
From Affinity to Proximity Techniques to Investigate Protein Complexes in Plants


Abstract
:1. Introduction
2. Affinity Purification Mass Spectrometry in Plants
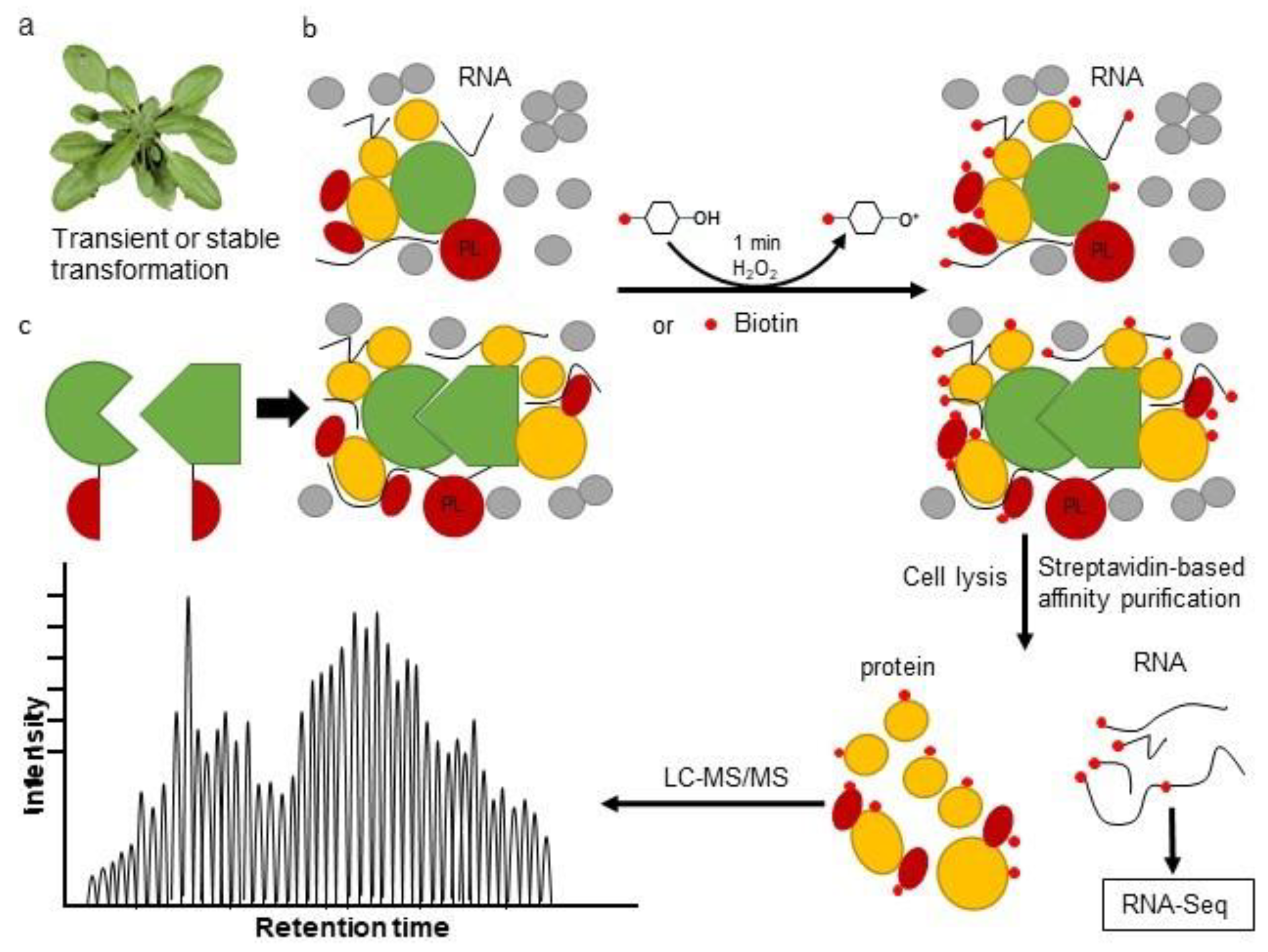
3. The Proximity Labeling Method
4. Combining Proximity Labeling and Affinity Purification-Mass Spectrometry
5. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.; Fernie, A.R. Stable and temporary enzyme complexes and metabolons involved in energy and redox metabolism. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]
- Chae, L.; Lee, I.; Shin, J.; Rhee, S.Y. Towards understanding how molecular networks evolve in plants. Curr. Opin. Plant Biol. 2012, 15, 177–184. [Google Scholar] [CrossRef]
- Lampugnani, E.R.; Wink, R.H.; Persson, S.; Somssich, M. The toolbox to study protein–protein interactions in plants. Crit. Rev. Plant Sci. 2018, 37, 308–334. [Google Scholar] [CrossRef]
- Struk, S.; Jacobs, A.; Sánchez Martín-Fontecha, E.; Gevaert, K.; Cubas, P.; Goormachtig, S. Exploring the protein–protein interaction landscape in plants. Plant Cell Environ. 2019, 42, 387–409. [Google Scholar] [CrossRef]
- Van Leene, J.; Eeckhout, D.; Cannoot, B.; De Winne, N.; Persiau, G.; Van De Slijke, E.; Vercruysse, L.; Dedecker, M.; Verkest, A.; Vandepoele, K.; et al. An improved toolbox to unravel the plant cellular machinery by tandem affinity purification of arabidopsis protein complexes. Nat. Protoc. 2015, 10, 169–187. [Google Scholar] [CrossRef]
- Dunham, W.H.; Mullin, M.; Gingras, A.C. Affinity-purification coupled to mass spectrometry: Basic principles and strategies. Proteomics 2012, 12, 1576–1590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, P.; Yuan, J.S. Plant protein-protein interaction network and interactome. Curr. Genom. 2010, 11, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, S.; Lau, V.; Song, R.; Ierullo, M.; Esteban, E.; Wu, Y.; Sivieng, T.; Nahal, H.; Gaudinier, A.; Pasha, A. Proteome-wide, structure-based prediction of protein-protein interactions/new molecular interactions viewer. Plant Physiol. 2019, 179, 1893–1907. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Niu, C.; Cao, J.; Ni, D.A.; Chu, Z. In silico-prediction of protein–protein interactions network about mapks and pp2cs reveals a novel docking site variants in brachypodium distachyon. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- De Bodt, S.; Proost, S.; Vandepoele, K.; Rouzé, P.; Van de Peer, Y. Predicting protein-protein interactions in arabidopsis thaliana through integration of orthology, gene ontology and co-expression. BMC Genom. 2009, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Barnouin, K. Two-dimensional gel electrophoresis for analysis of protein complexes. Protein-Protein Interact. 2004, 261, 479–497. [Google Scholar]
