
DNA G-Quadruplexes Contribute to CTCF Recruitment

 ,
,  ,
,  , , , , and
, , , , and 
Abstract
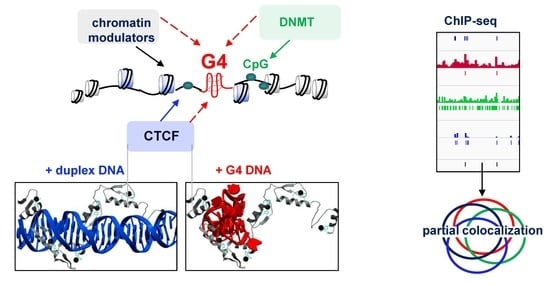
:
1. Introduction
2. Results
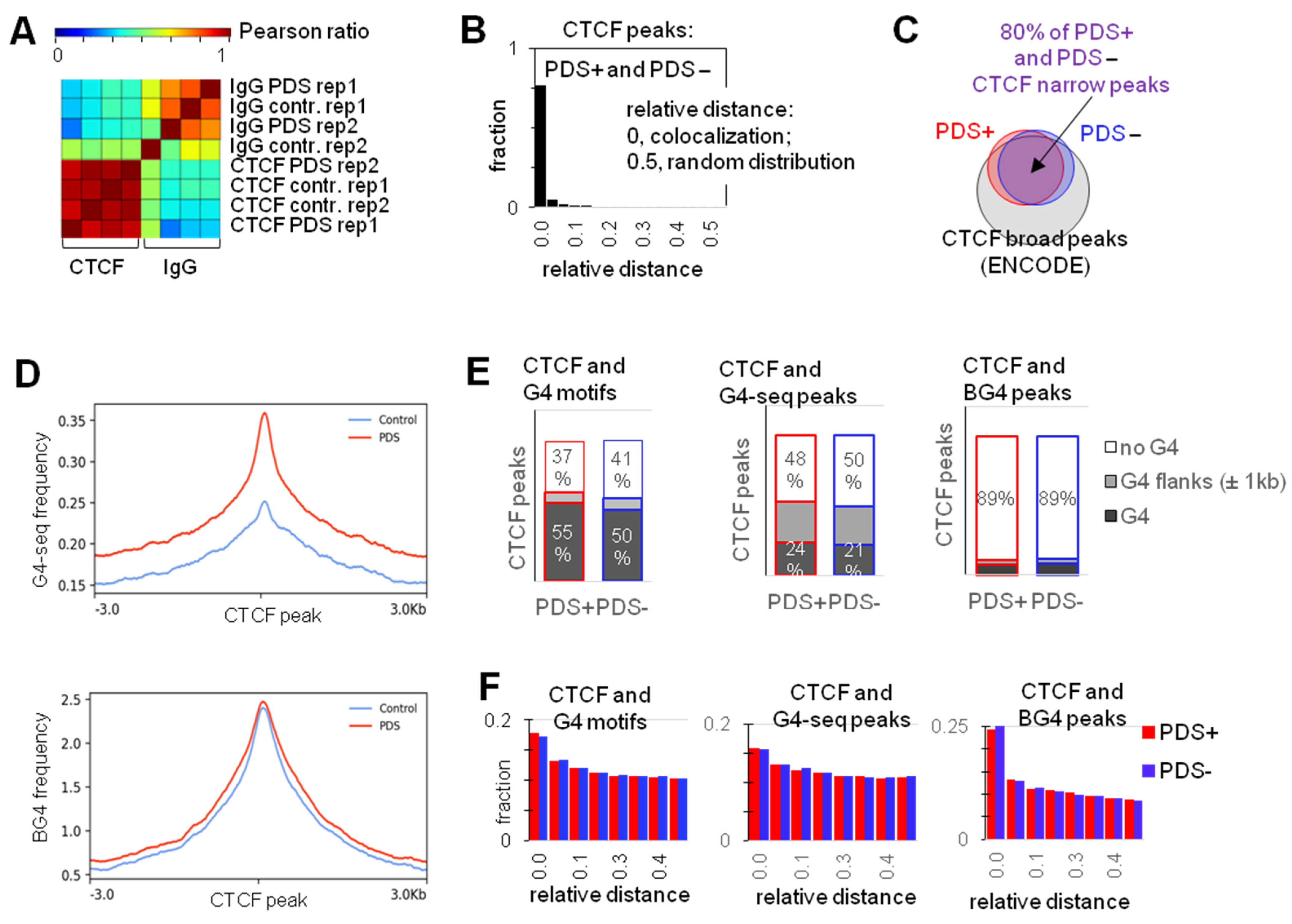
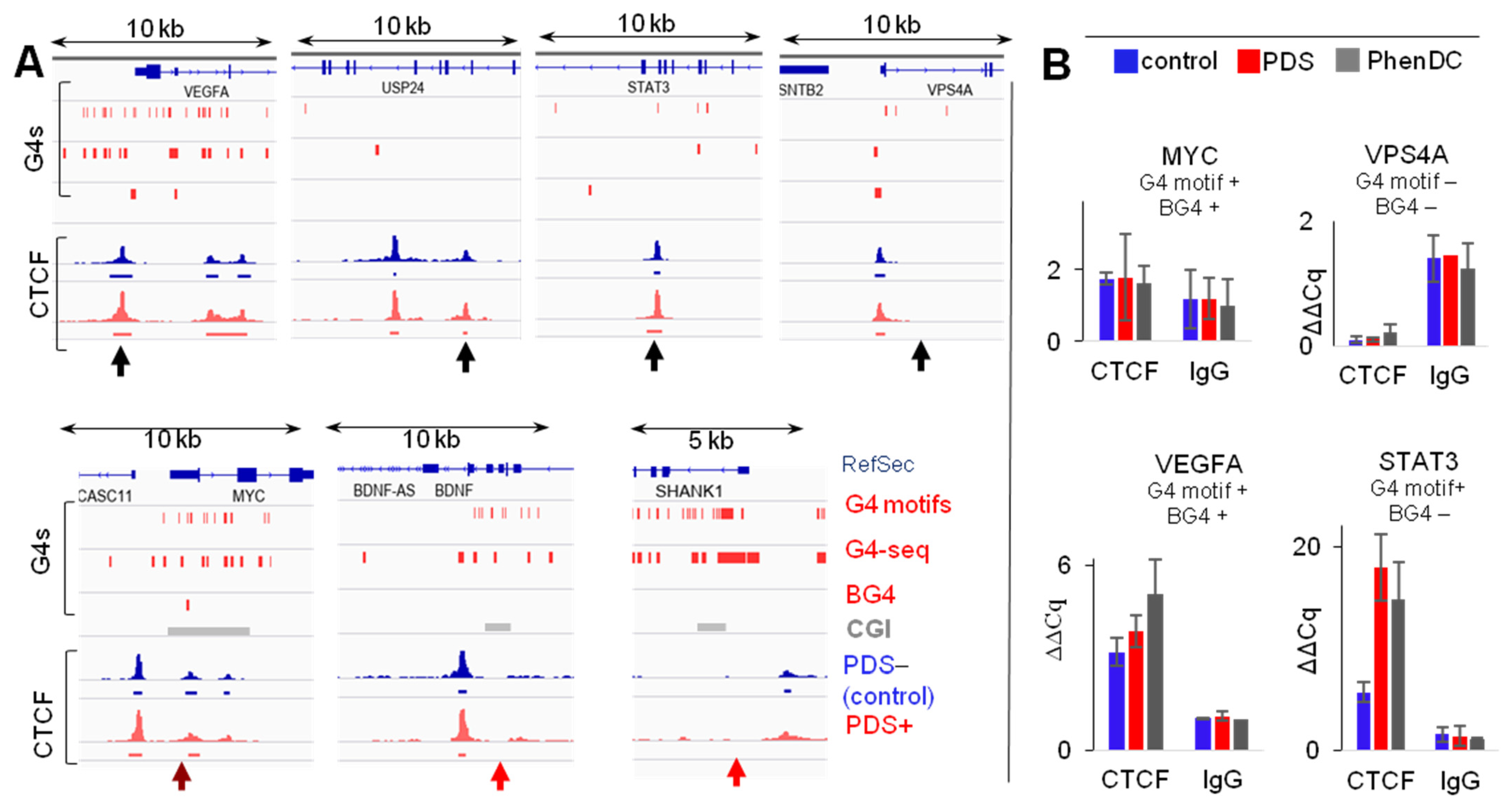
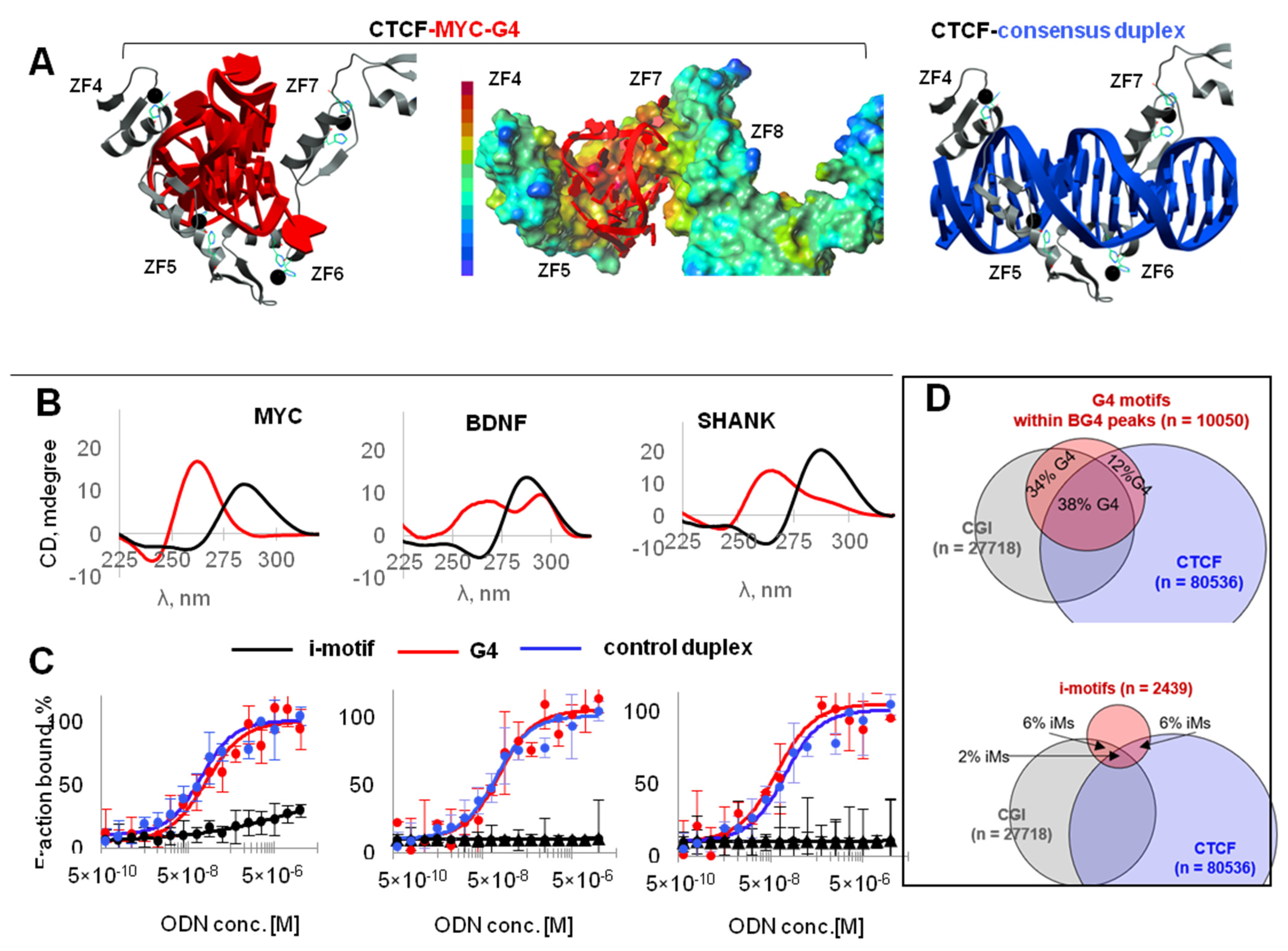
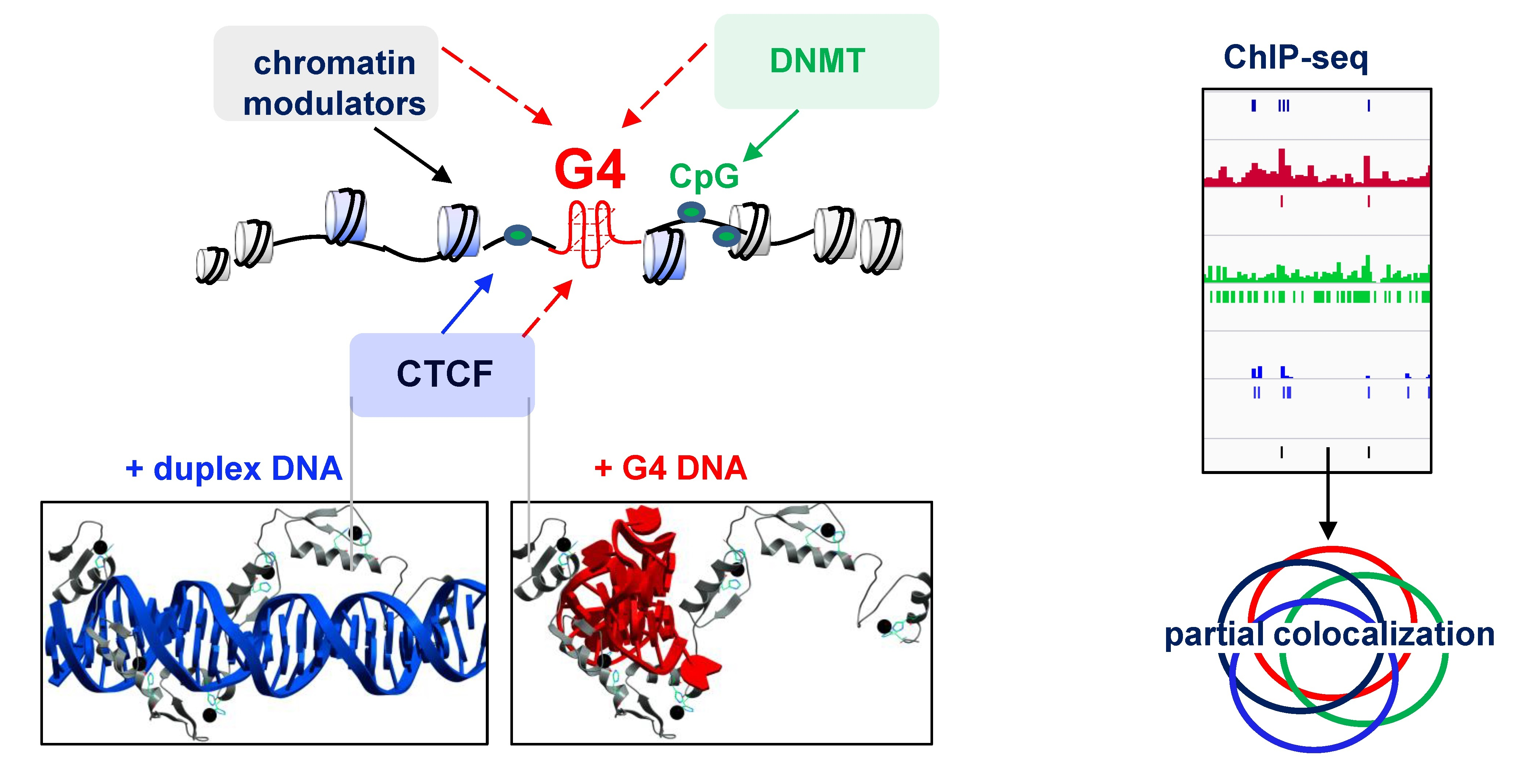
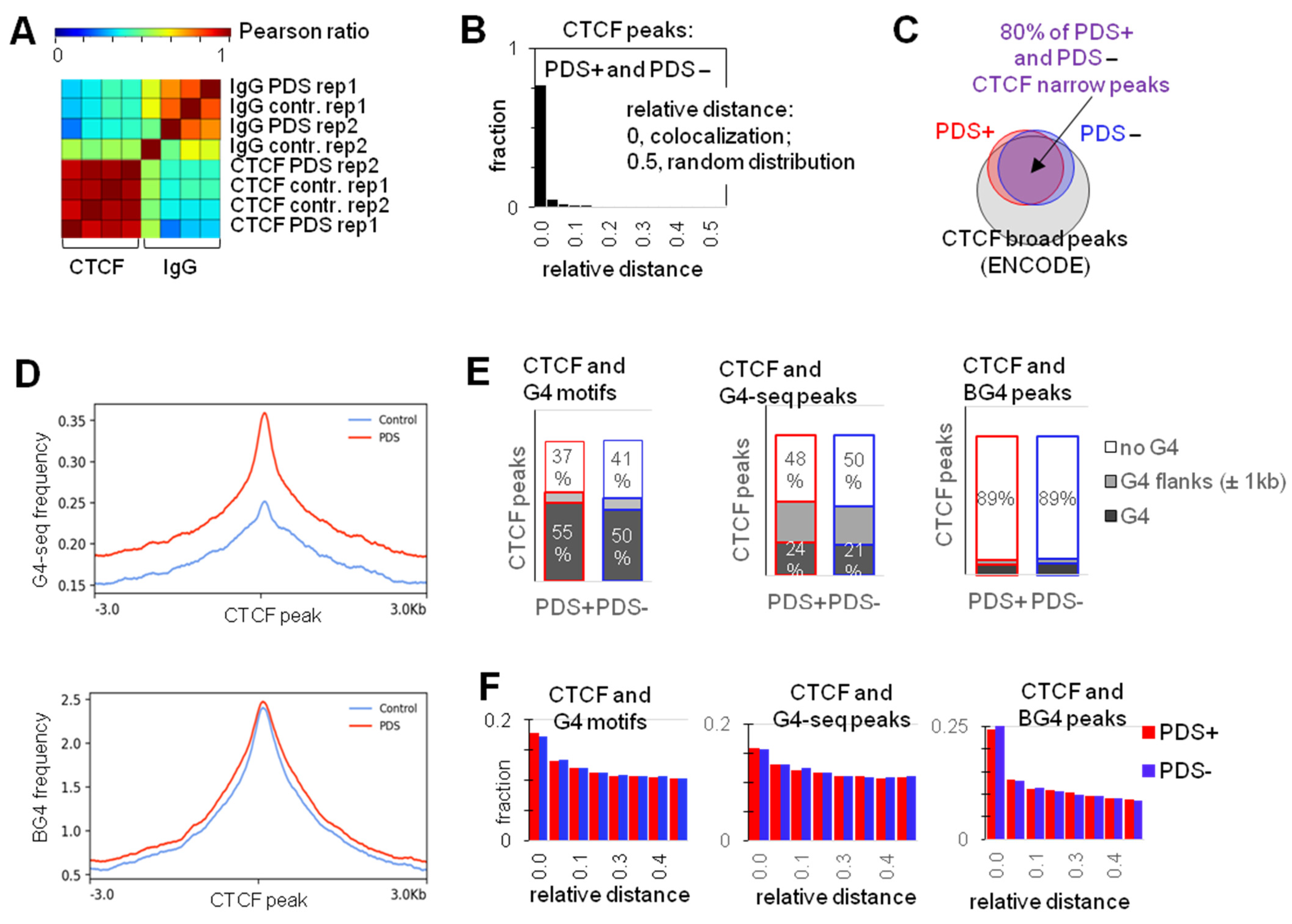
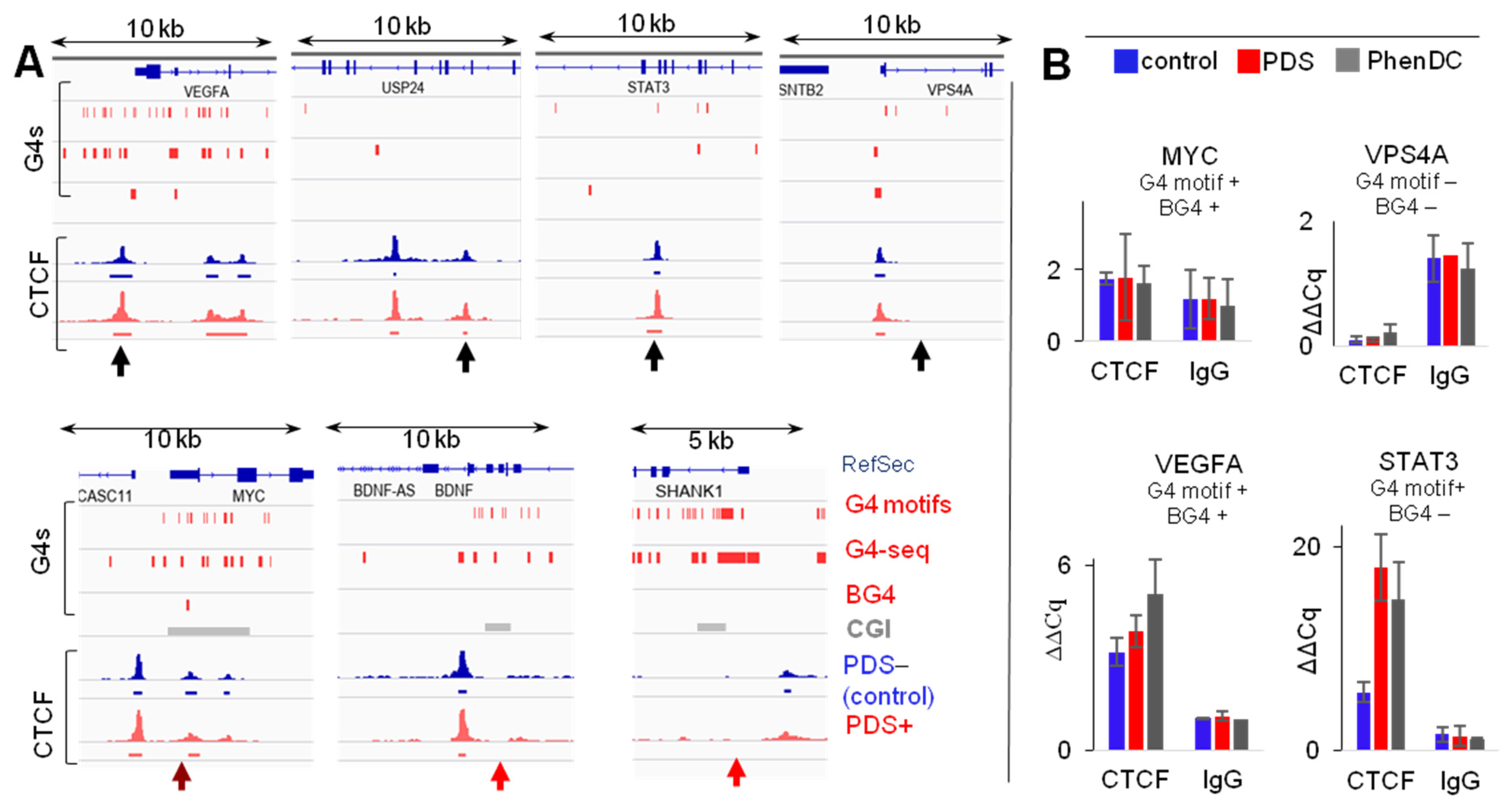
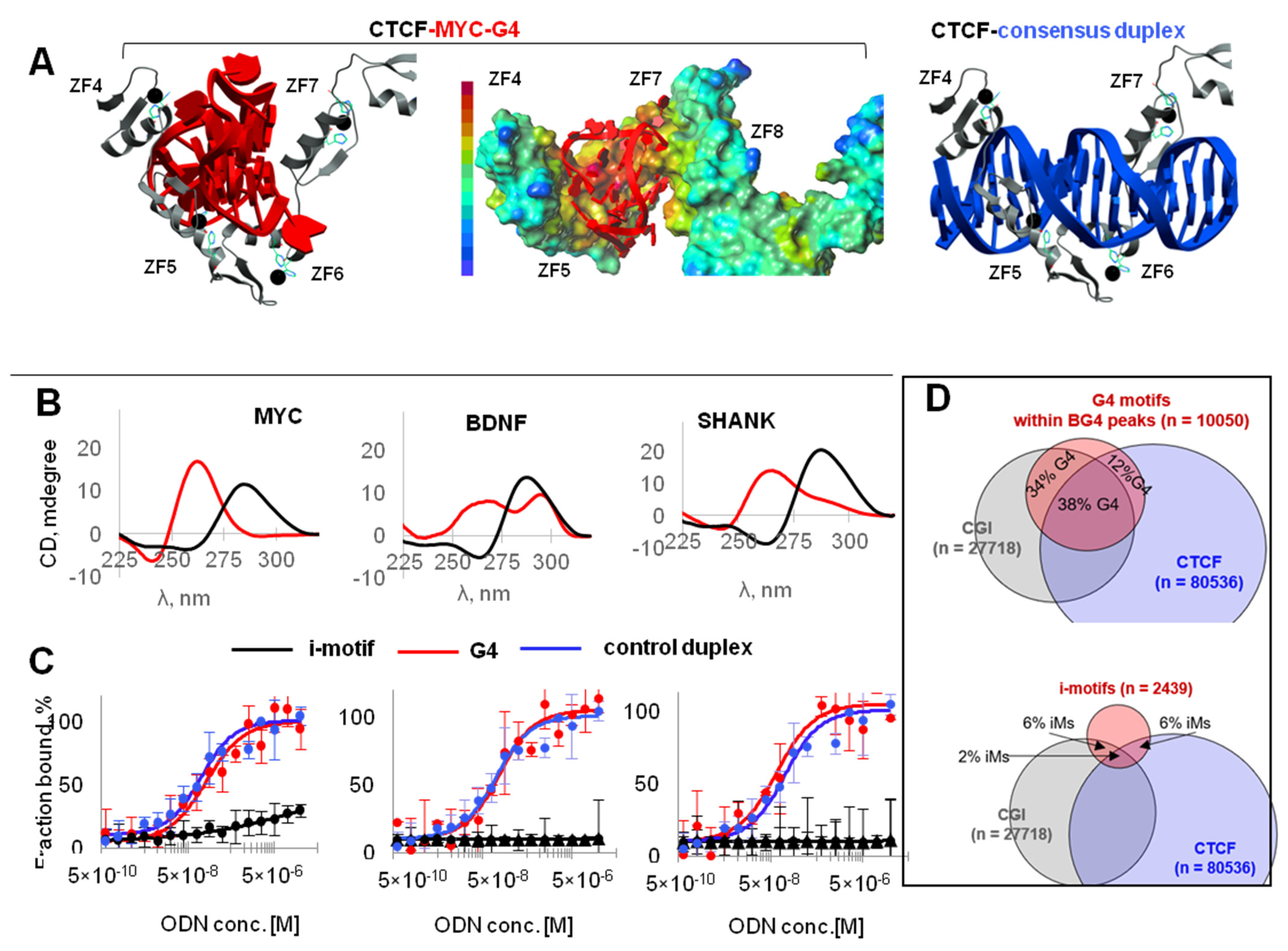
2.1. G4s Colocalize with CTCF-Bound Sites in the Genome and Interact with CTCF In Vitro
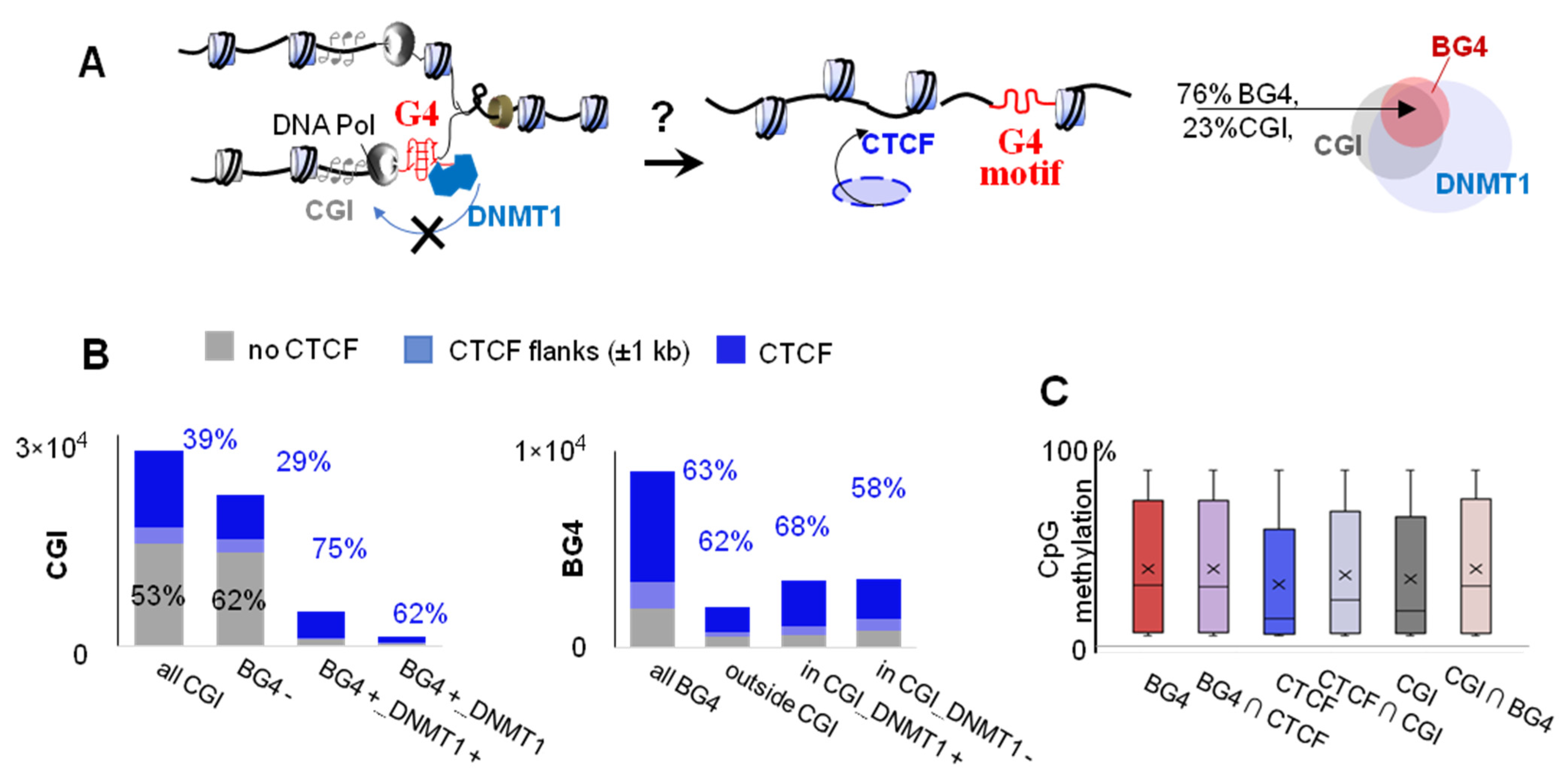
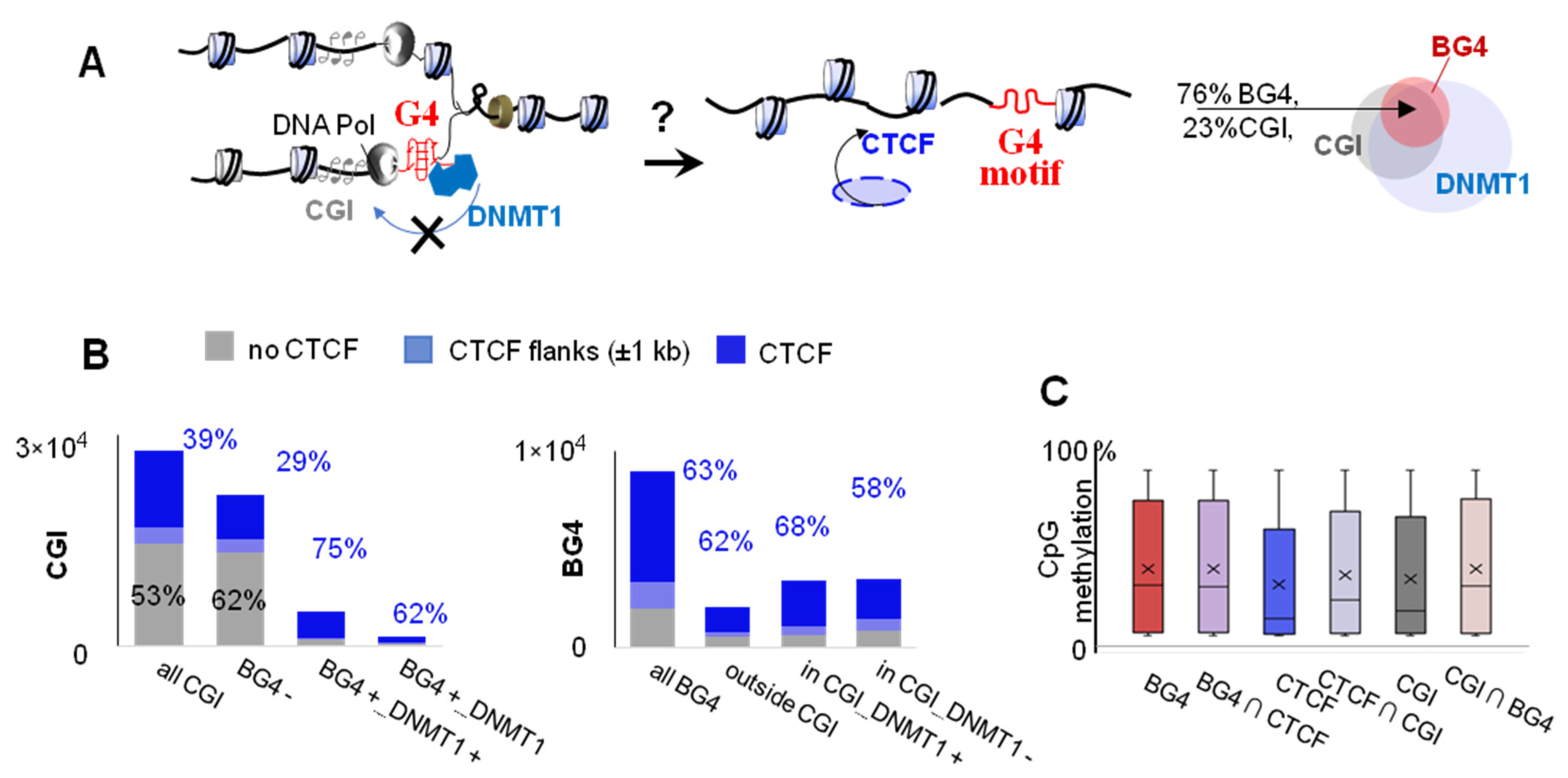
2.2. G4s Account for CTCF Occupancy in CGIs Irrespectively of DNMT1 Inhibition
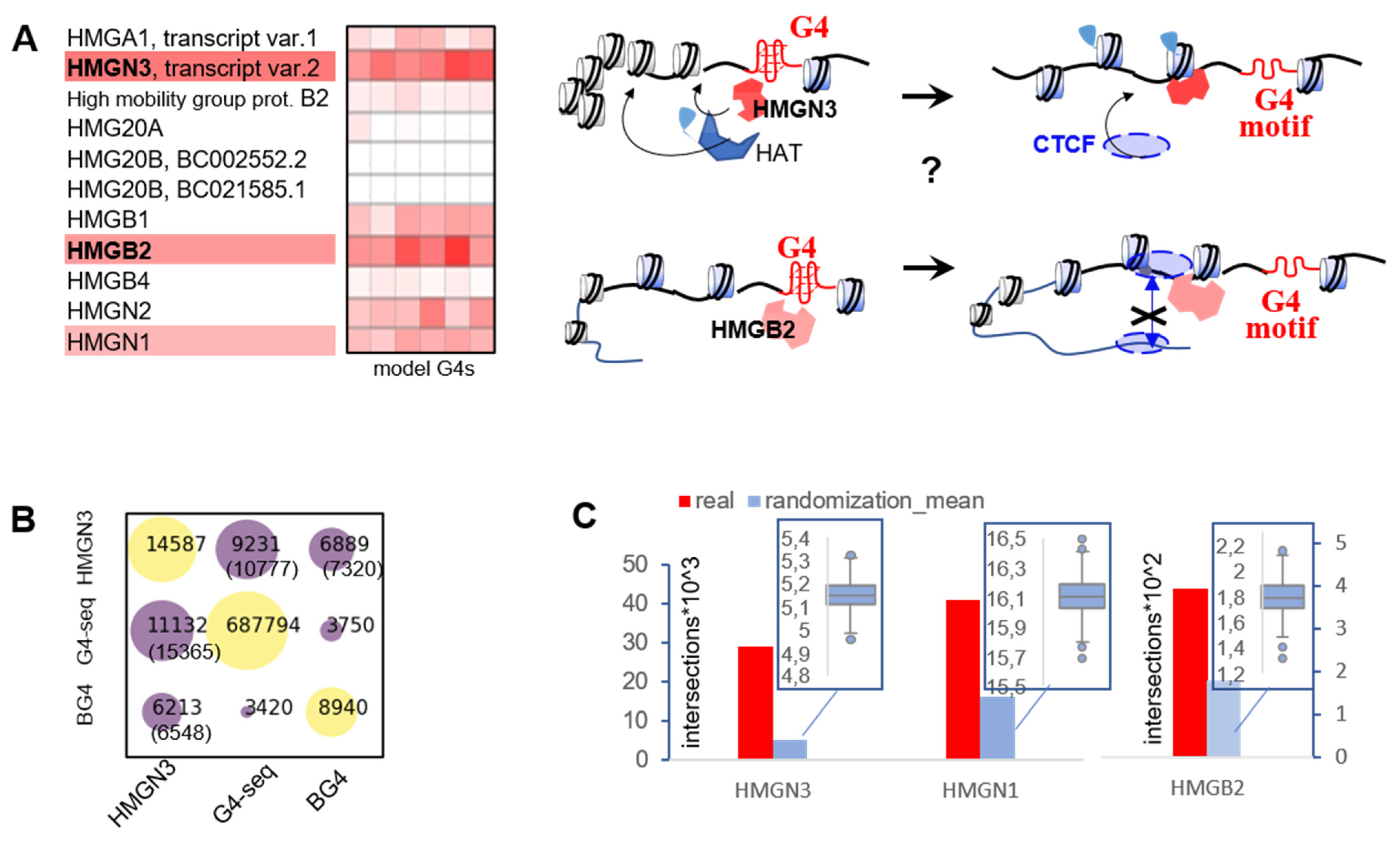
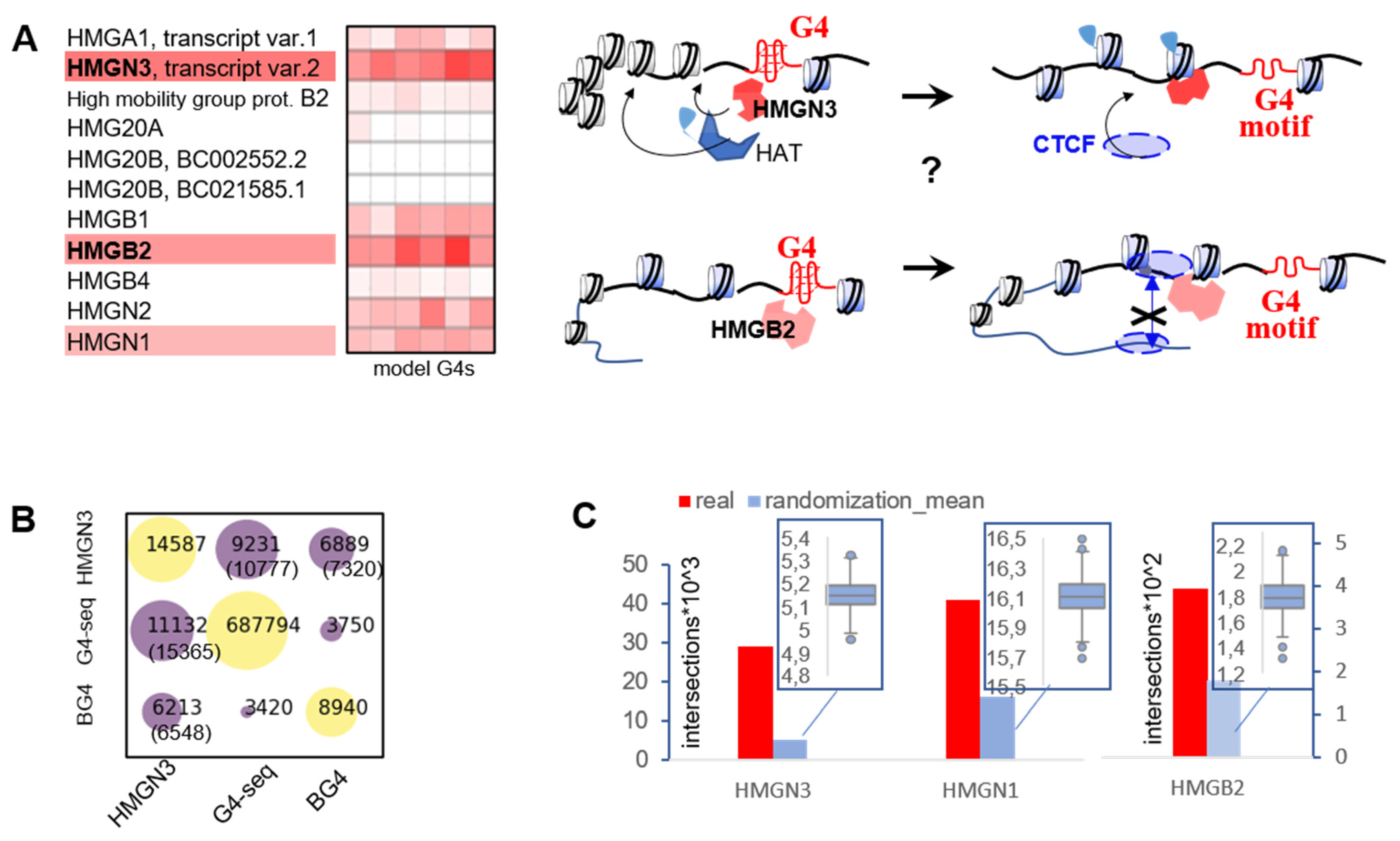
2.3. G4s May Recruit HMG Proteins That Prevent Chromatin Condensation and CTCF Aggregation
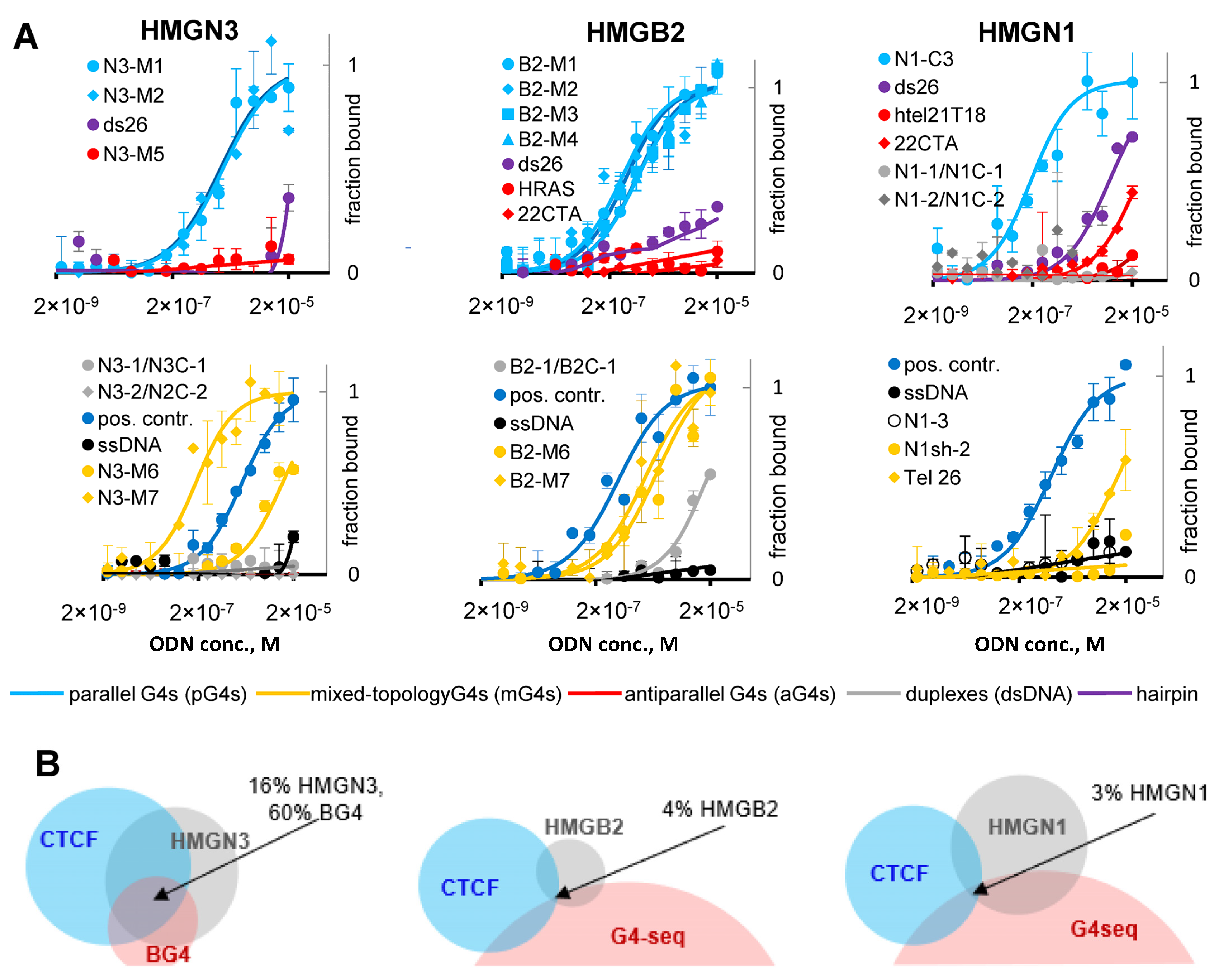
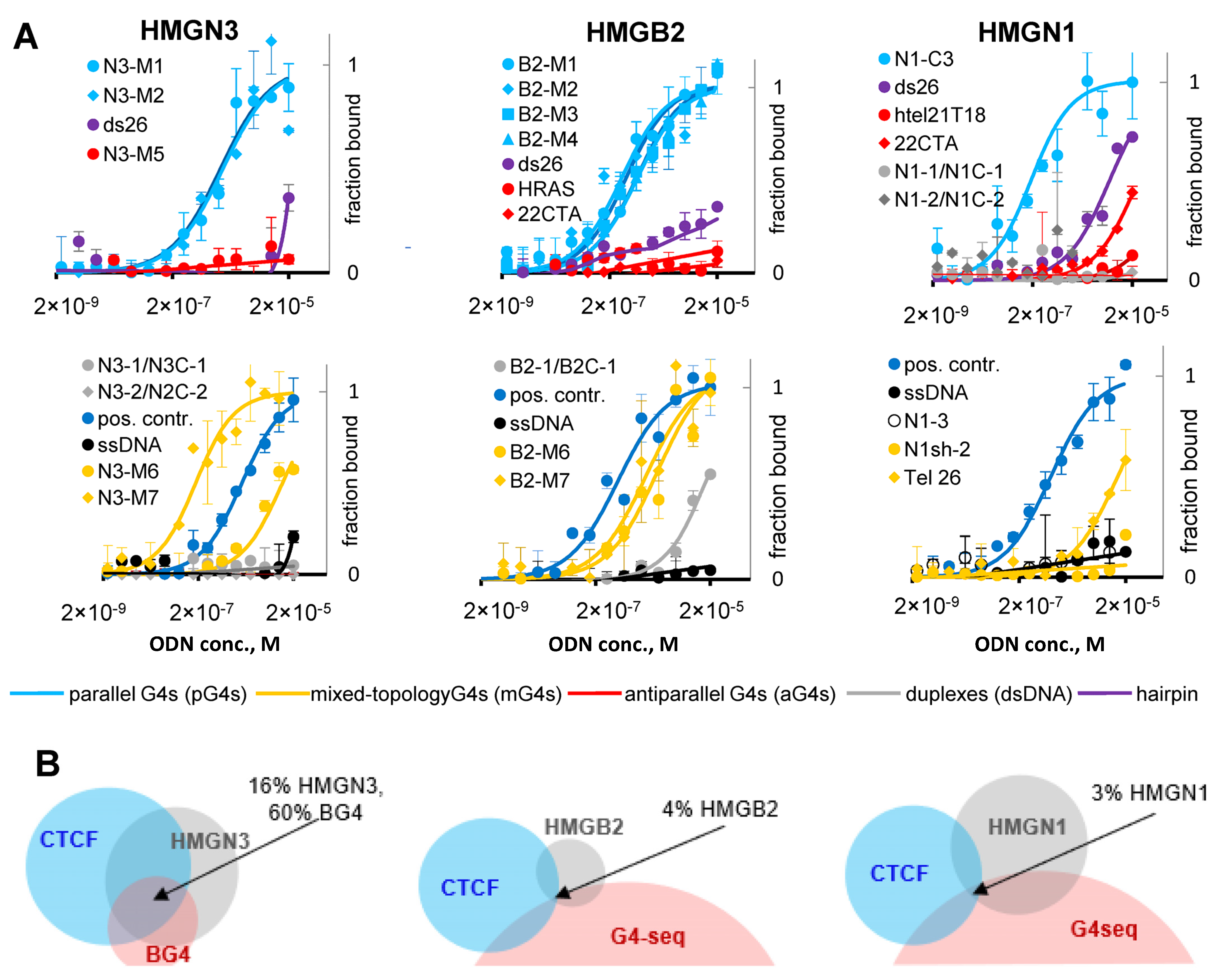
2.4. G4s Interact with HMG Proteins and Are Enriched in HMGN3- and CTCF-Bound Sites
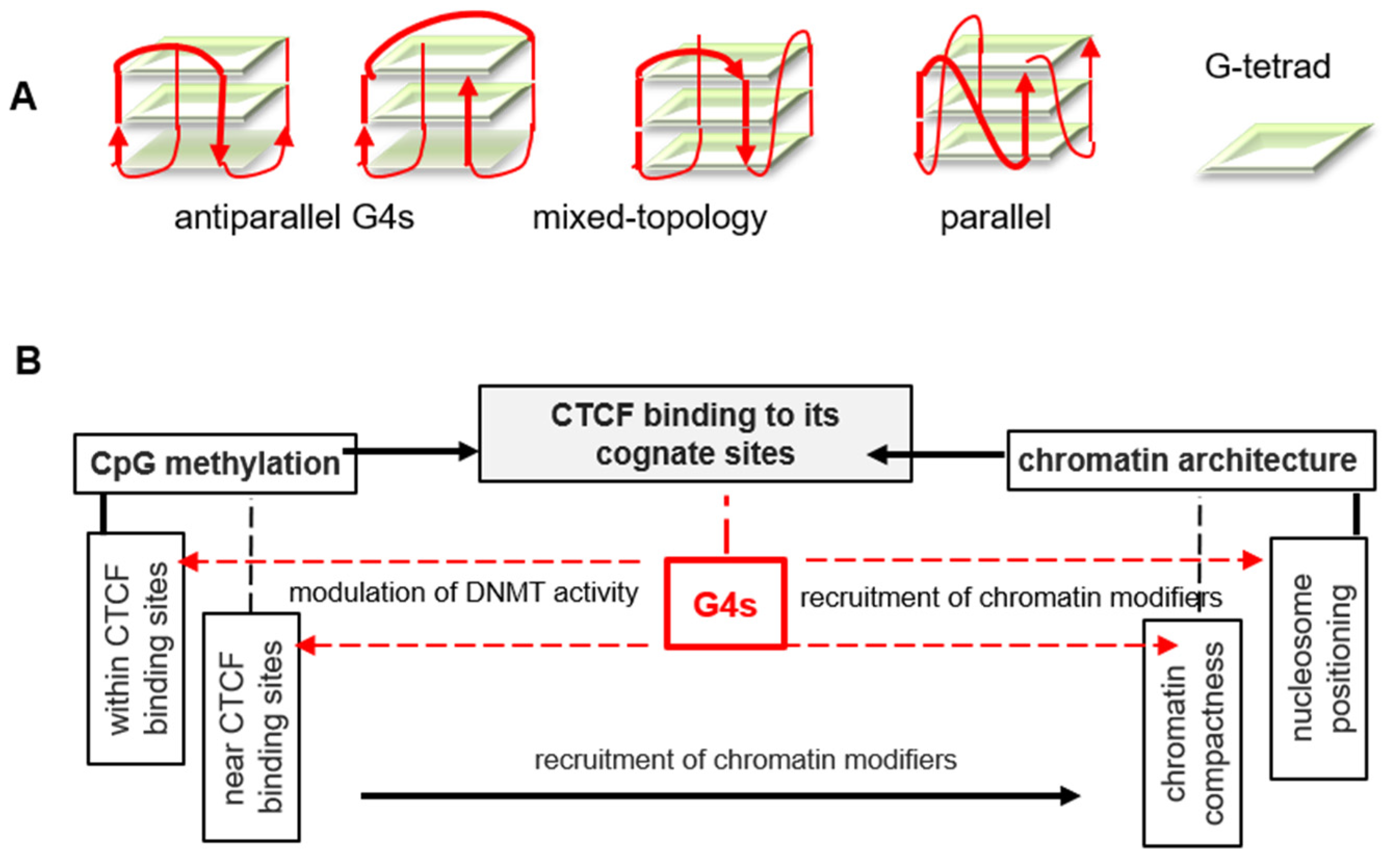
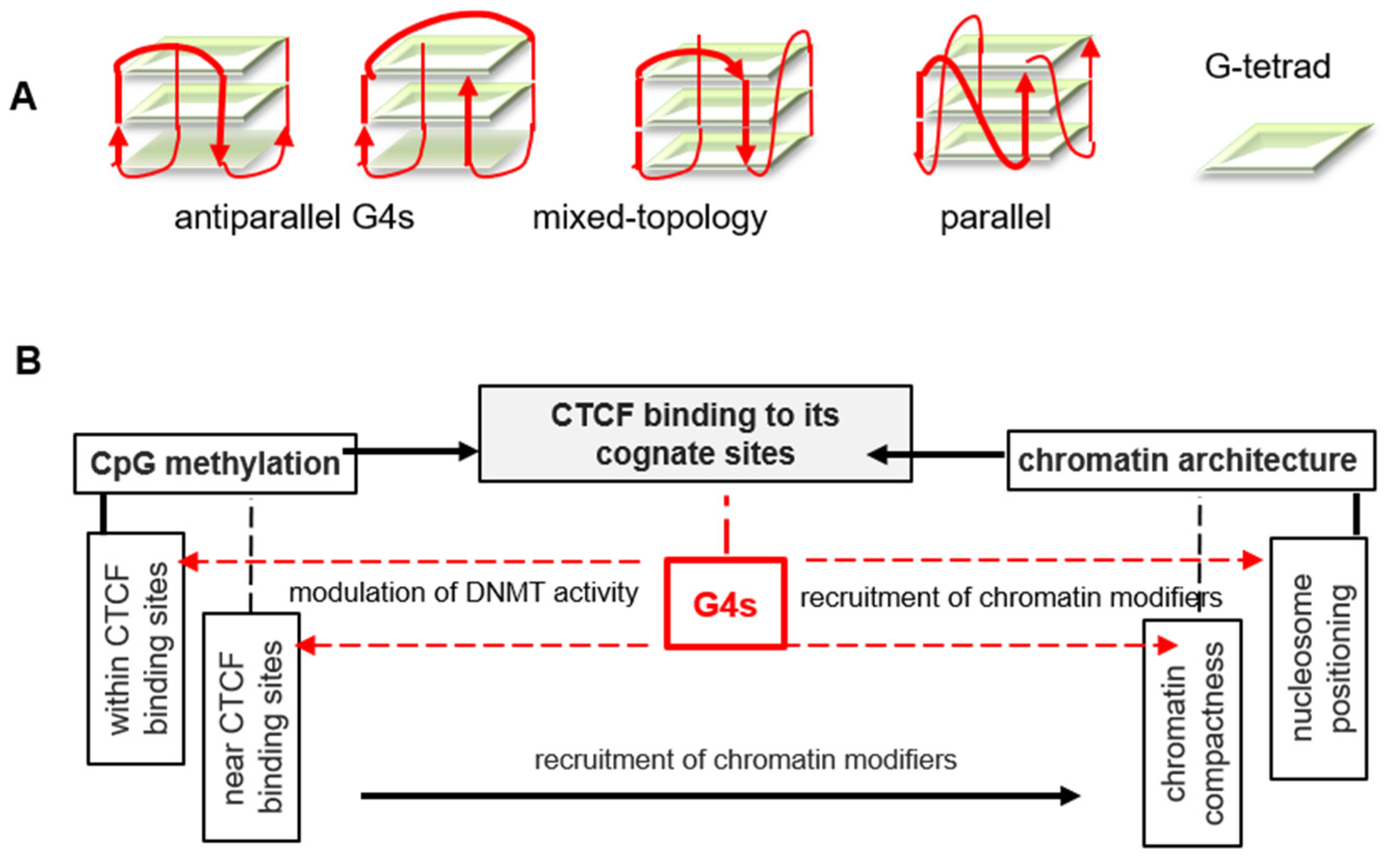
3. Discussion
4. Materials and Methods
4.1. Cell Culture Treatment with G4 Ligands and Chromatin Immunoprecipitation
4.2. ChIP-Seq and Data Analysis
4.3. ChIP-qPCR
4.4. Molecular Modeling
4.5. Bioinformatics
4.6. Oligonucleotides, Recombinant Proteins, and Small-Molecule Ligands
4.7. Circular Dichroism Spectroscopy and Electrophoresis
4.8. Microscale Thermophoresis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.K.; Sharma, S.; Chowdhury, S. Non-duplex G-Quadruplex Structures Emerge as Mediators of Epigenetic Modifications. Trends Genet. 2019, 35, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Varizhuk, A.; Isaakova, E.; Pozmogova, G. DNA G-Quadruplexes (G4s) Modulate Epigenetic (Re)Programming and Chromatin Remodeling Transient Genomic G4s Assist in the Establishment and Maintenance of Epigenetic Marks, While Persistent G4s May Erase Epigenetic Marks. Bioessays 2019, 41, e1900091. [Google Scholar] [CrossRef] [Green Version]
