
Aberrant Alternative Splicing in U2af1/Tet2 Double Mutant Mice Contributes to Major Hematological Phenotypes

 , , , ,
, , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
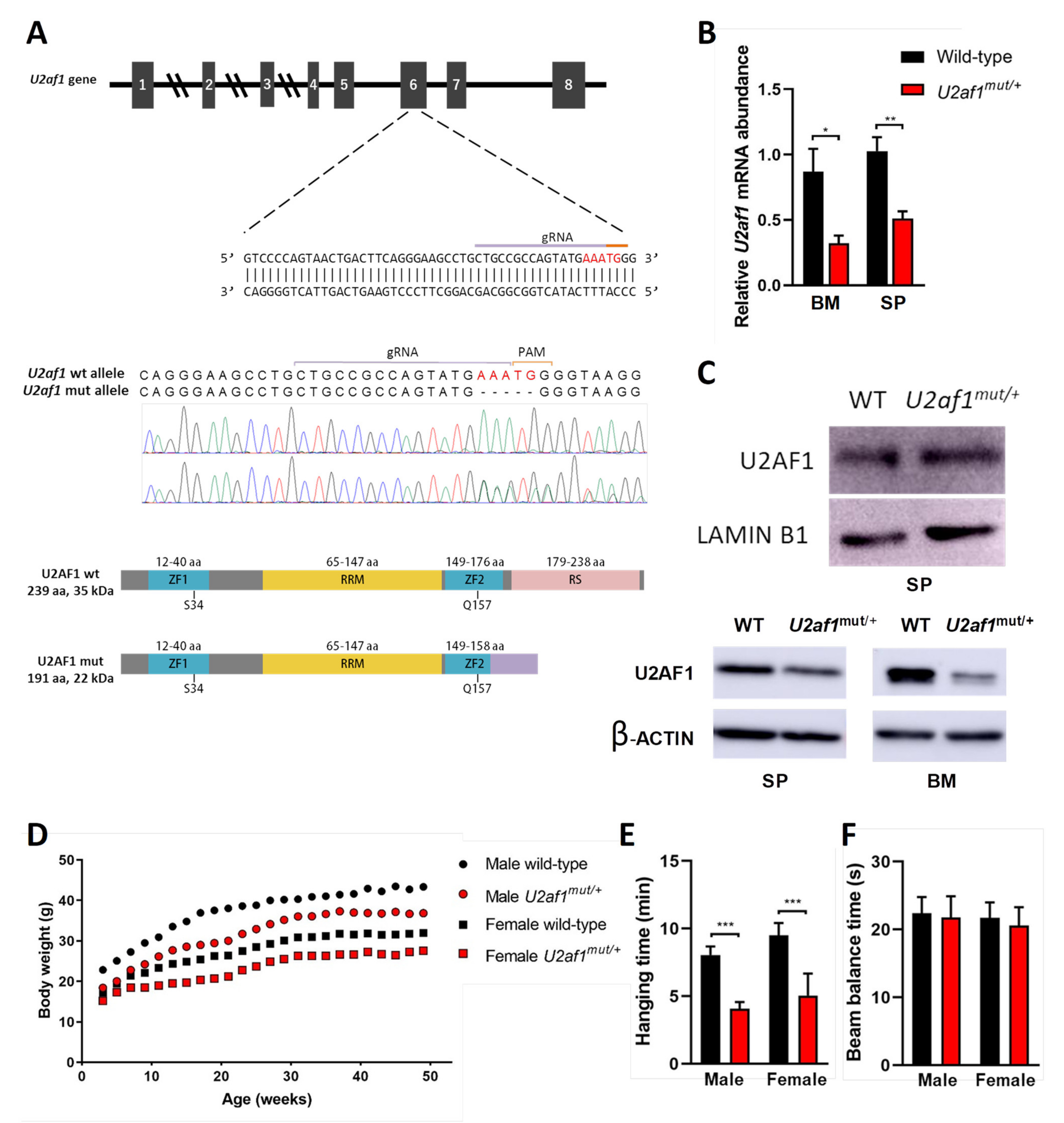
2.1. Development of U2af1 Mutant Allele
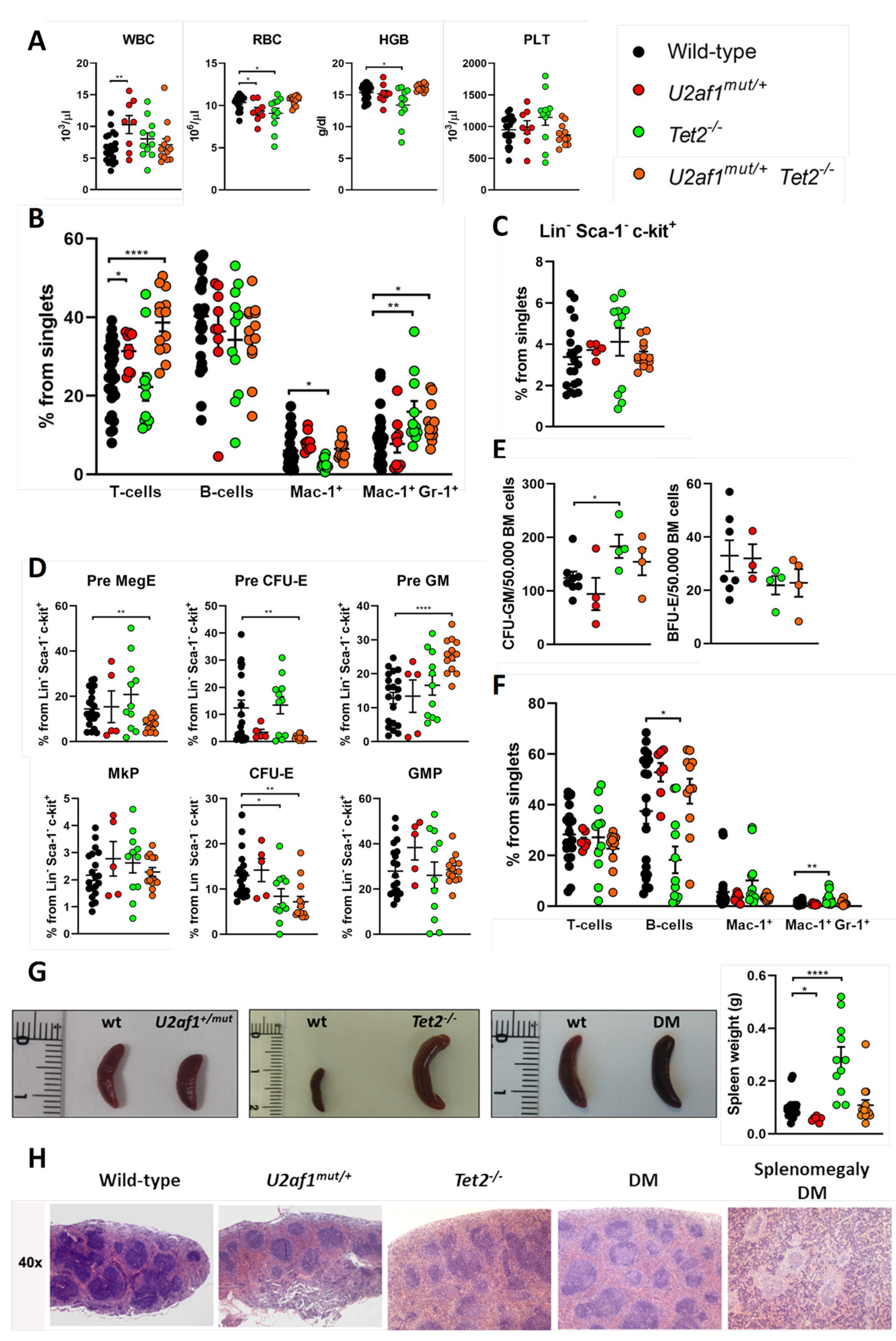
2.2. Old U2af1 Mutant Mice Show Normal Hematopoiesis and Uneven Splenomegaly
2.3. Monogranulocytic Progenitors Are Increased in U2af1mut/+ Tet2−/− Mutant Mice
2.4. U2af1mut/+ and U2af1mut/+ Tet2−/− Mutant HSPC Show a Defect in Reconstitution Capacity
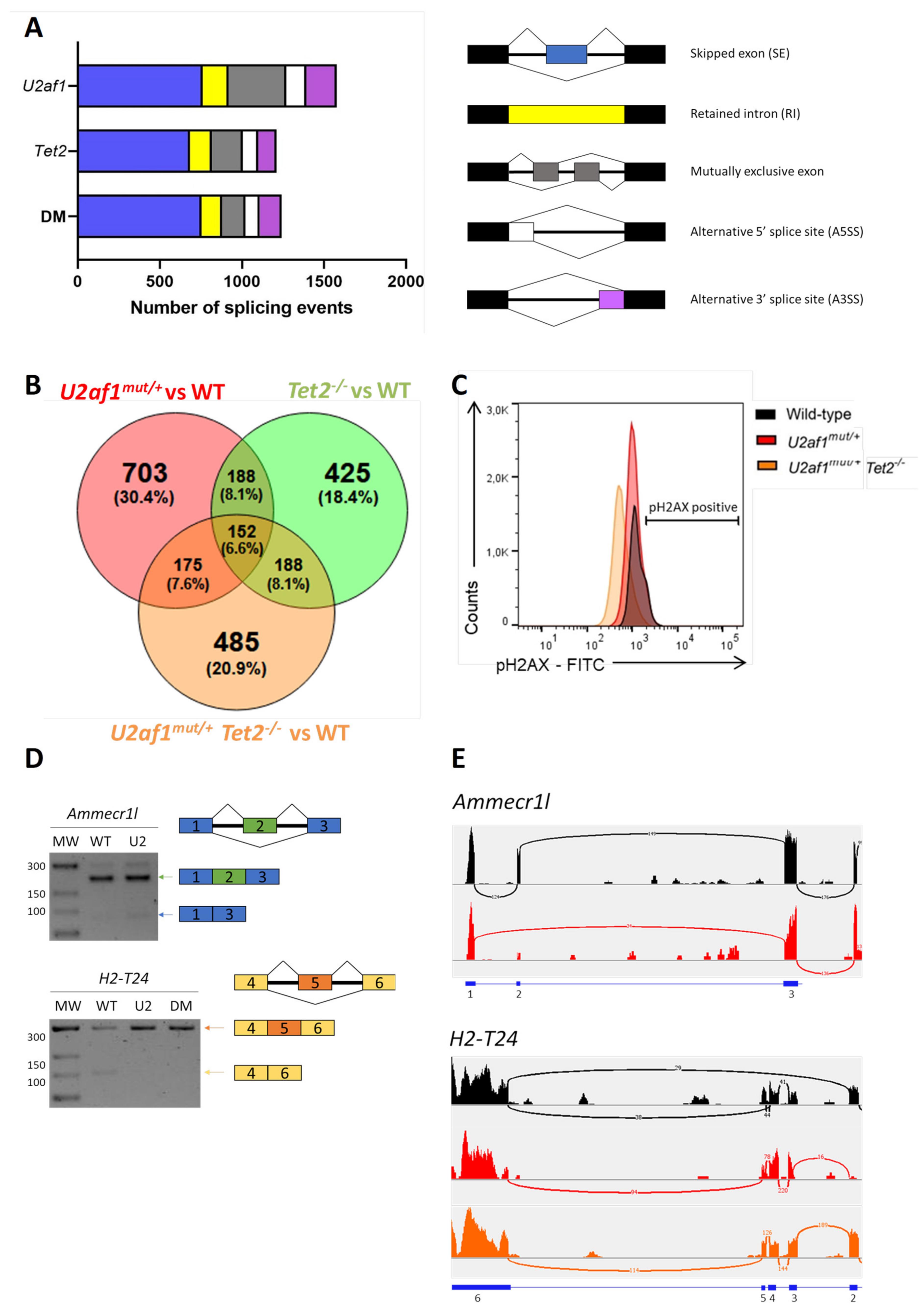
2.5. Exon Skipping Is the Alternatively Spliced Event More Frequently Observed
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. RT-qPCR
4.3. Western Blotting
4.4. Blood Count Analysis
4.5. Flow Cytometry Analysis
4.6. Histological Analysis
4.7. Cytospin Preparation
4.8. Colony-Forming Unit Assays
4.9. DNA Damage
4.10. LSK FACS-Sorting
4.11. Competitive Repopulation Assay
4.12. Behaviour Tests
4.13. RNA Sequencing Library Construction
4.14. RNA Sequencing Analysis
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA Splicing Factors as Oncoproteins and Tumour Suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Inoue, D.; Bradley, R.K.; Abdel-Wahab, O. Spliceosomal Gene Mutations in Myelodysplasia: Molecular Links to Clonal Abnormalities of Hematopoiesis. Genes Dev. 2016, 30, 989–1001. [Google Scholar] [CrossRef] [Green Version]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 Mutations Alter Splice Site Recognition in Hematological Malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef]
- Pangallo, J.; Kiladjian, J.J.; Cassinat, B.; Renneville, A.; Taylor, J.; Polaski, J.T.; North, K.; Abdel-Wahab, O.; Bradley, R.K. Rare and Private Spliceosomal Gene Mutations Drive Partial, Complete, and Dual Phenocopies of Hotspot Alterations. Blood 2020, 135, 1032–1043. