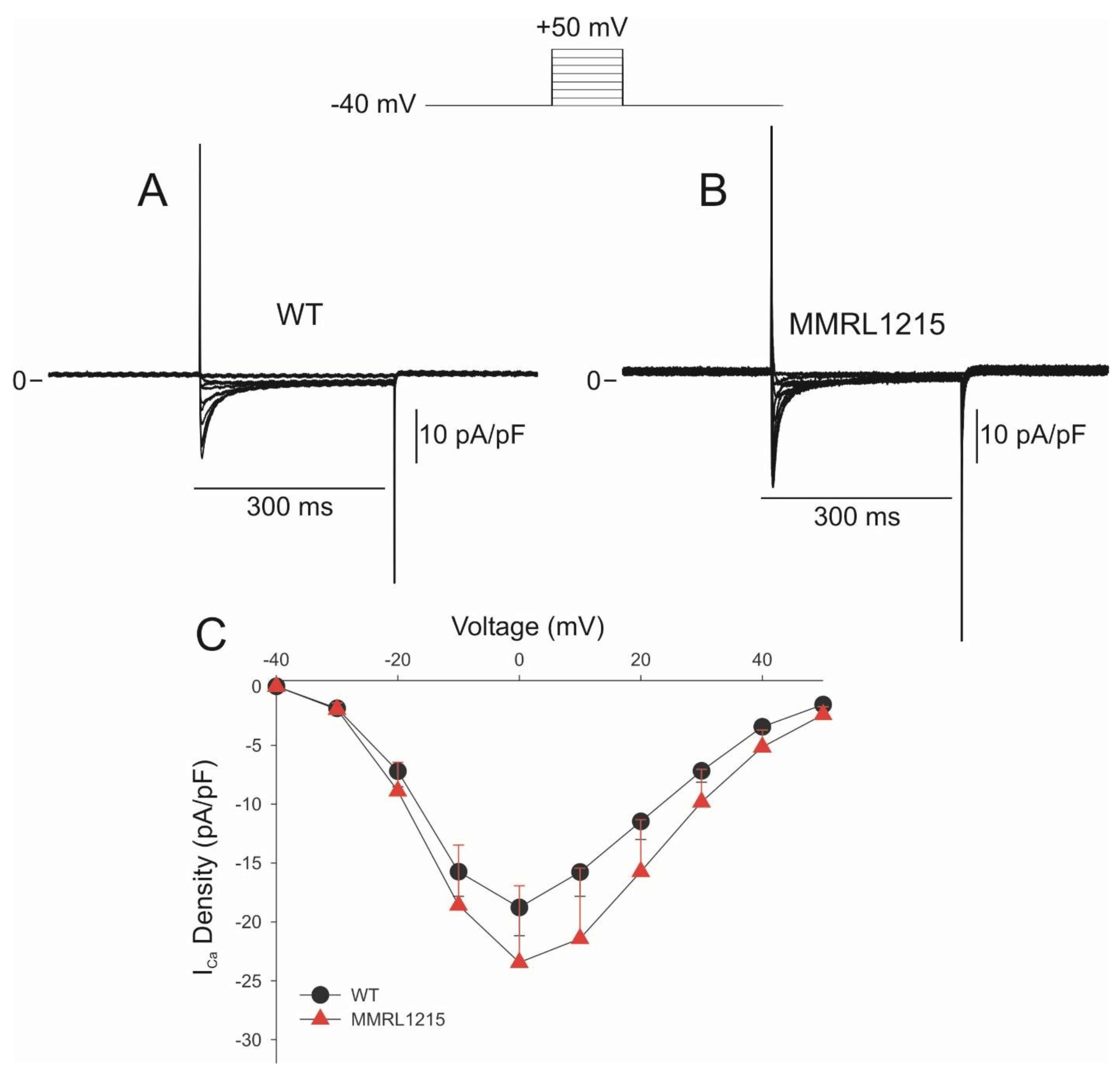
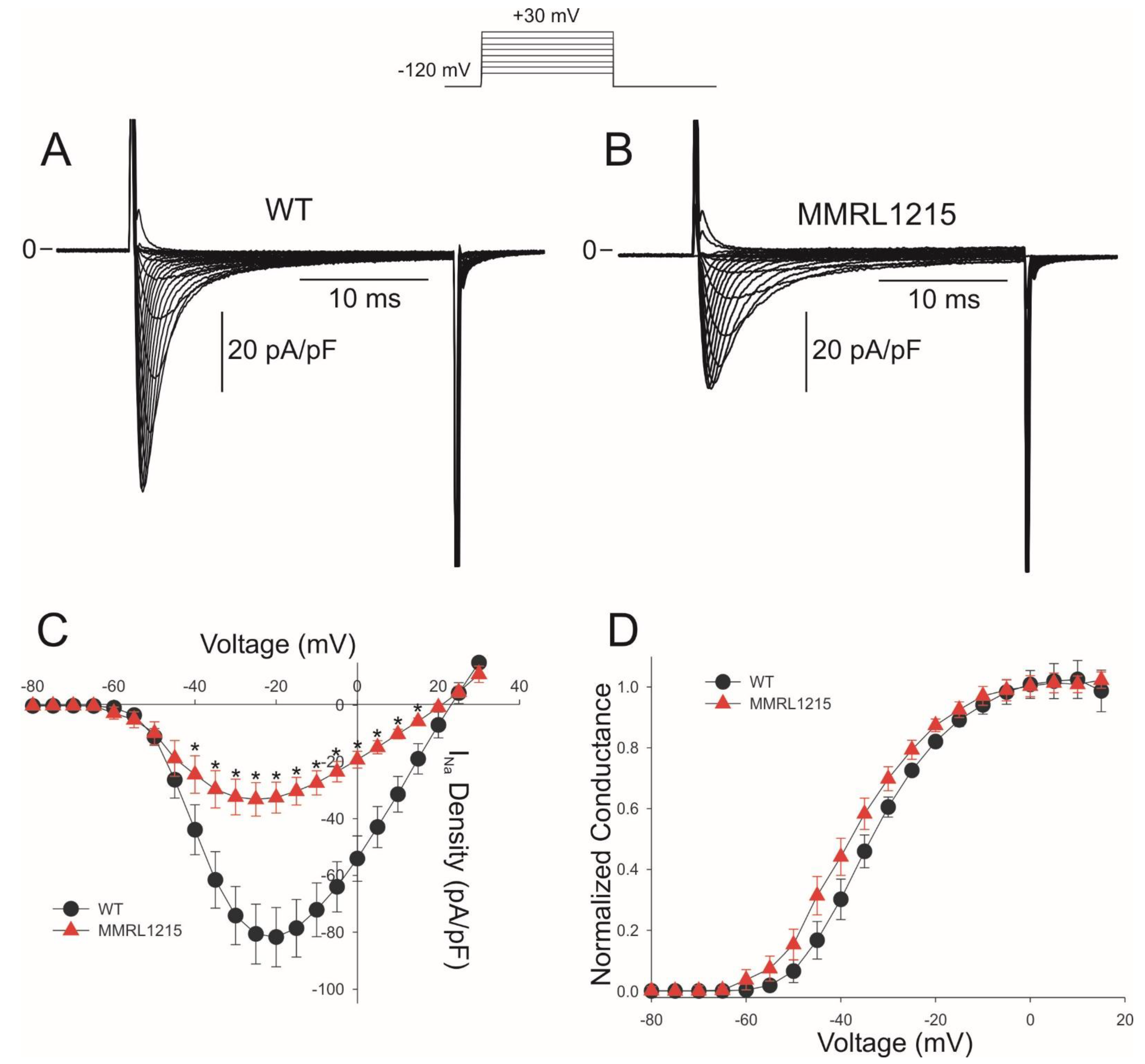
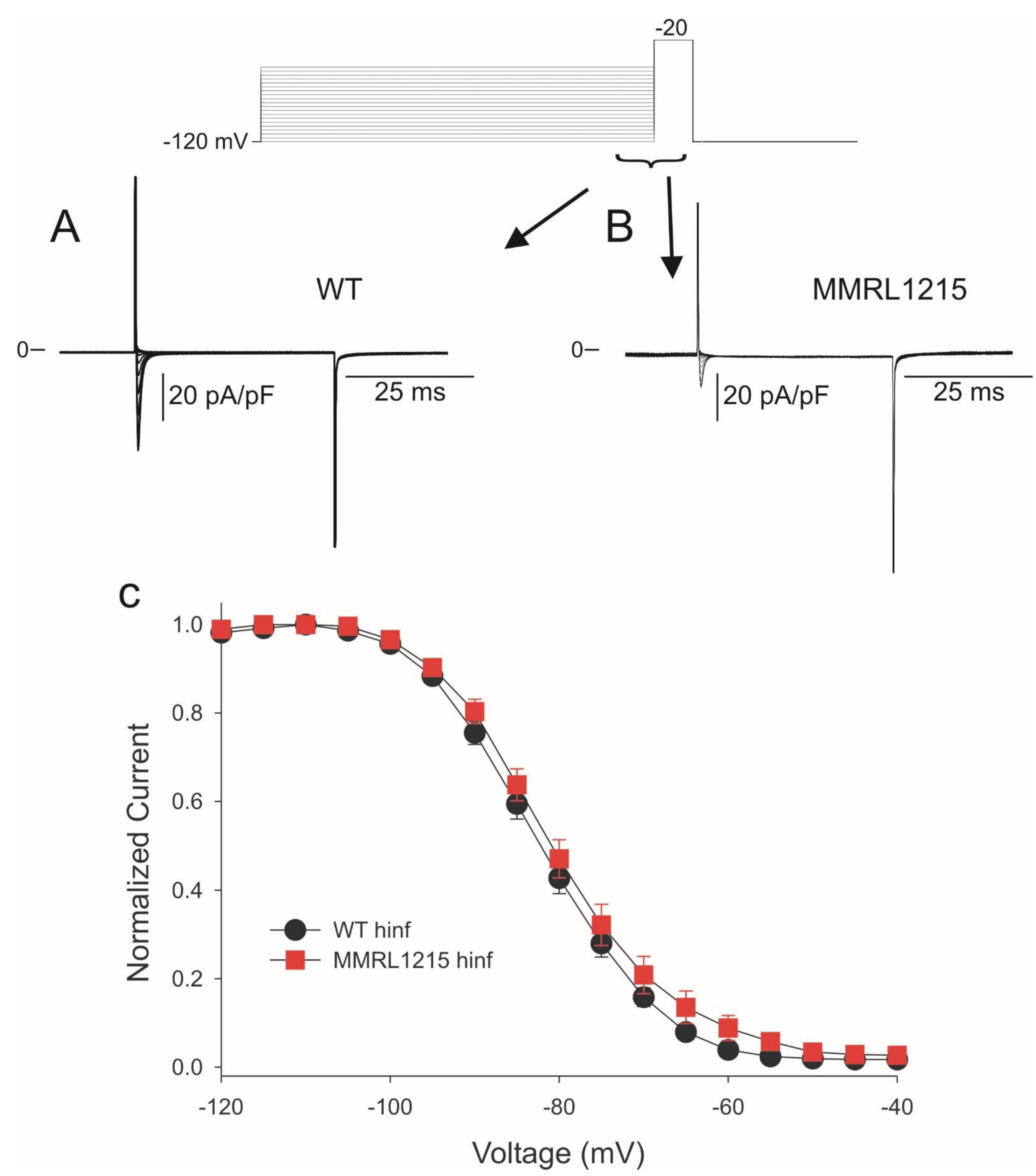
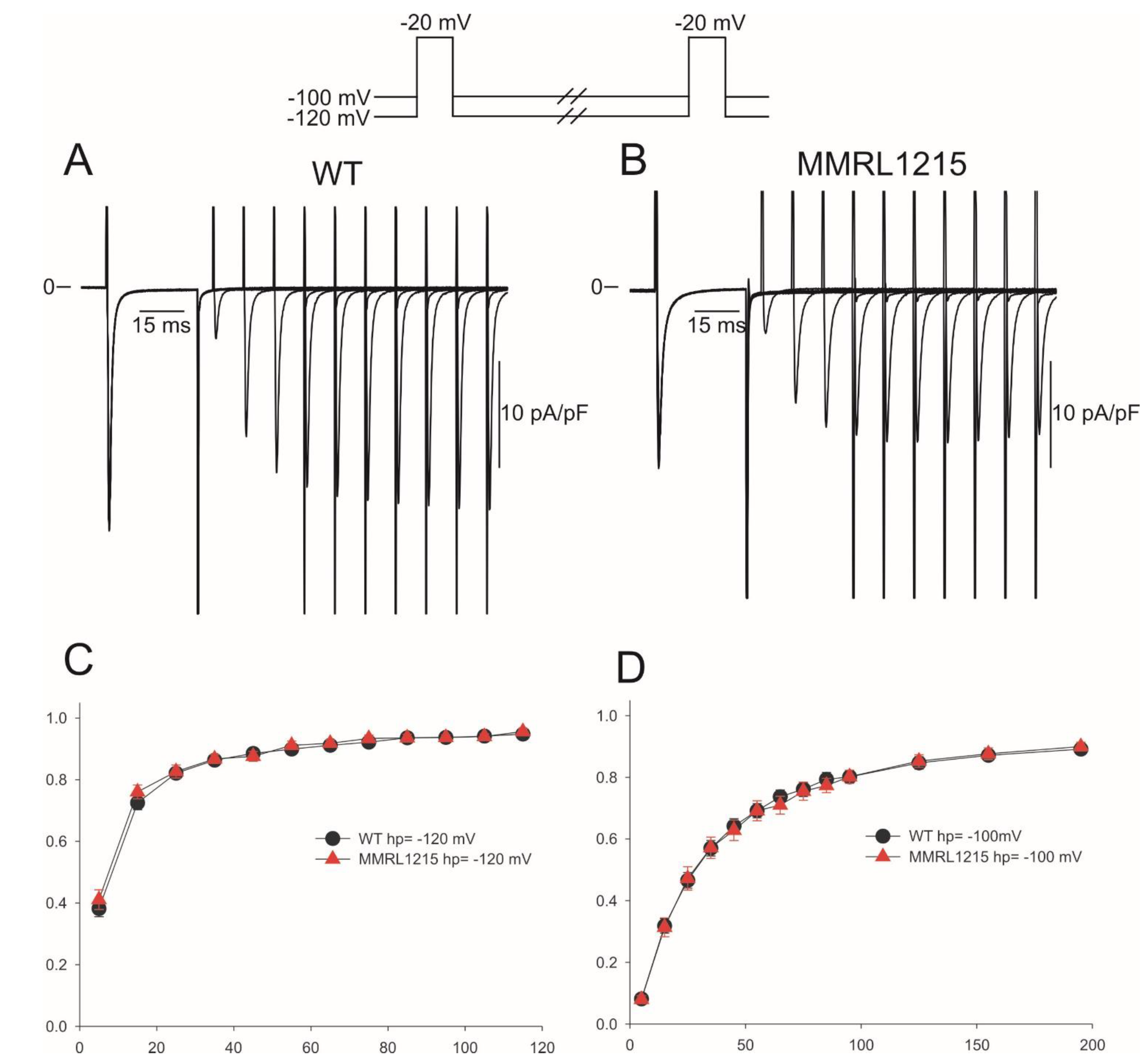
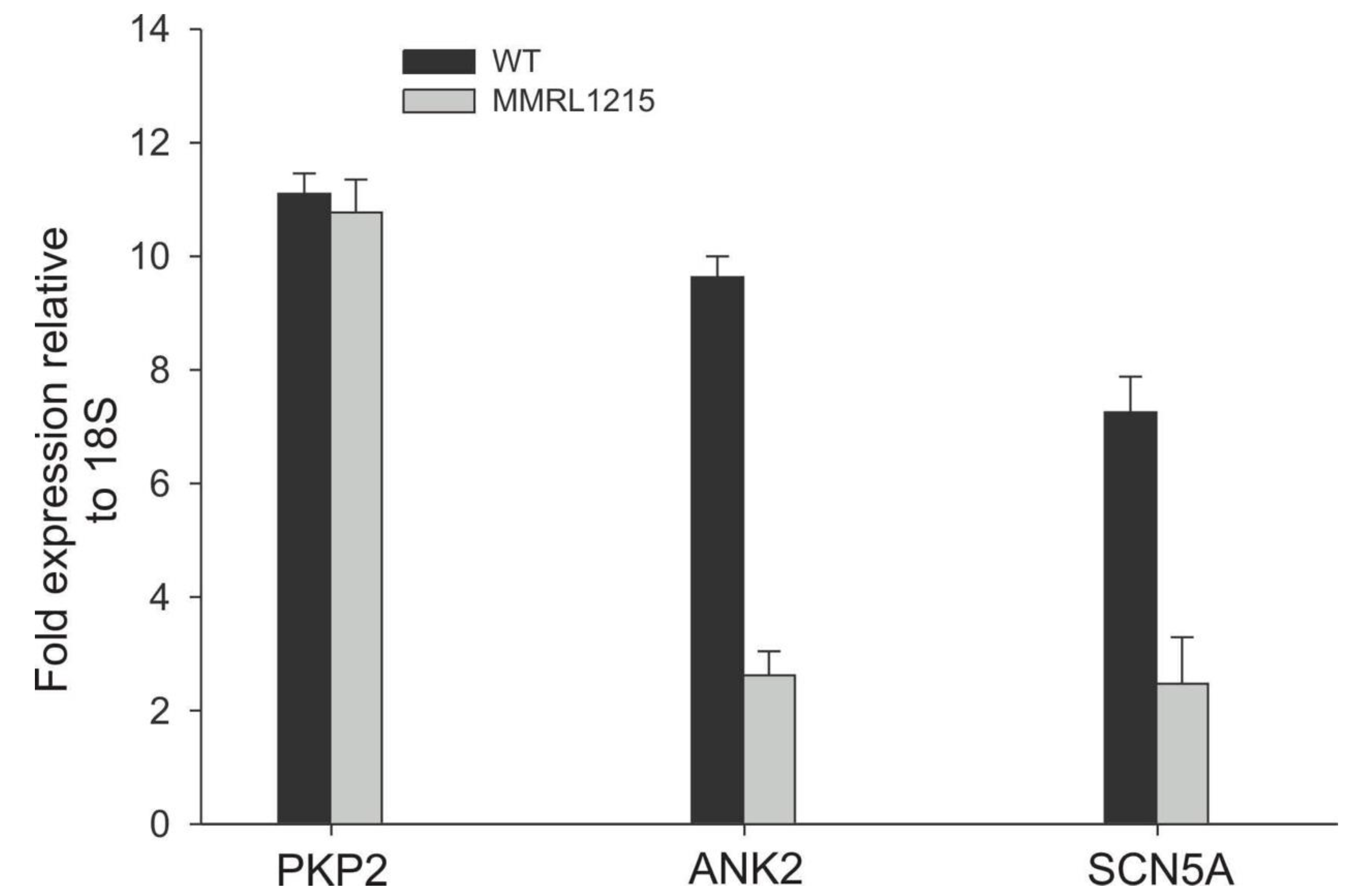
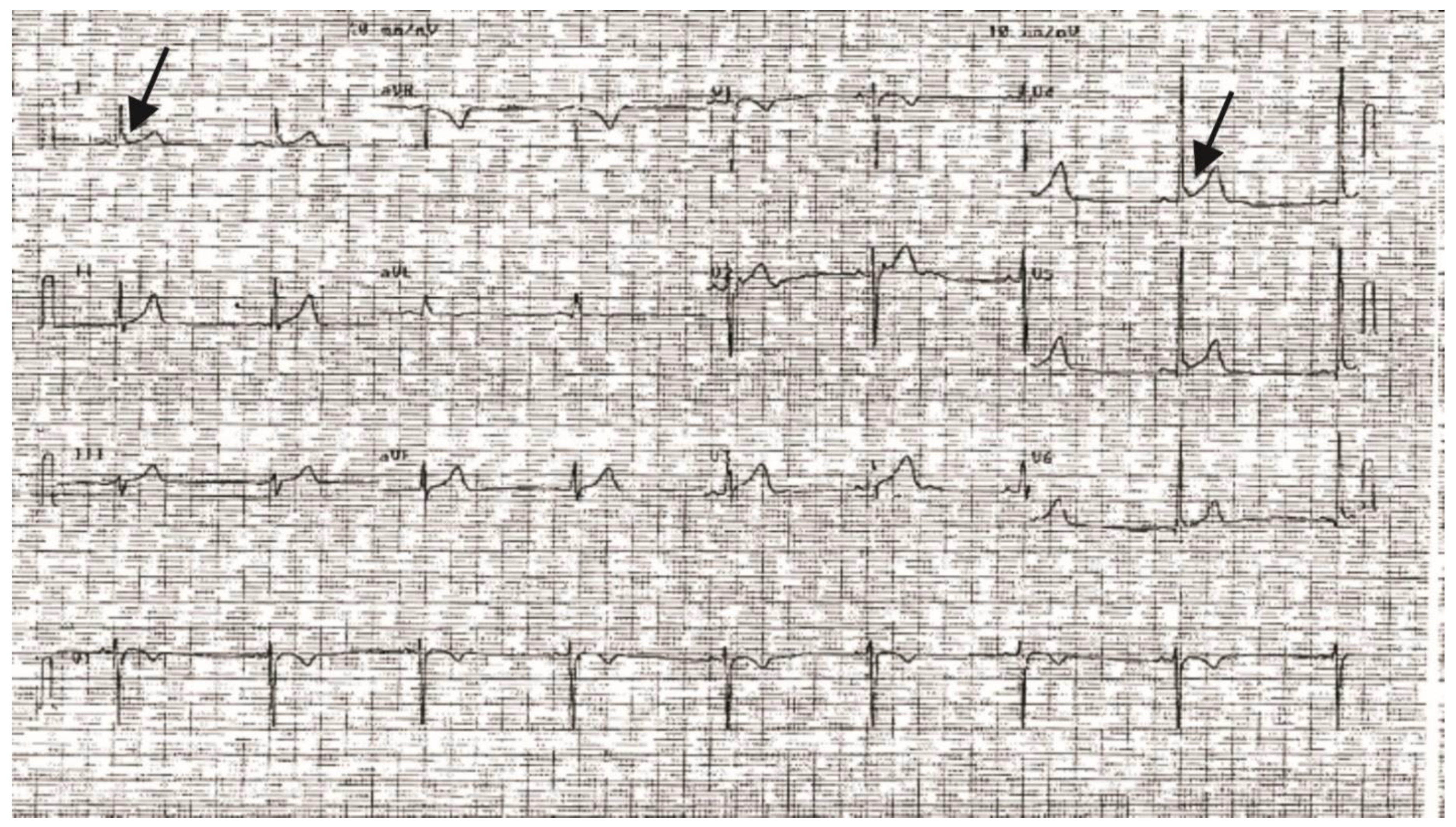
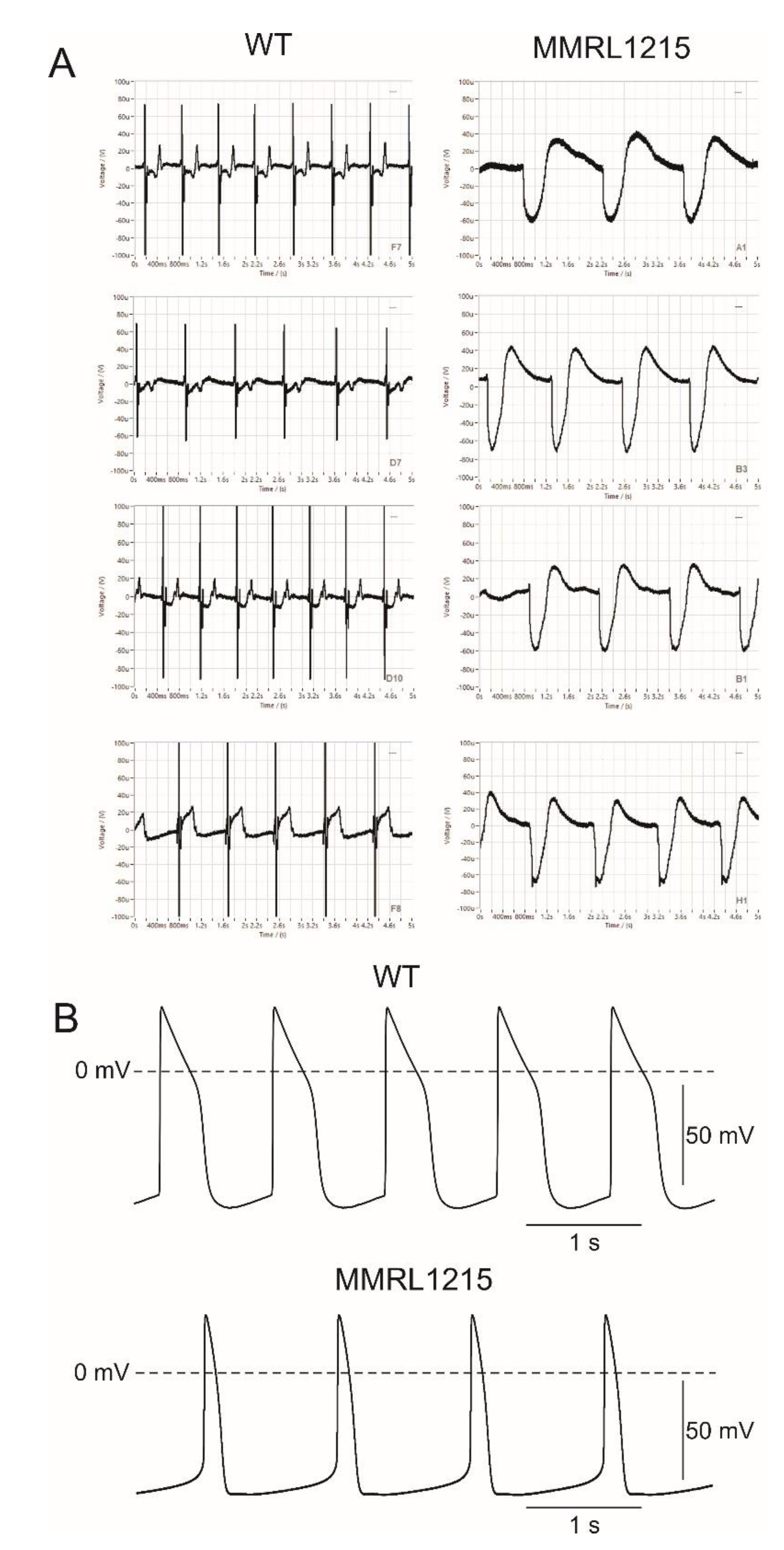
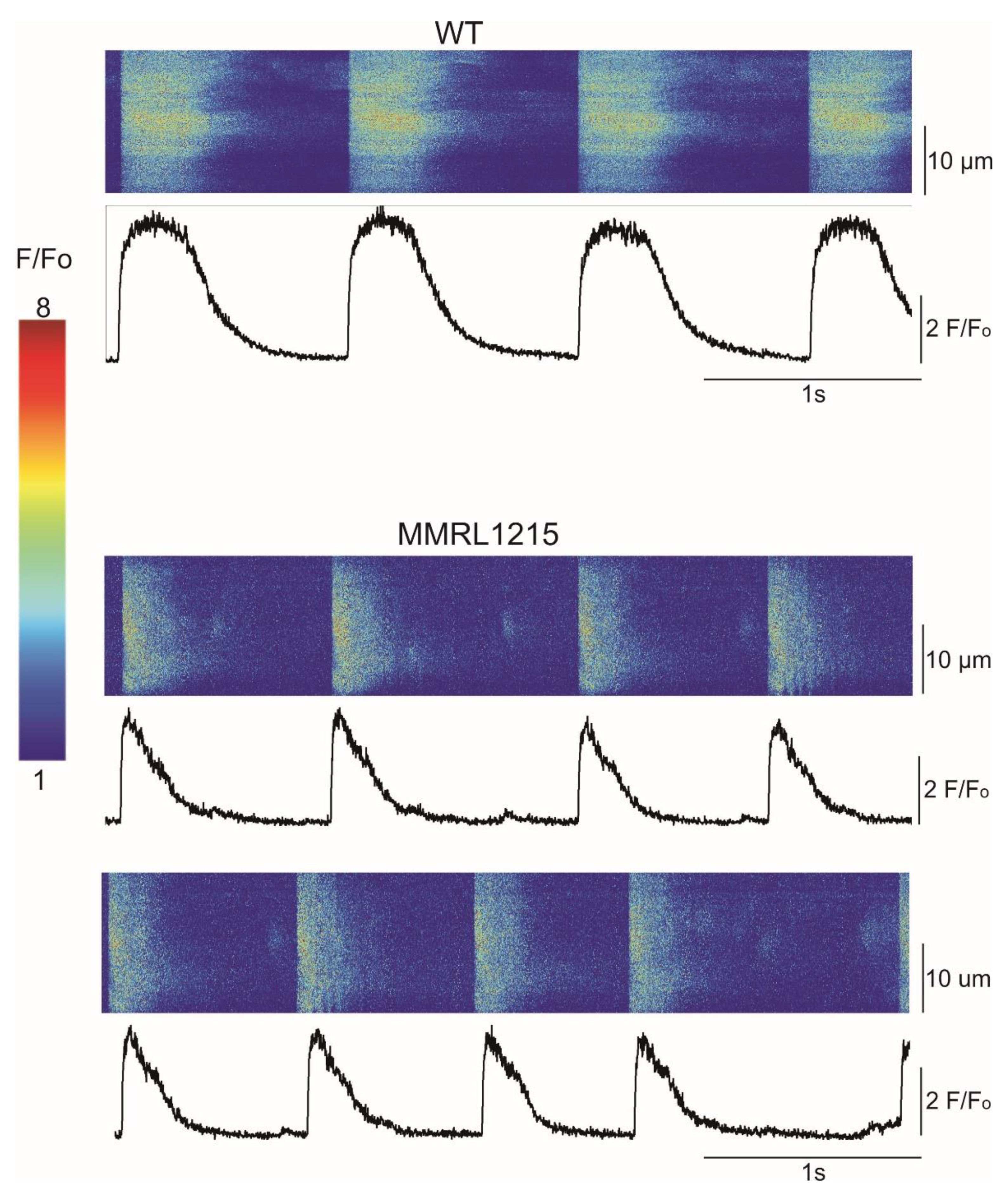
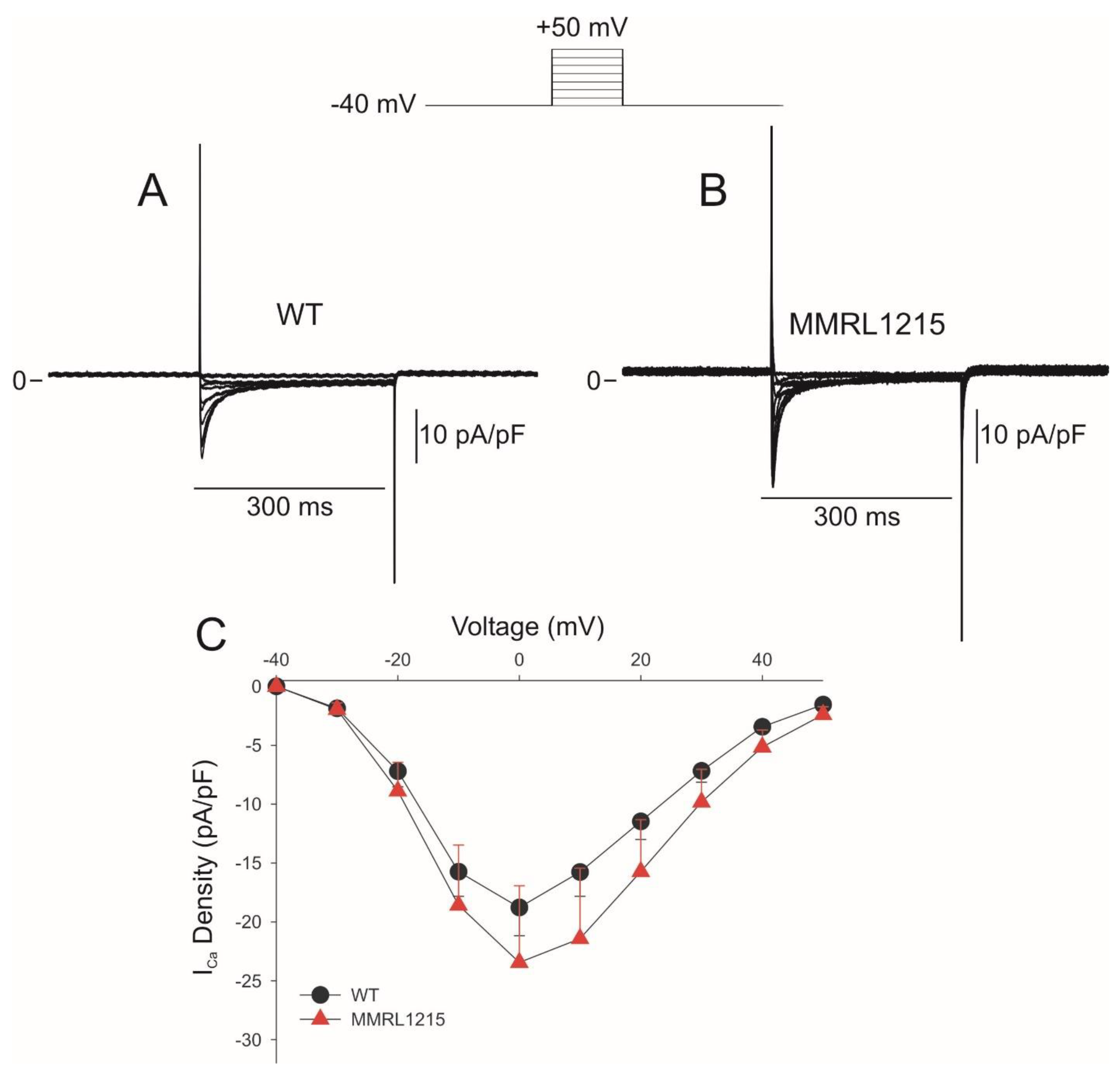
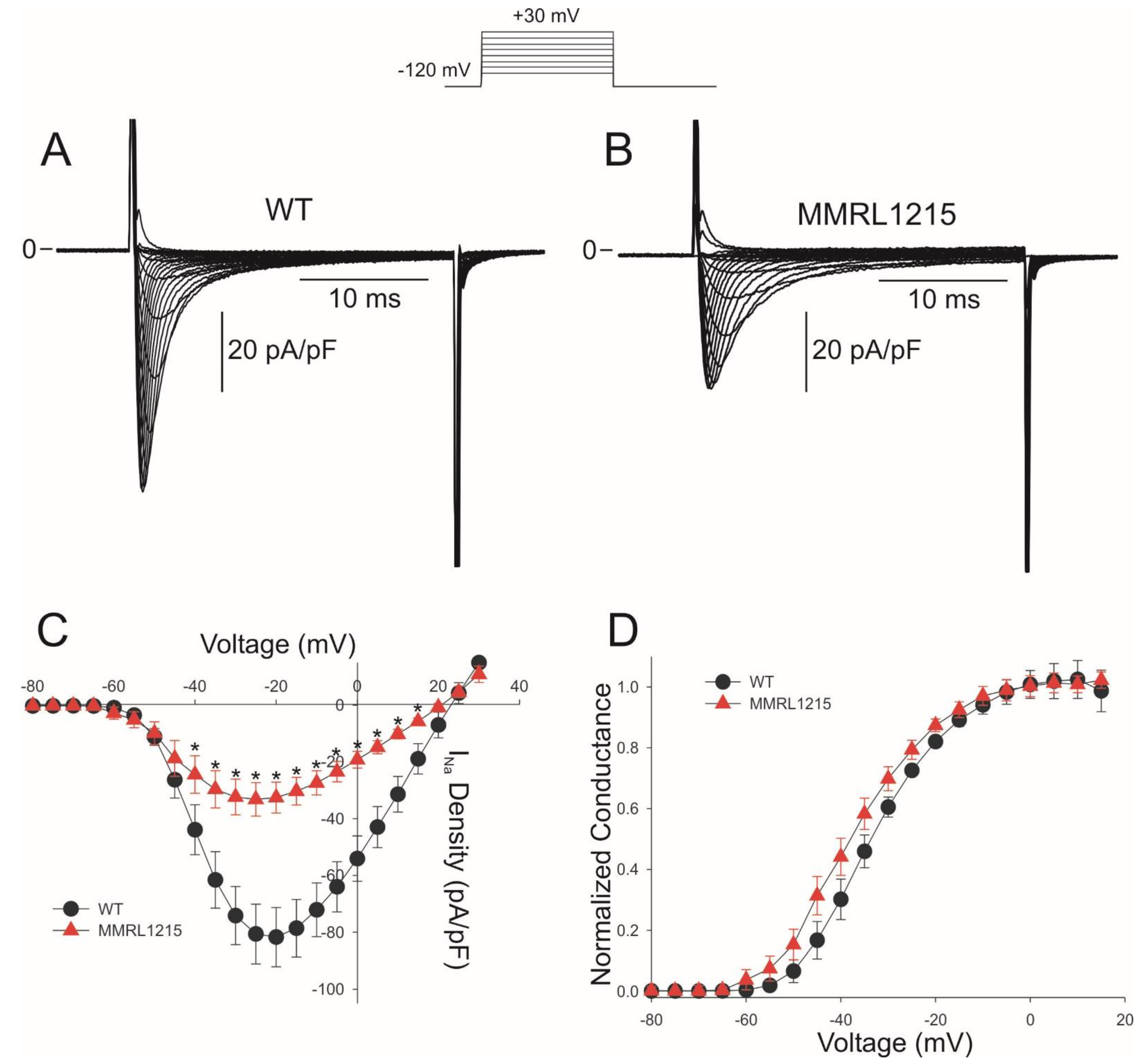
2. Results
3. Discussion
3.1. Summary of Main Findings
3.2. Conclusions
3.3. Limitations of the Study
4. Methods
4.1. Subjects
4.2. Next Generation Sequencing
4.3. Mutation Confirmation
4.4. Bioinformatics Analysis
4.5. hiPSC-CM Generation
4.6. Extracellular Field Potential and Impedance Recordings
4.7. Electrophysiological Recordings
4.8. Fluorescence Imaging
4.9. RNA Isolation and Quantitative Real-Time PCR
- SCN5A—Hs00165693_m1 (Life Technologies)
- PKP2—Hs00428040 m1 (Life Technologies)
- ANK2—00153998 m1 (Life Technologies)
- 18s-Hs03003631_g1 (Life Technologies)
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.C.; Garg, P.; Yoshida, Y.; Yamanaka, S.; Gepstein, L.; Hulot, J.-S.; Knollmann, B.C.; Schwartz, P.J. Towards Precision Medicine with Human iPSCs for Cardiac Channelopathies. Circ. Res. 2019, 125, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Terrenoire, C.; Wang, K.; Tung, K.W.C.; Chung, W.K.; Pass, R.H.; Lu, J.T.; Jean, J.-C.; Omari, A.; Sampson, K.J.; Kotton, D.; et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J. Gen. Physiol. 2012, 141, 61–72. [Google Scholar] [CrossRef] [Green Version]
- El-Battrawy, I.; Lan, H.; Cyganek, L.; Zhao, Z.; Li, X.; Buljubasic, F.; Lang, S.; Yücel, G.; Sattler, K.; Zimmermann, W.; et al. Modeling Short QT Syndrome Using Human-Induced Pluripotent Stem Cell–Derived Cardiomyocytes. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sendfeld, F.; Selga, E.; Scornik, F.S.; Pérez, G.J.; Mills, N.L.; Brugada, R. Experimental Models of Brugada syndrome. Int. J. Mol. Sci. 2019, 20, 2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, P.J.; Priori, S.G.; Dumaine, R.; Napolitano, C.; Antzelevitch, C.; Stramba-Badiale, M.; Richard, T.A.; Berti, M.R.; Bloise, R. A Molecular Link between the Sudden Infant Death Syndrome and the Long-QT Syndrome. N. Engl. J. Med. 2000, 343, 262–267. [Google Scholar] [CrossRef]
- Burashnikov, E.; Pfeiffer, R.; Barajas-Martinez, H.; Delpón, E.; Hu, D.; Desai, M.; Borggrefe, M.; Häissaguerre, M.; Kanter, R.; Pollevick, G.D.; et al. Mutations in the cardiac L-type calcium channel associated J wave sydnrome and sudden cardiac death. Heart Rhythm 2010, 7, 1872–1882. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Barajas-Martínez, H.; Terzic, A.; Park, S.; Pfeiffer, R.; Burashnikov, E.; Wu, Y.; Borggrefe, M.; Veltmann, C.; Schimpf, R.; et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int. J. Cardiol. 2014, 171, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, O.; Fernandez-Falgueras, A.; Lemus, X.; Sarquella-Brugada, G.; Cesar, S.; Coll, M.; Mates, J.; Arbelo, E.; Jordà, P.; Perez-Serra, A.; et al. Short QT Syndrome: A Comprehensive Genetic Interpretation and Clinical Translation of Rare Variants. J. Clin. Med. 2019, 8, 1035. [Google Scholar] [CrossRef] [Green Version]
- Antzelevitch, C. Genetic, Molecular and Cellular Mechanisms Underlying the J Wave Syndromes. Circ. J. 2012, 76, 1054–1065. [Google Scholar] [CrossRef] [Green Version]
- Bourier, F.; Denis, A.; Cheniti, G.; Lam, A.; Vlachos, K.; Takigawa, M.; Kitamura, T.; Frontera, A.; Duchateau, J.; Pambrun, T.; et al. Early Repolarization Syndrome: Diagnostic and Therapeutic Approach. Front. Cardiovasc. Med. 2018, 5. [Google Scholar] [CrossRef]