- Mao, S.Q.; Ghanbarian, A.T.; Spiegel, J.; Cuesta, S.M.; Beraldi, D.; Di Antonio, M.; Marsico, G.; Hansel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. DNA G-quadruplex structures mold the DNA methylome. Nat. Struct. Mol. Biol. 2018, 25, 951–957. [Google Scholar] [CrossRef]
- Li, L.; Williams, P.; Ren, W.; Wang, M.Y.; Gao, Z.; Miao, W.; Huang, M.; Song, J.; Wang, Y. YY1 interacts with guanine quadruplexes to regulate DNA looping and gene expression. Nat. Chem. Biol. 2021, 17, 161–168. [Google Scholar] [CrossRef]
- Hou, Y.; Li, F.Y.; Zhang, R.X.; Li, S.; Liu, H.D.; Qin, Z.H.S.; Sun, X. Integrative characterization of G-Quadruplexes in the three-dimensional chromatin structure. Epigenetics 2019, 14, 894–911. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, R.; Renkawitz, R.; Lobanenkov, V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001, 17, 520–527. [Google Scholar] [CrossRef]
- Kim, T.H.; Abdullaev, Z.K.; Smith, A.D.; Ching, K.A.; Loukinov, D.I.; Green, R.D.; Zhang, M.Q.; Lobanenkov, V.V.; Ren, B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 2007, 128, 1231–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, H.S.; Pugh, B.F. Comprehensive Genome-wide Protein-DNA Interactions Detected at Single-Nucleotide Resolution. Cell 2011, 147, 1408–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, H.; Wang, D.; Horton, J.R.; Zhang, X.; Corces, V.G.; Cheng, X. Structural Basis for the Versatile and Methylation-Dependent Binding of CTCF to DNA. Mol. Cell 2017, 66, 711–720.e3. [Google Scholar] [CrossRef] [Green Version]
- Wiehle, L.; Thorn, G.J.; Raddatz, G.; Clarkson, C.T.; Rippe, K.; Lyko, F.; Breiling, A.; Teif, V.B. DNA (de)methylation in embryonic stem cells controls CTCF-dependent chromatin boundaries. Genome Res. 2019, 29, 750–761. [Google Scholar] [CrossRef]