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu, K.S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, M.; Afable, M.G.; et al. Mutations in the Spliceosome Machinery, A Novel and Ubiquitous Pathway in Leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zou, D.; Yang, S.; Ouyang, G.; Mu, Q. Prognostic Significance of U2AF1 Mutations in Myelodysplastic Syndromes: A Meta-Analysis. J. Int. Med. Res. 2020, 48, 300060519891013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent Mutations in the U2AF1 Splicing Factor in Myelodysplastic Syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Wadugu, B.A.; Heard, A.; Srivatsan, S.N.; Alberti, M.O.; Ndonwi, M.; Grieb, S.; Bradley, J.; Shao, J.; Ahmed, T.; Shirai, C.L.; et al. U2AF1 Is a Haplo-Essential Gene Required for Cancer Cell Survival. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Fei, D.L.; Motowski, H.; Chatrikhi, R.; Prasad, S.; Yu, J.; Gao, S.; Kielkopf, C.L.; Bradley, R.K.; Varmus, H. Wild-Type U2AF1 Antagonizes the Splicing Program Characteristic of U2AF1-Mutant Tumors and Is Required for Cell Survival. PLoS Genet. 2016, 12, e1006384. [Google Scholar] [CrossRef]
- Palangat, M.; Anastasakis, D.G.; Liang Fei, D.; Lindblad, K.E.; Bradley, R.; Hourigan, C.S.; Hafner, M.; Larson, D.R. The Splicing Factor U2af1 Contributes to Cancer Progression through a Noncanonical Role in Translation Regulation. Genes Dev. 2019, 33, 482–497. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.; Huang, Y.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.D.; Yee Leong, W.; Weiling, L.; Reddy, P.N.G.; Sullivan, J.D.; Walter, M.J.; Zou, L.; Graubert, T.A. Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res. 2018, 78, 5363–5374. [Google Scholar] [CrossRef] [Green Version]
- Przychodzen, B.; Jerez, A.; Guinta, K.; Sekeres, M.A.; Padgett, R.; Maciejewski, J.P.; Makishima, H. Patterns of Missplicing Due to Somatic U2af1 Mutations in Myeloid Neoplasms. Blood 2013, 122, 999–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiozawa, Y.; Malcovati, L.; Gallì, A.; Sato-Otsubo, A.; Kataoka, K.; Sato, Y.; Watatani, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K. Aberrant Splicing and Defective mRNA Production Induced by Somatic Spliceosome Mutations in Myelodysplasia. Nat. Commun. 