- Zhang, Y.; Fernie, A.R. On the detection and functional significance of the protein–protein interactions of mitochondrial transport proteins. Biomolecules 2020, 10, 1107. [Google Scholar] [CrossRef]
- Zhang, Y.; Natale, R.; Domingues, A.P.J.; Toleco, M.R.; Siemiatkowska, B.; Fàbregas, N.; Fernie, A.R. Rapid identification of protein-protein interactions in plants. Curr. Protoc. Plant Biol. 2019, 4, e20099. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wen, Z.; Zhang, D.; Li, Z.; Li, D.; Nagalakshmi, U.; Dinesh-Kumar, S.P.; Zhang, Y. Proximity labeling: An emerging tool for probing in planta molecular interactions. Plant Commun. 2020, 100137. [Google Scholar] [CrossRef]
- Mair, A.; Xu, S.-L.; Branon, T.C.; Ting, A.Y.; Bergmann, D.C. Proximity labeling of protein complexes and cell-type-specific organellar proteomes in arabidopsis enabled by turboid. Elife 2019, 8, e47864. [Google Scholar] [CrossRef]
- Liu, X.; Salokas, K.; Weldatsadik, R.G.; Gawriyski, L.; Varjosalo, M. Combined proximity labeling and affinity purification− mass spectrometry workflow for mapping and visualizing protein interaction networks. Nat. Protoc. 2020, 15, 3182–3211. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Sacco, R.; Stukalov, A.; Van Drogen, A.; Planyavsky, M.; Hauri, S.; Aebersold, R.; Bennett, K.L.; Colinge, J.; Gstaiger, M. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized ap-ms. Nat. Methods 2013, 10, 307. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Birendra, K.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461. [Google Scholar] [CrossRef] [Green Version]
- Kwak, C.; Shin, S.; Park, J.-S.; Jung, M.; Nhung, T.T.M.; Kang, M.-G.; Lee, C.; Kwon, T.-H.; Park, S.K.; Mun, J.Y. Contact-id, a tool for profiling organelle contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation. Proc. Natl. Acad. Sci. USA 2020, 117, 12109–12120. [Google Scholar] [CrossRef]
- Bauer, A.; Kuster, B. Affinity purification-mass spectrometry. Eur. J. Biochem. 2003, 270, 570–578. [Google Scholar] [CrossRef]
- Li, Y. The tandem affinity purification technology: An overview. Biotechnol. Lett. 2011, 33, 1487–1499. [Google Scholar] [CrossRef]
- Rohila, J.S.; Chen, M.; Cerny, R.; Fromm, M.E. Improved tandem affinity purification tag and methods for isolation of protein heterocomplexes from plants. Plant J. 2004, 38, 172–181. [Google Scholar] [CrossRef]