- Brugada, R.; Hong, K.; Cordeiro, J.; Dumaine, R. Short QT syndrome. Can. Med. Assoc. J. 2005, 173, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Koncz, I.; Gurabi, Z.; Patocskai, B.; Panama, B.K.; Szél, T.; Hu, D.; Barajas-Martínez, H.; Antzelevitch, C. Mechanisms underlying the development of the electrocardiographic and arrhythmic manifestations of early repolarization syndrome. J. Mol. Cell. Cardiol. 2014, 68, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugada, R.; Hong, K.; Dumaine, R.; Cordeiro, J.; Gaita, F.; Borggrefe, M.; Menendez, T.M.; Brugada, J.; Pollevick, G.D.; Wolpert, C.; et al. Sudden Death Associated With Short-QT Syndrome Linked to Mutations in HERG. Circulation 2004, 109, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellocq, C.; Van Ginneken, A.C.G.; Bezzina, C.R.; Alders, M.; Escande, D.; Mannens, M.M.A.M.; Baró, I.; Wilde, A.A.M. Mutation in the KCNQ1 Gene Leading to the Short QT-Interval Syndrome. Circulation 2004, 109, 2394–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priori, S.G.; Pandit, S.V.; Rivolta, I.; Berenfeld, O.; Ronchetti, E.; Dhamoon, A.; Napolitano, C.; Anumonwo, J.; di Barletta, M.R.; Gudapakkam, S.; et al. A Novel Form of Short QT Syndrome (SQT3) Is Caused by a Mutation in the KCNJ2 Gene. Circ. Res. 2005, 96, 800–807. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Remme, C.A.; Tadros, R.; Bezzina, C.R.; Delmar, M. Beyond the One Gene-One Disease Paradigm: Complex Genetics and Pleiotropy in Inheritable Cardiac Disorders. Circulation 2019, 140, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Barajas-Martinez, H.; Smith, M.; Hu, D.; Goodrow, R.J.; Puleo, C.; Hasdemir, C.; Antzelevitch, C.; Pfeiffer, R.; Treat, J.A.; Cordeiro, J.M. Susceptibility to Ventricular Arrhythmias Resulting from Mutations in FKBP1B, PXDNL, and SCN9A Evaluated in hiPSC Cardiomyocytes. Stem Cells Int. 2020, 2020, 8842398. [Google Scholar] [CrossRef]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Remme, C.A.; Schumacher, C.A.; Scicluna, B.P.; Wolswinkel, R.; de