- Heberle, E.; Bardet, A.F. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 2019, 63, 727–741. [Google Scholar] [PubMed] [Green Version]
- Vavouri, T.; Lehner, B. Human genes with CpG island promoters have a distinct transcription-associated chromatin organization. Genome Biol. 2012, 13, R110. [Google Scholar] [CrossRef] [Green Version]
- Teif, V.B.; Beshnova, D.A.; Vainshtein, Y.; Marth, C.; Mallm, J.P.; Hofer, T.; Rippe, K. Nucleosome repositioning links DNA (de)methylation and differential CTCF binding during stem cell development. Genome Res. 2014, 24, 1285–1295. [Google Scholar] [CrossRef] [Green Version]
- Barutcu, A.R.; Lian, J.B.; Stein, J.L.; Stein, G.S.; Imbalzano, A.N. The connection between BRG1, CTCF and topoisomerases at TAD boundaries. Nucleus-Phila 2017, 8, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Barisic, D.; Stadler, M.B.; Iurlaro, M.; Schubeler, D. Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature 2019, 569, 136–140. [Google Scholar] [CrossRef]
- Owens, N.; Papadopoulou, T.; Festuccia, N.; Tachtsidi, A.; Gonzalez, I.; Dubois, A.; Vandormael-Pournin, S.; Nora, E.P.; Bruneau, B.G.; Cohen-Tannoudji, M.; et al. CTCF confers local nucleosome resiliency after DNA replication and during mitosis. Elife 2019, 8, 8. [Google Scholar] [CrossRef]
- Makowski, M.M.; Grawe, C.; Foster, B.M.; Nguyen, N.V.; Bartke, T.; Vermeulen, M. Global profiling of protein-DNA and protein-nucleosome binding affinities using quantitative mass spectrometry. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Vlasenok, M.; Levchenko, O.; Basmanov, D.; Klinov, D.; Varizhuk, A.; Pozmogova, G. Data set on G4 DNA interactions with human proteins. Data Brief 2018, 18, 348–359. [Google Scholar] [CrossRef]