- Bontinck, M.; Van Leene, J.; Gadeyne, A.; De Rybel, B.; Eeckhout, D.; Nelissen, H.; De Jaeger, G. Recent trends in plant protein complex analysis in a developmental context. Front. Plant Sci. 2018, 9, 640. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.-C.; Wong, S.-L. Structure-guided design of an engineered streptavidin with reusability to purify streptavidin-binding peptide tagged proteins or biotinylated proteins. PLoS ONE 2013, 8, e69530. [Google Scholar] [CrossRef] [Green Version]
- Née, G.; Tilak, P.; Finkemeier, I. A versatile workflow for the identification of protein–protein interactions using gfp-trap beads and mass spectrometry-based label-free quantification. In Plant Proteomics; Springer: Heidelberg, Germany, 2020; pp. 257–271. [Google Scholar]
- Besbrugge, N.; Van Leene, J.; Eeckhout, D.; Cannoot, B.; Kulkarni, S.R.; De Winne, N.; Persiau, G.; Van De Slijke, E.; Bontinck, M.; Aesaert, S. Gsyellow, a multifaceted tag for functional protein analysis in monocot and dicot plants. Plant Physiol. 2018, 177, 447–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.G.; Skerra, A. The strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007, 2, 1528. [Google Scholar] [CrossRef]
- Rubio, V.; Shen, Y.; Saijo, Y.; Liu, Y.; Gusmaroli, G.; Dinesh-Kumar, S.P.; Deng, X.W. An alternative tandem affinity purification strategy applied to arabidopsis protein complex isolation. Plant J. 2005, 41, 767–778. [Google Scholar] [CrossRef]
- Struk, S.; Braem, L.; Walton, A.; De Keyser, A.; Boyer, F.-D.; Persiau, G.; De Jaeger, G.; Gevaert, K.; Goormachtig, S. Quantitative tandem affinity purification, an effective tool to investigate protein complex composition in plant hormone signaling: Strigolactones in the spotlight. Front. Plant Sci. 2018, 9, 528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sampathkumar, A.; Kerber, S.M.; Swart, C.; Hille, C.; Seerangan, K.; Graf, A.; Sweetlove, L.; Fernie, A.R. A moonlighting role for enzymes of glycolysis in the co-localization of mitochondria and chloroplasts. Nat. Commun. 2020, 11, 4509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Beard, K.F.M.; Swart, C.; Bergmann, S.; Krahnert, I.; Nikoloski, Z.; Graf, A.; Ratcliffe, R.G.; Sweetlove, L.J.; Fernie, A.R.; et al. Protein-protein interactions and metabolite channelling in the plant tricarboxylic acid cycle. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y. The crapome: A contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 2013, 10, 730. [Google Scholar] [CrossRef] [Green Version]
- Nesvizhskii, A.I. Computational and informatics strategies for identification of specific protein interaction partners in affinity purification mass spectrometry experiments. Proteomics 2012, 12, 1639–1655. [Google Scholar] [CrossRef] [Green Version]