Jonge, B.; Bezzina, C.R.; Veldkamp, M.W. Functional Nav1.8 channels in intracardiac neurons: The link between SCN10A and cardiac electrophysiology. Circ. Res. 2012, 111, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterfi, Z.; Tóth, Z.E.; Kovács, H.A.; Lázár, E.; Sum, A.; Donkó, Á.; Sirokmány, G.; Shah, A.; Geiszt, M. Peroxidasin-like protein: A novel peroxidase homologue in the human heart. Cardiovasc. Res. 2013, 101, 393–399. [Google Scholar] [CrossRef]
- Masia, R.; Enkvetchakul, D.; Nichols, C. Differential nucleotide regulation of KATP channels by SUR1 and SUR2A. J. Mol. Cell. Cardiol. 2005, 39, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Noma, A. ATP-regulated K+ channels in cardiac muscle. Nat. Cell Biol. 1983, 305, 147–148. [Google Scholar] [CrossRef]
- Nichols, C.G.; Ripoll, C.; Lederer, W.J. ATP-sensitive potassium channel modulation of the guinea pig ventricular action potential and contraction. Circ. Res. 1991, 68, 280–287. [Google Scholar] [CrossRef]
- Hu, D.; Barajas-Martinez, H.; Terzic, A.; Borggrefe, M.; Veltmann, C.; Schimpf, R. Compound mutations in ABCC9 and SCN5A associated with a malignant form of overlap syndrome: Brugada, long QT and early repolarization syndromes. Heart Rhythm 2011, 8, S463. [Google Scholar]
- Mohler, P.J.; Schott, J.-J.; Gramolini, A.O.; Dilly, K.W.; Guatimosim, S.; Dubell, W.H.; Song, L.-S.; Haurogné, K.; Kyndt, F.; Ali, M.E.; et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nat. Cell Biol. 2003, 421, 634–639. [Google Scholar] [CrossRef]
- Lowe, J.S.; Palygin, O.; Bhasin, N.; Hund, T.J.; Boyden, P.A.; Shibata, E.; Anderson, M.E.; Mohler, P.J. Voltage-gated Nav channel targeting in the heart requires an ankyrin-G–dependent cellular pathway. J. Cell Biol. 2008, 180, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Gusky, H.C.; Novelli, V.; Kim, C.; Tirasawasdichai, T.; Judge, D.; et al. Missense