- Li, P.T.; Wang, Z.F.; Chu, I.T.; Kuan, Y.M.; Li, M.H.; Huang, M.C.; Chiang, P.C.; Chang, T.C.; Chen, C.T. Expression of the human telomerase reverse transcriptase gene is modulated by quadruplex formation in its first exon due to DNA methylation. J. Biol. Chem. 2017, 292, 20859–20870. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, R.; Muller, S.; Yeoman, J.A.; Trentesaux, C.; Riou, J.F.; Balasubramanian, S. A novel small molecule that alters shelterin integrity and triggers a DNA-damage response at telomeres. J. Am. Chem Soc. 2008, 130, 15758–15759. [Google Scholar] [CrossRef] [Green Version]
- De Cian, A.; DeLemos, E.; Mergny, J.L.; Teulade-Fichou, M.P.; Monchaud, D. Highly efficient G-quadruplex recognition by bisquinolinium compounds. Am. Chem. Soc. 2007, 129, 1856–1857. [Google Scholar] [CrossRef]
- Muller, S.; Sanders, D.A.; Di Antonio, M.; Matsis, S.; Riou, J.F.; Rodriguez, R.; Balasubramanian, S. Pyridostatin analogues promote telomere dysfunction and long-term growth inhibition in human cancer cells. Org. Biomol. Chem. 2012, 10, 6537–6546. [Google Scholar] [CrossRef] [PubMed]
- Le, H.T.; Miller, M.C.; Buscaglia, R.; Dean, W.L.; Holt, P.A.; Chaires, J.B.; Trent, J.O. Not all G-quadruplexes are created equally: An investigation of the structural polymorphism of the c-Myc G-quadruplex-forming sequence and its interaction with the porphyrin TMPyP4. Org. Biomol. Chem. 2012, 10, 9393–9404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, A.T.; Modi, Y.S.; Patel, D.J. Propeller-type parallel-stranded g-quadruplexes in the human c-myc promoter. J. Am. Chem. Soc. 2004, 126, 8710–8716. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.P.; Huppert, J.L.; Waller, Z.A.E. Identification of multiple genomic DNA sequences which form i-motif structures at neutral pH. Nucleic Acids Res. 2017, 45, 2951. [Google Scholar] [CrossRef] [PubMed]