- Bürckstümmer, T.; Bennett, K.L.; Preradovic, A.; Schütze, G.; Hantschel, O.; Superti-Furga, G.; Bauch, A. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat. Methods 2006, 3, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Batelli, G.; Verslues, P.E.; Agius, F.; Qiu, Q.; Fujii, H.; Pan, S.; Schumaker, K.S.; Grillo, S.; Zhu, J.-K. SOS2 promotes salt tolerance in part by interacting with the vacuolar h+-atpase and upregulating its transport activity. Mol. Cell. Biol. 2007, 27, 7781–7790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, I.F.; Curran, A.; Woolsey, R.; Quilici, D.; Cushman, J.C.; Mittler, R.; Harmon, A.; Harper, J.F. Proteomic profiling of tandem affinity purified 14-3-3 protein complexes in arabidopsis thaliana. Proteomics 2009, 9, 2967–2985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Fukao, Y.; Iwamoto, M.; Haraguchi, T.; Hara-Nishimura, I. Identification and characterization of nuclear pore complex components in arabidopsis thaliana. Plant Cell 2010, 22, 4084–4097. [Google Scholar] [CrossRef] [Green Version]
- Miteva, Y.V.; Budayeva, H.G.; Cristea, I.M. Proteomics-based methods for discovery, quantification, and validation of protein–protein interactions. Anal. Chem. 2013, 85, 749–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Giese, J.; Kerbler, S.M.-L.; Siemiatkowska, B.; de Souza, L.P.; Alpers, J.; Medeiros, D.B.; Hincha, D.K.; Daloso, D.M.; Stitt, M.; et al. Two mitochondrial phosphatases, pp2c63 and sal2, are required for posttranslational regulation of the tca cycle in arabidopsis. Mol. Plant 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Swart, C.; Alseekh, S.; Scossa, F.; Jiang, L.; Obata, T.; Graf, A.; Fernie, A.R. The extra-pathway interactome of the tca cycle: Expected and unexpected metabolic interactions. Plant Physiol. 2018, 177, 966–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, C.E.; Mocanu, V.; Mocanu, M.; Dicheva, N.; Warren, M.R. Mass spectrometry for post-translational modifications. Neuroproteomics 2010, 2010. [Google Scholar]
- Makowski, M.M.; Willems, E.; Jansen, P.W.; Vermeulen, M. Cross-linking immunoprecipitation-ms (xip-ms): Topological analysis of chromatin-associated protein complexes using single affinity purification. Mol. Cell. Proteom. 2016, 15, 854–865. [Google Scholar] [CrossRef] [Green Version]
- Bellati, J.; Champeyroux, C.; Hem, S.; Rofidal, V.; Krouk, G.; Maurel, C.; Santoni, V. Novel aquaporin regulatory mechanisms revealed by interactomics. Mol. Cell. Proteom. 2016, 15, 3473–3487. [Google Scholar] [CrossRef] [Green Version]