Mutations in Plakophilin-2 Cause Sodium Current Deficit and Associate with a Brugada Syndrome Phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Viskin, S.; Oliva, A.; Carrier, T.; Cordeiro, J.M.; Barajas-Martinez, H.; Wu, Y.; Burashnikov, E.; Sicouri, S.; Brugada, R.; et al. Novel mutation in the SCN5A gene associated with arrhythmic storm development during acute myocardial infarction. Heart Rhythm 2007, 4, 1072–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, A.; Hu, D.; Viskin, S.; Carrier, T.; Cordeiro, J.M.; Barajas-Martínez, H.; Wu, Y.; Burashnikov, E.; Brugada, R.; Rosso, R.; et al. SCN5A Mutation associated with acute myocardial infarction. Leg. Med. 2009, 11, S206–S209. [Google Scholar] [CrossRef] [Green Version]
- Fatovich, D.M. Delayed lethal arrhythmia after an electrical injury. Emerg. Med. J. 2007, 24, 743. [Google Scholar] [CrossRef] [Green Version]
- Cordeiro, J.M.; Nesterenko, V.V.; Sicouri, S.; Goodrow Jr, R.J.; Treat, J.A.; Desai, M.; Wu, Y.; Doss, M.X.; Antzelevitch, C.; Di Diego, J.M. Identification and characterization of a transient outward K+ current in human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2013, 60, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Vaidyanathan, R.; Markandeya, Y.S.; Kamp, T.J.; Makielski, J.C.; January, C.T.; Eckhardt, L.L. IK1-enhanced human-induced pluripotent stem cell-derived cardiomyocytes: An improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am. J. Physiol. Circ. Physiol. 2016, 310, H1611–H1621. [Google Scholar] [CrossRef] [Green Version]
- Calloe, K.; Chlus, N.; Nof, E.; Jespersen, T.; Olesen, S.-P.; Antzelevitch, C.; Cordeiro, J.M. Comparison of the Effects of the Transient Outward Potassium Channel Activator NS5806 on Canine Atrial and Ventricular Cardiomyocytes. Biophys. J. 2010, 98, 334a. [Google Scholar] [CrossRef] [Green Version]