- Postnikov, Y.V.; Bustin, M. Functional interplay between histone H1 and HMG proteins in chromatin. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1859, 462–467. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.J.; Cutter, A.R.; Fang, H.; Postnikov, Y.V.; Bustin, M.; Hayes, J.J. HMGN1 and 2 remodel core and linker histone tail domains within chromatin. Nucleic Acids Res. 2017, 45, 9917–9930. [Google Scholar] [CrossRef]
- Barkess, G.; Postnikov, Y.; Campos, C.D.; Mishra, S.; Mohan, G.; Verma, S.; Bustin, M.; West, K.L. The chromatin-binding protein HMGN3 stimulates histone acetylation and transcription across the Glyt1 gene. Biochem. J. 2012, 442, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Zirkel, A.; Nikolic, M.; Sofiadis, K.; Mallm, J.P.; Brackley, C.A.; Gothe, H.; Drechsel, O.; Becker, C.; Altmuller, J.; Josipovic, N.; et al. HMGB2 Loss upon Senescence Entry Disrupts Genomic Organization and Induces CTCF Clustering across Cell Types. Mol. Cell 2018, 70, 730–744.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Tanizawa, H.; Fatkhutdinov, N.; Bitler, B.G.; Le, L.; Alicea, G.; Yang, T.L.; Johnson, B.; et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J. Cell Biol. 2016, 215, 325–334. [Google Scholar] [CrossRef]
- Hansel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.W.; Alberti, P.; Guedin, A.; Lacroix, L.; Riou, J.F.; Royle, N.J.; Mergny, J.L.; Phan, A.T. Sequence variant (CTAGGG)(n) in the human telomere favors a G-quadruplex structure containing a G center dot C center dot G center dot C tetrad. Nucleic Acids Res. 2009, 37, 6239–6248. [Google Scholar] [CrossRef]
- Membrino, A.; Cogoi, S.; Pedersen, E.B.; Xodo, L.E. G4-DNA Formation in the HRAS Promoter and Rational Design of Decoy Oligonucleotides for Cancer Therapy. PLoS ONE 2011, 6, e24421. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.D.; Zhou, B.; Geng, Y.Y.; Tam, D.Y.; Feng, R.; Miao, H.T.; Xu, N.N.; Shi, X.; You, Y.Y.; Hong, Y.N.; et al. A chair-type G-quadruplex structure formed by a human telomeric variant DNA in K+ solution. Chem. Sci. 2019, 10, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anselmet, A.; Mayat, E.; Wietek, S.; Layer, P.G.; Payrastre, B.; Massoulie, J. Non-antisense cellular responses to oligonucleotides. Febs Lett. 2002, 510, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Varizhuk, A.; Ischenko, D.; Tsvetkov, V.; Novikov, R.; Kulemin, N.; Kaluzhny, D.; Vlasenok, M.; Naumov, V.; Smirnov, I.; Pozmogova, G. The expanding repertoire of G4 DNA structures. Biochimie 2017, 135, 54–62. [Google Scholar] [CrossRef]
- Raiber, E.A.; Kranaster, R.; Lam, E.; Nikan, M.; Balasubramanian, S. A non-canonical DNA structure is a binding motif for the transcription factor SP1 in vitro. Nucleic Acids Res. 2011, 40, 1499–1508. [Google Scholar] [CrossRef] [Green Version]
- Da Ros, S.; Nicoletto, G.; Rigo, R.; Ceschi, S.; Zorzan, E.; Dacasto, M.; Giantin, M.; Sissi, C. G-Quadruplex Modulation of SP1 Functional Binding Sites at the KIT Proximal Promoter. Int. J. Mol. Sci. 2020, 22, 329. [Google Scholar] [CrossRef]
- Xiao, T.J.; Li, X.; Felsenfeld, G. The Myc-associated zinc finger protein (MAZ) works together with CTCF to control cohesin positioning and genome organization. Proc. Natl. Acad. Sci. USA 2021, 118, 7. [Google Scholar] [CrossRef]
- Valton, A.L.; Prioleau, M.N. G-Quadruplexes in DNA Replication: A Problem or a Necessity? Trends Genet. 2016, 32, 697–706. [Google Scholar] [CrossRef]
- Reina, C.; Cavalieri, V. Epigenetic Modulation of Chromatin States and Gene Expression by G-Quadruplex Structures. Int. J. Mol. Sci. 2020, 21, 4172. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, H. Enhancer-promoter interaction facilitated by transiently forming G-quadruplexes. Sci. Rep. 2015, 5, 9165. [Google Scholar] [CrossRef] [Green Version]