- Glatter, T.; Schittenhelm, R.B.; Rinner, O.; Roguska, K.; Wepf, A.; Jünger, M.A.; Köhler, K.; Jevtov, I.; Choi, H.; Schmidt, A. Modularity and hormone sensitivity of the drosophila melanogaster insulin receptor/target of rapamycin interaction proteome. Mol. Syst. Biol. 2011, 7, 547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, K.; Selbach, M. Quantitative affinity purification mass spectrometry: A versatile technology to study protein–protein interactions. Front. Genet. 2015, 6, 237. [Google Scholar] [CrossRef] [Green Version]
- Pagliuca, F.W.; Collins, M.O.; Lichawska, A.; Zegerman, P.; Choudhary, J.S.; Pines, J. Quantitative proteomics reveals the basis for the biochemical specificity of the cell-cycle machinery. Mol. Cell 2011, 43, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Mosbech, A.; Gibbs-Seymour, I.; Kagias, K.; Thorslund, T.; Beli, P.; Povlsen, L.; Nielsen, S.V.; Smedegaard, S.; Sedgwick, G.; Lukas, C. Dvc1 (c1orf124) is a DNA damage–targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nat. Struct. Mol. Biol. 2012, 19, 1084. [Google Scholar] [CrossRef]
- Satpathy, S.; Wagner, S.A.; Beli, P.; Gupta, R.; Kristiansen, T.A.; Malinova, D.; Francavilla, C.; Tolar, P.; Bishop, G.A.; Hostager, B.S. Systems-wide analysis of bcr signalosomes and downstream phosphorylation and ubiquitylation. Mol. Syst. Biol. 2015, 11, 810. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-Q.; Chien, C.-W.; Ai, L.-F.; Zhao, J.; Zhang, Z.; Li, K.H.; Burlingame, A.L.; Sun, Y.; Wang, Z.-Y. The arabidopsis b-box protein bzs1/bbx20 interacts with hy5 and mediates strigolactone regulation of photomorphogenesis. J. Genet. Genom. 2016, 43, 555–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thelen, J.J.; Peck, S.C. Quantitative proteomics in plants: Choices in abundance. Plant Cell 2007, 19, 3339–3346. [Google Scholar] [CrossRef] [Green Version]
- Ramisetty, S.R.; Washburn, M.P. Unraveling the dynamics of protein interactions with quantitative mass spectrometry. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 216–228. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed maxlfq. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Cox, J. The maxquant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301. [Google Scholar] [CrossRef]
- Smaczniak, C.; Li, N.; Boeren, S.; America, T.; Van Dongen, W.; Goerdayal, S.S.; De Vries, S.; Angenent, G.C.; Kaufmann, K. Proteomics-based identification of low-abundance signaling and regulatory protein complexes in native plant tissues. Nat. Protoc. 2012, 7, 2144. [Google Scholar] [CrossRef]
- Nelissen, H.; Eeckhout, D.; Demuynck, K.; Persiau, G.; Walton, A.; Van Bel, M.; Vervoort, M.; Candaele, J.; De Block, J.; Aesaert, S. Dynamic changes in angustifolia3 complex composition reveal a growth regulatory mechanism in the maize leaf. Plant Cell 2015, 27, 1605–1619. [Google Scholar] [CrossRef] [Green Version]