- Veerman, C.C.; Mengarelli, I.; Lodder, E.M.; Kosmidis, G.; Bellin, M.; Zhang, M.; Dittmann, S.; Guan, K.; Wilde, A.A.M.; Schulze-Bahr, E.; et al. Switch From Fetal to Adult SCN5A Isoform in Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes Unmasks the Cellular Phenotype of a Conduction Disease–Causing Mutation. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Calloe, K.; Aistrup, G.L.; Di Diego, J.M.; Goodrow, R.J.; Treat, J.A.; Cordeiro, J.M. Interventricular differences in sodium current and its potential role in Brugada syndrome. Physiol. Rep. 2018, 6, e13787. [Google Scholar] [CrossRef] [PubMed]
- Treat, J.A.; Goodrow, R.J.; Bot, C.T.; Haedo, R.J.; Cordeiro, J.M. Pharmacological enhancement of repolarization reserve in human induced pluripotent stem cells derived cardiomyocytes. Biochem. Pharmacol. 2019, 169, 113608. [Google Scholar] [CrossRef] [PubMed]
- Calloe, K.; Goodrow, R.; Olesen, S.-P.; Antzelevitch, C.; Cordeiro, J.M. Tissue-specific effects of acetylcholine in the canine heart. Am. J. Physiol. Circ. Physiol. 2013, 305, H66–H75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barajas-Martinez, H.; Goodrow, R.J.; Hu, D.; Patel, P.; Desai, M.; Panama, B.K.; Treat, J.A.; Aistrup, G.L.; Cordeiro, J.M. Biophysical and molecular comparison of sodium current in cells isolated from canine atria and pulmonary vein. Pflügers Arch. Eur. J. Physiol. 2017, 469, 703–712. [Google Scholar] [CrossRef]
- Goodrow, R.J.; Desai, S.; Treat, J.A.; Panama, B.K.; Desai, M.; Nesterenko, V.V.; Cordeiro, J.M. Biophysical comparison of sodium currents in native cardiac myocytes and human induced pluripotent stem cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 2018, 90, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, J.M.; Bridge, J.; Spitzer, K. Early and delayed afterdepolarizations in rabbit heart Purkinje cells viewed by confocal microscopy. Cell Calcium 2001, 29, 289–297. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # Locus | Gene | Exon | Protein | Coding | Sift | Dbsnp |