2018, 9, 3649. [Google Scholar] [CrossRef] [PubMed]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Alberti, M.O.; Zhu, M.; Srivatsan, S.N.; Ahmed, T.; Shao, J.J.; Wadugu, B.; Shirai, C.L.; Walter, M.J. Mutant U2AF1S34F and U2AF1Q157P Induce Distinct RNA Splicing and Hematopoietic Phenotypes In Vivo. Blood 2019, 134, 770. [Google Scholar] [CrossRef]
- Bapat, A.; Keita, N.; Martelly, W.; Kang, P.; Seet, C.; Jacobsen, J.R.; Stoilov, P.; Hu, C.; Crooks, G.M.; Sharma, S. Disease Mutations of Splicing Factor SRSF2 Cause G2-M Arrest and Skewed Differentiation of Human Hematopoietic Stem and Progenitor Cells. Stem Cells 2018, 36, 1663–1675. [Google Scholar] [CrossRef]
- Yip, B.H.; Steeples, V.; Repapi, E.; Armstrong, R.N.; Llorian, M.; Roy, S.; Shaw, J.; Dolatshad, H.; Taylor, S.; Verma, A.; et al. The U2AF1S34F Mutation Induces Lineage-Specific Splicing Alterations in Myelodysplastic Syndromes. J. Clin. Investig. 2017, 127, 2206–2221. [Google Scholar] [CrossRef] [Green Version]
- Fei, D.L.; Zhen, T.; Durham, B.; Ferrarone, J.; Zhang, T.; Garrett, L.; Yoshimi, A.; Abdel-Wahab, O.; Bradley, R.K.; Liu, P.; et al. Impaired Hematopoiesis and Leukemia Development in Mice with a Conditional Knock-In Allele of a Mutant Splicing Factor Gene U2af1. Proc. Natl. Acad. Sci. USA 2018, 115, E10437–E10446. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.; Yang, Y.; Le, B.T.; Zhang, Y.; Abdel-Wahab, O.; Zang, C.; Mohi, G. U2af1 is Required for Survival and Function of Hematopoietic Stem/Progenitor Cells. Leukemia 2021, 1–17. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and Biological Implications of Driver Mutations in Myelodysplastic Syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; Mi, X.; North, K.; Binder, M.; Penson, A.; Lasho, T.; Knorr, K.; Haddadin, M.; Liu, B.; Pangallo, J.; et al. Single-Cell Genomics Reveals the Genetic and Molecular Bases for Escape from Mutational Epistasis in Myeloid Neoplasms. Blood 2020, 136, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Langemeijer, S.M.C.; Kuiper, R.P.; Berends, M.; Knops, R.; Aslanyan, M.G.; Massop, M.; Stevens-Linders, E.; van Hoogen, P.; van Kessel, A.G.; Raymakers, R.A.P.; et al. Acquired Mutations in TET2 Are Common in Myelodysplastic Syndromes. Nat. Genet. 