- Kantidze, O.L.; Gurova, K.V.; Studitsky, V.M.; Razin, S.V. The 3D Genome as a Target for Anticancer Therapy. Trends Mol. Med. 2020, 26, 141–149. [Google Scholar] [CrossRef]
- Schuijers, J.; Manteiga, J.C.; Weintraub, A.S.; Day, D.S.; Zamudio, A.V.; Hnisz, D.; Lee, T.I.; Young, R.A. Transcriptional Dysregulation of MYC Reveals Common Enhancer-Docking Mechanism. Cell Rep. 2018, 23, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Lutz, M.; Burke, L.J.; Barreto, G.; Goeman, F.; Greb, H.; Arnold, R.; Schultheiss, H.; Brehm, A.; Kouzarides, T.; Lobanenkov, V.; et al. Transcriptional repression by the insulator protein CTCF involves histone deacetylases. Nucleic Acids Res. 2000, 28, 1707–1713. [Google Scholar] [CrossRef] [Green Version]
- Filippova, G.N.; Fagerlie, S.; Klenova, E.M.; Myers, C.; Dehner, Y.; Goodwin, G.; Neiman, P.E.; Collins, S.J.; Lobanenkov, V.V. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell. Biol. 1996, 16, 2802–2813. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.X.; Lin, Y.H.; Johnson, T.D.; Rozek, L.S.; Sartor, M.A. PePr: A peak-calling prioritization pipeline to identify consistent or differential peaks from replicated ChIP-Seq data. Bioinformatics 2014, 30, 2568–2575. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, I.I.; Tsvetkov, V.B.; Isaakova, E.A.; Severov, V.V.; Khomyakova, E.A.; Lacis, I.A.; Lazarev, V.N.; Lagarkova, M.A.; Pozmogova, G.E.; Varizhuk, A.M. Transcription-facilitating histone chaperons interact with genomic and synthetic G4 structures. Int. J. Biol. Macromol. 2020, 160, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Macindoe, G.; Mavridis, L.; Venkatraman, V.; Devignes, M.D.; Ritchie, D.W. HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010, 38, W445–W449. [Google Scholar] [CrossRef]