- Keilhauer, E.C.; Hein, M.Y.; Mann, M. Accurate protein complex retrieval by affinity enrichment mass spectrometry (ae-ms) rather than affinity purification mass spectrometry (ap-ms). Mol. Cell. Proteom. 2015, 14, 120–135. [Google Scholar] [CrossRef] [Green Version]
- Smaczniak, C.; Immink, R.G.; Muiño, J.M.; Blanvillain, R.; Busscher, M.; Busscher-Lange, J.; Dinh, Q.P.; Liu, S.; Westphal, A.H.; Boeren, S. Characterization of mads-domain transcription factor complexes in arabidopsis flower development. Proc. Natl. Acad. Sci. USA 2012, 109, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Wendrich, J.R.; Boeren, S.; Möller, B.K.; Weijers, D.; De Rybel, B. In vivo identification of plant protein complexes using ip-ms/ms. In Plant Hormones; Springer: Heidelberg, Germany, 2017; pp. 147–158. [Google Scholar]
- Mravec, J.; Petrášek, J.; Li, N.; Boeren, S.; Karlova, R.; Kitakura, S.; Pařezová, M.; Naramoto, S.; Nodzyński, T.; Dhonukshe, P. Cell plate restricted association of drp1a and pin proteins is required for cell polarity establishment in arabidopsis. Current Biology 2011, 21, 1055–1060. [Google Scholar] [CrossRef]
- De Rybel, B.; Möller, B.; Yoshida, S.; Grabowicz, I.; de Reuille, P.B.; Boeren, S.; Smith, R.S.; Borst, J.W.; Weijers, D. A bhlh complex controls embryonic vascular tissue establishment and indeterminate growth in arabidopsis. Dev. Cell 2013, 24, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.I.; Roux, K.J. Filling the void: Proximity-based labeling of proteins in living cells. Trends Cell Biol. 2016, 26, 804–817. [Google Scholar] [CrossRef] [Green Version]
- Kido, K.; Yamanaka, S.; Nakano, S.; Motani, K.; Shinohara, S.; Nozawa, A.; Kosako, H.; Ito, S.; Sawasaki, T. Airid, a novel proximity biotinylation enzyme, for analysis of protein–protein interactions. Elife 2020, 9, e54983. [Google Scholar] [CrossRef]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for bioid proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef]
- Rhee, H.-W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of apex2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with turboid. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef]
- Smith, F.D.; Omar, M.H.; Nygren, P.J.; Soughayer, J.; Hoshi, N.; Lau, H.-T.; Snyder, C.G.; Branon, T.C.; Ghosh, D.; Langeberg, L.K. Single nucleotide polymorphisms alter kinase anchoring and the subcellular targeting of a-kinase anchoring proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E11465–E11474. [Google Scholar] [CrossRef] [Green Version]
- Paek, J.; Kalocsay, M.; Staus, D.P.; Wingler, L.; Pascolutti, R.; Paulo, J.A.; Gygi, S.P.; Kruse, A.C. Multidimensional tracking of gpcr signaling via peroxidase-catalyzed proximity labeling. Cell 2017, 169, 338–349.e311. [Google Scholar] [CrossRef] [Green Version]