|---|---|---|---|---|---|---|
| chr1:237947931 | RYR2 | 90 | p.Arg4307Cys in Ryanodine receptor 2 | c.12919C>T | Tolerated | rs200092869 |
| chr4:114286207 | ANK2 | 41 | p.Val3634Asp in Ankyrin-B | c.10901T>A | Damaging | rs66785829 |
| chr12:5154064 | KCNA5 | 1 | p.Ala251Thr in Kv1.5 | c.751G>A | Tolerated | rs12720442 |
| chr12:22035732 | ABCC9 | 14 | p.Arg663Cys in ATP Binding Cassette C9 | c.1987C>T | Damaging | rs200349671 |
| chr12:33049590 | PKP2 | 1 | p.Asp26Asn in Plakophilin 2 | c.76G>A | Damaging | rs143004808 |
| chr19:615946 | HCN2 | 8 | p.Pro715_Pro717del in Hyperpolarization activated Cyclic Nucleotide channel 2 | c.2156_2164delCGCCGCCGC | rs879255365 |
| Spontaneous Cycle Length (bpm) | AP Amplitude (mV) | APD50 (ms) | APD90 (ms) | Vmax (V/s) | Maximum Diastolic Potential (mV) | |
|---|---|---|---|---|---|---|
| CONTROL | 61.9 ± 4.2 (n = 14) | 105.7 ± 4.4 (n = 14) | 380.6 ± 31.9 (n = 14) | 456.2 ± 29.4 (n = 14) | 26.9 ± 2.8 (n = 14) | −72.5 ± 1.6 (n = 14) |
| MMRL1215 | 48.7 ± 4.0 (n = 16) * | 102 ± 2.2 (n = 16) | 206.6 ± 33.8 (n = 16) * | 267.5 ± 38.6 (n = 16) * | 22.6 ± 3.3 (n = 16) | −71.9 ± 1.4 (n = 16) |
| Spontaneous Cycle Length (bpm) | AP Amplitude (mV) | APD50 (ms) | APD90 (ms) | Vmax V/s) | Maximum Diastolic Potential (mV) | |
|---|---|---|---|---|---|---|
| MMRL1215 –glybenclamide | 43.3 ± 4.8 (n = 6) | 100.2 ± 5.0 (n = 6) | 240.9 ± 42.8 (n = 6) | 295.7 ± 44.4 (n = 6) | 33.4 ± 10.9 (n = 6) | −67.1 ± 2.8 (n = 6) |
| MMRL1215 +glybenclamide | 54.3 ± 12.3 (n = 6) | 91.3 ± 10.4 (n = 6) | 262.5 ± 33.8 (n = 6) | 403.5 ± 50.6 (n = 6) * | 24.8 ± 10.5 (n = 6) | −61.9 ± 5.7 (n = 6) |
| WT –glybenclamide | 56.2 ± 5.7 (n = 10) | 105.4 ± 1.7 (n = 10) | 354.69 ± 31.9 (n = 10) | 464.7 ± 34.3 (n = 10) | 29.4 ± 3.4 (n = 10) | −67.1 ± 2.8 (n = 10) |