- Belmonte-Reche, E.; Morales, J.C. G4-iM Grinder: When size and frequency matter. G-Quadruplex, i-Motif and higher order structure search and analysis tool. NAR Genom. Bioinform. 2020, 2, lqz005. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.Y.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ODN | Kd, µM (HMGN3) | Kd, µM (HMGB2) | Kd, µM (HMGN1) |
|---|---|---|---|
| pG4s | 1.5 ± 0.6 | 0.6 ± 0.3 | 0.2 ± 0.1 (N1–C3); 1.6 ± 0.3 (pos. contr.) |
| aG4s | >>20 | >>20 | ≥20 |
| mG4s | ≥10 (M6); 0.15 ± 0.06 (M7) | 4 ± 2 (M6); 1.6 ± 0.6 (M7) | ≥10 (Tel 26); >20 (N1sh-2) |
| hairpin | ≥20 | ≥20 | ≥10 |
| dsDNA | >>20 | >>20 | >>20 |
| ssDNA | >>20 | >>20 | >>20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tikhonova, P.; Pavlova, I.; Isaakova, E.; Tsvetkov, V.; Bogomazova, A.; Vedekhina, T.; Luzhin, A.V.; Sultanov, R.; Severov, V.; Klimina, K.; et al. DNA G-Quadruplexes Contribute to CTCF Recruitment. Int. J. Mol. Sci. 2021, 22, 7090. https://doi.org/10.3390/ijms22137090
Tikhonova P, Pavlova I, Isaakova E, Tsvetkov V, Bogomazova A, Vedekhina T, Luzhin AV, Sultanov R, Severov V, Klimina K, et al. DNA G-Quadruplexes Contribute to CTCF Recruitment. International Journal of Molecular Sciences. 2021; 22(13):7090. https://doi.org/10.3390/ijms22137090
Chicago/Turabian StyleTikhonova, Polina, Iulia Pavlova, Ekaterina Isaakova, Vladimir Tsvetkov, Alexandra Bogomazova, Tatjana Vedekhina, Artem V. Luzhin, Rinat Sultanov, Vjacheslav Severov, Ksenia Klimina, and et al. 2021. "DNA G-Quadruplexes Contribute to CTCF Recruitment" International Journal of Molecular Sciences 22, no. 13: 7090. https://doi.org/10.3390/ijms22137090
APA StyleTikhonova, P., Pavlova, I., Isaakova, E., Tsvetkov, V., Bogomazova, A., Vedekhina, T., Luzhin, A. V., Sultanov, R., Severov, V., Klimina, K., Kantidze, O. L., Pozmogova, G., Lagarkova, M., & Varizhuk, A. (2021). DNA G-Quadruplexes Contribute to CTCF Recruitment. International Journal of Molecular Sciences, 22(13), 7090. https://doi.org/10.3390/ijms22137090