- Lobingier, B.T.; Hüttenhain, R.; Eichel, K.; Miller, K.B.; Ting, A.Y.; von Zastrow, M.; Krogan, N.J. An approach to spatiotemporally resolve protein interaction networks in living cells. Cell 2017, 169, 350–360.e312. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.M.T.; Kim, J.; Doan, T.T.; Lee, M.-W.; Lee, M. Apex proximity labeling as a versatile tool for biological research. Biochemistry 2019, 59, 260–269. [Google Scholar] [CrossRef]
- Varnaitė, R.; MacNeill, S.A. Meet the neighbors: Mapping local protein interactomes by proximity-dependent labeling with bioid. Proteomics 2016, 16, 2503–2518. [Google Scholar] [CrossRef] [Green Version]
- Béganton, B.; Solassol, I.; Mangé, A.; Solassol, J. Protein interactions study through proximity-labeling. Expert Rev. Proteom. 2019, 16, 717–726. [Google Scholar] [CrossRef]
- Dumont, A.-A.; Dumont, L.; Berthiaume, J.; Auger-Messier, M. P38α mapk proximity assay reveals a regulatory mechanism of alternative splicing in cardiomyocytes. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 118557. [Google Scholar] [CrossRef]
- Liu, G.; Papa, A.; Katchman, A.N.; Zakharov, S.I.; Roybal, D.; Hennessey, J.A.; Kushner, J.; Yang, L.; Chen, B.-X.; Kushnir, A. Mechanism of adrenergic ca v 1.2 stimulation revealed by proximity proteomics. Nature 2020, 577, 695–700. [Google Scholar] [CrossRef]
- Arora, D.; Abel, N.B.; Liu, C.; Van Damme, P.; Yperman, K.; Eeckhout, D.; Dai Vu, L.; Wang, J.; Tornkvist, A.; Impens, F. Establishment of proximity-dependent biotinylation approaches in different plant model systems. Plant Cell 2020, 32, 3388–3407. [Google Scholar] [CrossRef]
- Kim, T.-W.; Park, C.H.; Hsu, C.-C.; Zhu, J.-Y.; Hsiao, Y.; Branon, T.; Xu, S.-L.; Ting, A.Y.; Wang, Z.-Y. Application of turboid-mediated proximity labeling for mapping a gsk3 kinase signaling network in arabidopsis. bioRxiv 2019, 636324. [Google Scholar]
- Zhang, Y.; Song, G.; Lal, N.K.; Nagalakshmi, U.; Li, Y.; Zheng, W.; Huang, P.J.; Branon, T.C.; Ting, A.Y.; Walley, J.W.; et al. Turboid-based proximity labeling reveals that ubr7 is a regulator of n nlr immune receptor-mediated immunity. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hesketh, G.G.; Youn, J.-Y.; Samavarchi-Tehrani, P.; Raught, B.; Gingras, A.-C. Parallel exploration of interaction space by bioid and affinity purification coupled to mass spectrometry. In Proteomics; Comai, L.K.J., Mallick, P., Eds.; Springer: New York, NY, USA, 2017; Volume 1550, pp. 115–136. [Google Scholar]