| WT +glybenclamide | 51.4 ± 9.1 (n = 10) | 105.5 ± 3.5 (n = 10) | 316.3 ± 35.0 (n = 10) | 424.2 ± 34.3 (n = 10) | 31.0 ± 4.8 (n = 10) | −61.9 ± 5.7 (n = 10) |
| Spontaneous Cycle Length | Number with Regular Spontaneous Cycle Length | % with Regular Spontaneous Cycle Length | (F-Fo)/Fo | |
|---|---|---|---|---|
| CONTROL | 1410.1 ± 42.3 ms (n = 77) | 77/78 | 98.7% | 5.76 ± 0.36 (n = 77) |
| MMRL 1215 | 1460.0 ± 58.0 ms (n = 82) | 82/86 | 95.3% | 4.81 ± 0.36 (n = 82) * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Treat, J.A.; Pfeiffer, R.; Barajas-Martinez, H.; Goodrow, R.J.; Bot, C.; Haedo, R.J.; Knox, R.; Cordeiro, J.M. Overlap Arrhythmia Syndromes Resulting from Multiple Genetic Variations Studied in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 7108. https://doi.org/10.3390/ijms22137108
Treat JA, Pfeiffer R, Barajas-Martinez H, Goodrow RJ, Bot C, Haedo RJ, Knox R, Cordeiro JM. Overlap Arrhythmia Syndromes Resulting from Multiple Genetic Variations Studied in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. International Journal of Molecular Sciences. 2021; 22(13):7108. https://doi.org/10.3390/ijms22137108
Chicago/Turabian StyleTreat, Jacqueline A., Ryan Pfeiffer, Hector Barajas-Martinez, Robert J. Goodrow, Corina Bot, Rodolfo J. Haedo, Ronald Knox, and Jonathan M. Cordeiro. 2021. "Overlap Arrhythmia Syndromes Resulting from Multiple Genetic Variations Studied in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes" International Journal of Molecular Sciences 22, no. 13: 7108. https://doi.org/10.3390/ijms22137108
APA StyleTreat, J. A., Pfeiffer, R., Barajas-Martinez, H., Goodrow, R. J., Bot, C., Haedo, R. J., Knox, R., & Cordeiro, J. M. (2021). Overlap Arrhythmia Syndromes Resulting from Multiple Genetic Variations Studied in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. International Journal of Molecular Sciences, 22(13), 7108. https://doi.org/10.3390/ijms22137108