2009, 41, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Awada, H.; Nagata, Y.; Goyal, A.; Asad, M.F.; Patel, B.; Hirsch, C.M.; Kuzmanovic, T.; Guan, Y.; Przychodzen, B.P.; Aly, M.; et al. Invariant Phenotype and Molecular Association of Biallelic TET2 Mutant Myeloid Neoplasia. Blood 2019, 3, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Helin, K. Role of TET Enzymes in DNA Methylation, Development, and Cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Cai, X.; Cai, C.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.; Xu, M. Deletion of Tet2 in Mice Leads to Dysregulated Hematopoietic Stem Cells and Subsequent Development of Myeloid Malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, T.R.; Gomes, A.Q.; Barbosa-Morais, N.L.; Benes, V.; Ansorge, W.; Wollerton, M.; Smith, C.W.; Valcárcel, J.; Carmo-Fonseca, M. Diversity of Vertebrate Splicing Factor U2AF35: Identification of Alternatively Spliced U2AF1 mRNAS. J. Biol. Chem. 2004, 279, 27039–27049. [Google Scholar] [CrossRef] [Green Version]
- Kralovicova, J.; Knut, M.; Cross, N.C.P.; Vorechovsky, I. Identification of U2AF(35)-Dependent Exons by RNA-Seq Reveals a Link Between 3′ Splice-Site Organization and Activity of U2AF-Related Proteins. Nucleic Acids Res. 2015, 43, 3747–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Zhou, B.; Thol, F.; Zhou, Y.; Chen, L.; Shao, C.; De Boever, C.; Hou, J.; Li, H.; Chaturvedi, A.; et al. Distinct Splicing Signatures Affect Converged Pathways in Myelodysplastic Syndrome Patients Carrying Mutations in Different Splicing Regulators. RNA 2016. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Capetillo, O.; Lee, A.; Nussenzweig, M.; Nussenzweig, A. H2AX: The Histone Guardian of the Genome. DNA Repair Amst. 2004, 3, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.J.; Yang, L.X. γ-H2AX- A Novel Biomaker for DNA Double-Strand Breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Malcovati, L.; Papaemmanuil, E.; Bowen, D.T.; Boultwood, J.; Della Porta, M.G.; Pascutto, C.; Travaglino, E.; Groves, M.J.; Godfrey, A.L.; Ambaglio, I.; et al. Clinical Significance of SF3B1 Mutations in Myelodysplastic Syndromes and Myelodysplastic/Myeloproliferative Neoplasms. Blood 2011, 118, 6239–6246. [Google Scholar] [CrossRef] [Green Version]
- Quivoron, C.; Couronne, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, F.; Ster, M.H.; et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event During Human Lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-Eleven-Translocation 2 (TET2) Negatively Regulates Homeostasis and Differentiation of Hematopoietic Stem Cells in Mice. Proc. Natl. Acad. Sci. USA 2011, 108, 14566–14571. [Google Scholar] [CrossRef] [Green Version]
- Yoshimi, A.; Lin, K.T.; Wiseman, D.H.; Rahman, M.A.; Pastore, A.; Wang, B.; Lee, S.C.W.; Micol, J.B.; Zhang, S.J.; de Botton, S.; et al. Coordinated Alterations in RNA Splicing and Epigenetic Regulation Drive Leukaemogenesis. Nature 2019, 574, 273–277. [Google Scholar] [CrossRef]
- Pellagatti, A.; Armstrong, R.N.; Steeples, V.; Sharma, E.; Repapi, E.; Singh, S.; Sanchi, A.; Radujkovic, A.; Horn, P.; Dolatshad, H.; et al. Impact of Spliceosome Mutations on RNA Splicing in Myelodysplasia: Dysregulated Genes/Pathways and Clinical Associations. Blood 2018, 132, 1225–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Pronk, C.J.H.; Rossi, D.J.; Månsson, R.; Attema, J.L.; Norddahl, G.L.; Kwok, F.C.C.; Sigvardsson, M.; Weissman, I.L.; Bryder, D. Elucidation of the Phenotypic, Functional, and Molecular Topography of a Myeloerythroid Progenitor Cell Hierarchy. Cell Stem Cell 2007, 1, 428–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, K.; Perez-Cerezales, S.; Papasaikas, P.; Ramos-Ibeas, P.; López-Cardona, A.P.; Laguna-Barraza, R.; Fonseca, B.N.; Pericuesta, E.; Fernández-González, R.; Planells, B.; et al. Impaired Spermatogenesis, Muscle, and Erythrocyte Function in U12 Intron Splicing-Defective Zrsr1 Mutant Mice. Cell Rep. 2018, 23, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and Flexible Detection of Differential Alternative Splicing from Replicate RNA-Seq Data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, 3. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R Package Rsubread Is Easier, Faster, Cheaper and Better for Alignment and Quantification of RNA Sequencing Reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential Expression Analysis for Sequence Count Data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Valiente, C.; Garcia-Ruiz, C.; Rosón, B.; Liquori, A.; González-Romero, E.; Fernández-González, R.; Gómez-Redondo, I.; Cervera, J.; Gutiérrez-Adán, A.; Sanjuan-Pla, A. Aberrant Alternative Splicing in U2af1/Tet2 Double Mutant Mice Contributes to Major Hematological Phenotypes. Int. J. Mol. Sci. 2021, 22, 6963. https://doi.org/10.3390/ijms22136963
Martínez-Valiente C, Garcia-Ruiz C, Rosón B, Liquori A, González-Romero E, Fernández-González R, Gómez-Redondo I, Cervera J, Gutiérrez-Adán A, Sanjuan-Pla A. Aberrant Alternative Splicing in U2af1/Tet2 Double Mutant Mice Contributes to Major Hematological Phenotypes. International Journal of Molecular Sciences. 2021; 22(13):6963. https://doi.org/10.3390/ijms22136963
Chicago/Turabian StyleMartínez-Valiente, Cristina, Cristian Garcia-Ruiz, Beatriz Rosón, Alessandro Liquori, Elisa González-Romero, Raúl Fernández-González, Isabel Gómez-Redondo, José Cervera, Alfonso Gutiérrez-Adán, and Alejandra Sanjuan-Pla. 2021. "Aberrant Alternative Splicing in U2af1/Tet2 Double Mutant Mice Contributes to Major Hematological Phenotypes" International Journal of Molecular Sciences 22, no. 13: 6963. https://doi.org/10.3390/ijms22136963
APA StyleMartínez-Valiente, C., Garcia-Ruiz, C., Rosón, B., Liquori, A., González-Romero, E., Fernández-González, R., Gómez-Redondo, I., Cervera, J., Gutiérrez-Adán, A., & Sanjuan-Pla, A. (2021). Aberrant Alternative Splicing in U2af1/Tet2 Double Mutant Mice Contributes to Major Hematological Phenotypes. International Journal of Molecular Sciences, 22(13), 6963. https://doi.org/10.3390/ijms22136963