- Lambert, J.-P.; Tucholska, M.; Go, C.; Knight, J.D.; Gingras, A.-C. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteom. 2015, 118, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, M.; Siemiatkowska, B.; Toleco, M.R.; Jing, Y.; Strotmann, V.; Zhang, J.; Stahl, Y.; Fernie, A.R. A highly efficient agrobacterium-mediated method for transient gene expression and functional studies in multiple plant species. Plant Commun. 2020, 1, 100028. [Google Scholar] [CrossRef]
- Choi, H.; Larsen, B.; Lin, Z.-Y.; Breitkreutz, A.; Mellacheruvu, D.; Fermin, D.; Qin, Z.S.; Tyers, M.; Gingras, A.-C.; Nesvizhskii, A.I. Saint: Probabilistic scoring of affinity purification–mass spectrometry data. Nat. Methods 2011, 8, 70–73. [Google Scholar] [CrossRef]
- Chen, C.; Hou, J.; Tanner, J.J.; Cheng, J. Bioinformatics methods for mass spectrometry-based proteomics data analysis. Int. J. Mol. Sci. 2020, 21, 2873. [Google Scholar] [CrossRef] [Green Version]
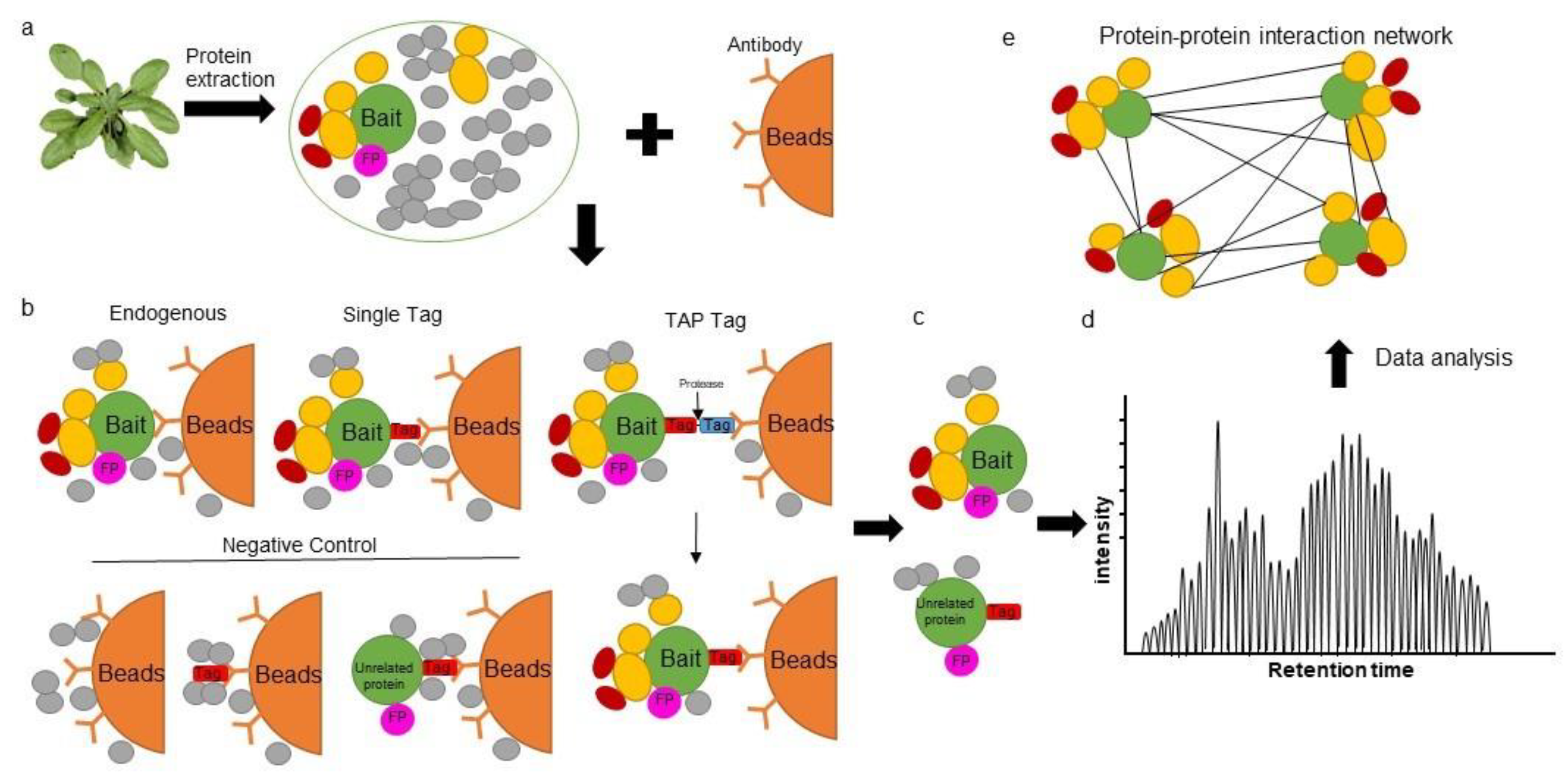

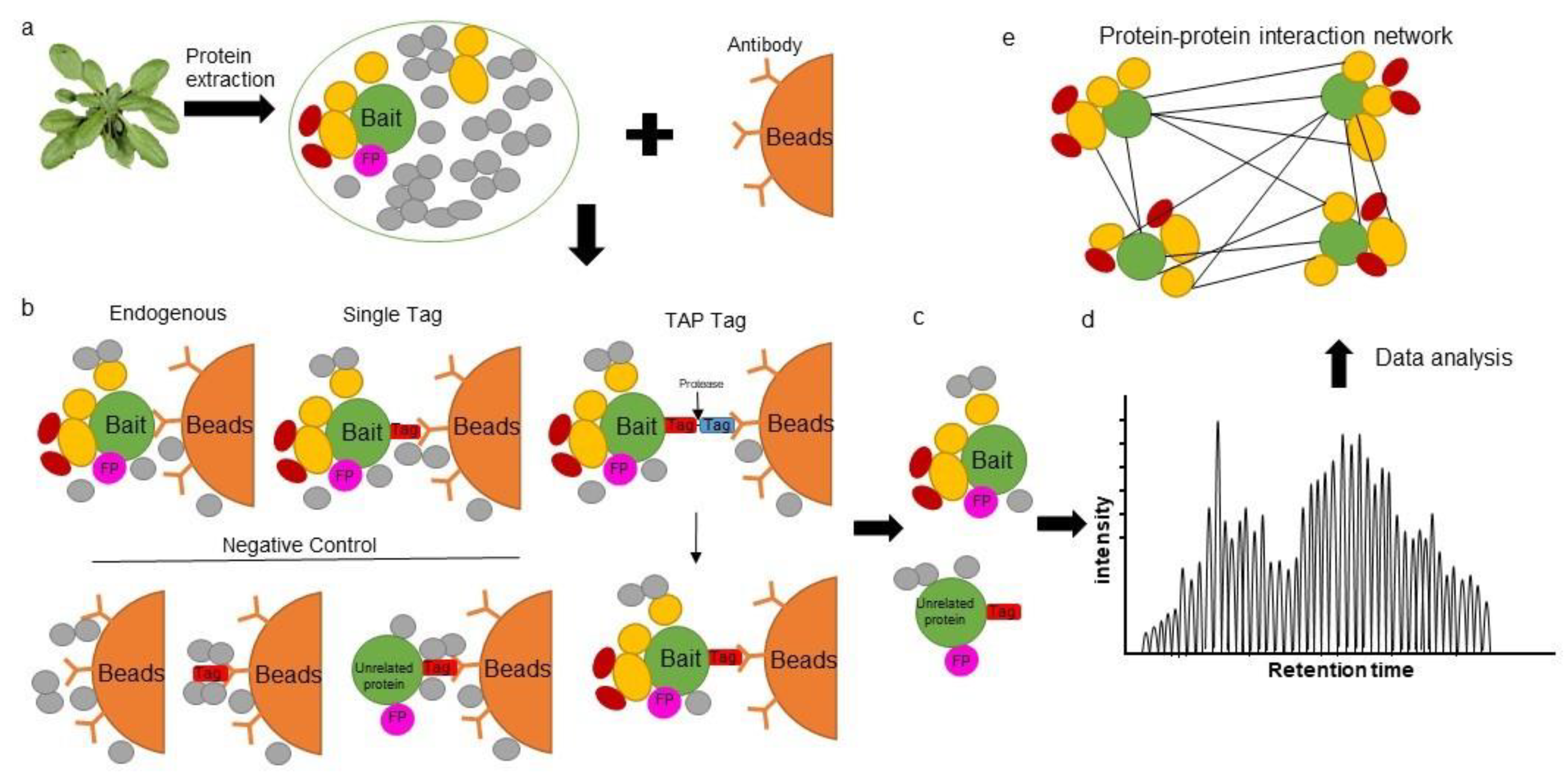{kind=link}
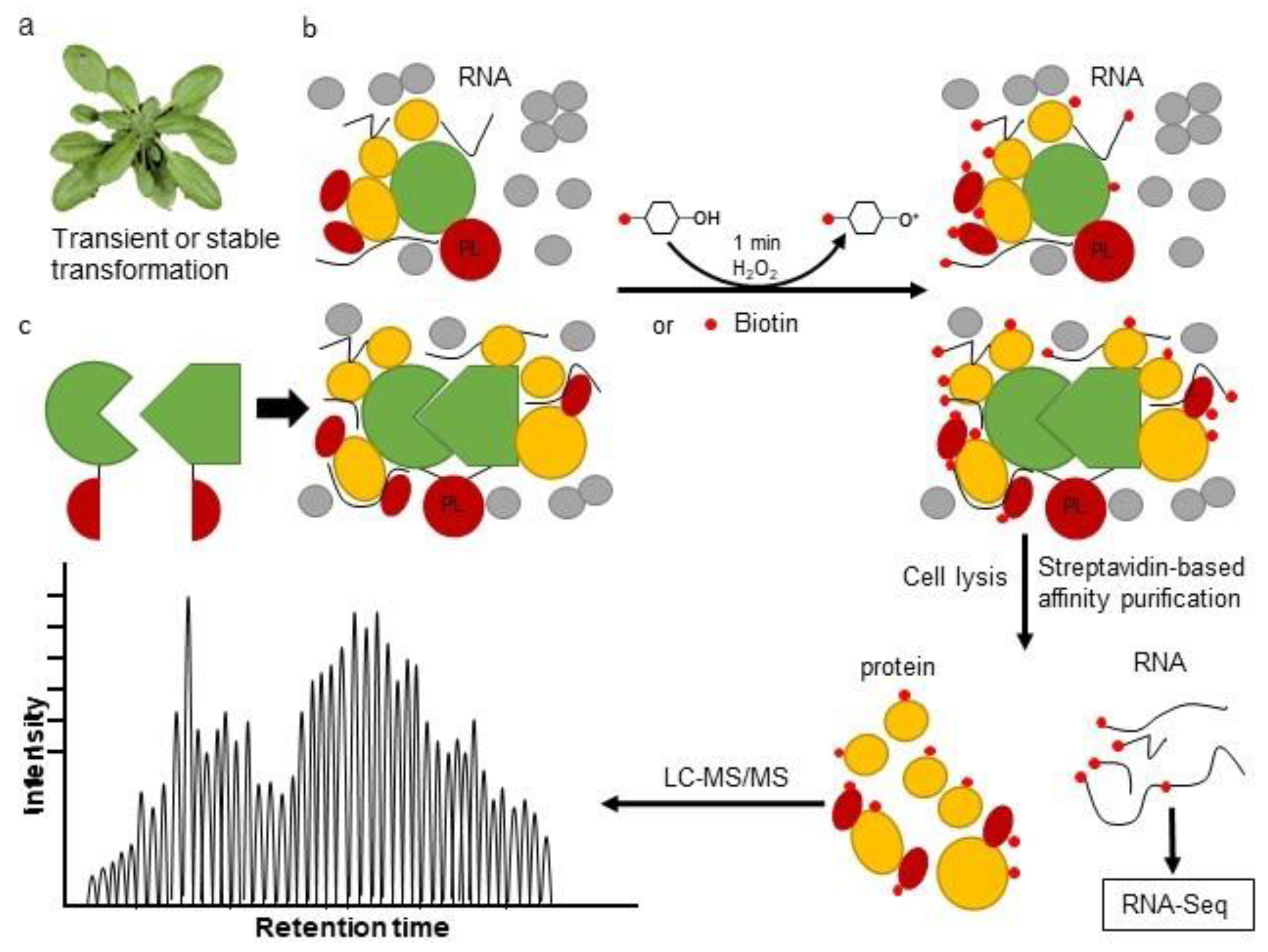{kind=link}
| Tag | Sequence/Size | Affinity Resin | Elution Conditions | Reference |
|---|---|---|---|---|
| TAPi tag | 45 kDa | Calmodulin binding peptide with two protein A domain | Protein A/low pH | [22,23] |
| Streptavidin binding peptide (SBP) | WSHPQFEK | Streptavidin | Desthiobiotin | [23,24] |
| GSyellow | 37 kDa | Streptavidin-binding peptide tag with citrine yellow fluorescent protein | Desthiobiotin/pH | [23,25,26] |
| Fluorescent protein (GFP, YFP) | 26.9 kDa | Anti-GFP | pH | [13,23,25,26] |
| GSrhino tag | 21.9 kDa | two IgG-binding domains of protein G and a SBP tag | Streptavidin elution buffer [5] | [5,23,27] |
| Alternative TAP (TAPa) | 26 kDa | 2 xIgG-BD with 6 XHis and 9 Xmyc | HR3C cleavage/Imidazole/low pH | [23,28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerbler, S.M.; Natale, R.; Fernie, A.R.; Zhang, Y. From Affinity to Proximity Techniques to Investigate Protein Complexes in Plants. Int. J. Mol. Sci. 2021, 22, 7101. https://doi.org/10.3390/ijms22137101
Kerbler SM, Natale R, Fernie AR, Zhang Y. From Affinity to Proximity Techniques to Investigate Protein Complexes in Plants. International Journal of Molecular Sciences. 2021; 22(13):7101. https://doi.org/10.3390/ijms22137101
Chicago/Turabian StyleKerbler, Sandra M., Roberto Natale, Alisdair R. Fernie, and Youjun Zhang. 2021. "From Affinity to Proximity Techniques to Investigate Protein Complexes in Plants" International Journal of Molecular Sciences 22, no. 13: 7101. https://doi.org/10.3390/ijms22137101
APA StyleKerbler, S. M., Natale, R., Fernie, A. R., & Zhang, Y. (2021). From Affinity to Proximity Techniques to Investigate Protein Complexes in Plants. International Journal of Molecular Sciences, 22(13), 7101. https://doi.org/10.3390/ijms22137101