
Effect of the Nucleophile’s Nature on Chloroacetanilide Herbicides Cleavage Reaction Mechanism. A DFT Study

 and
and
Abstract
:1. Introduction
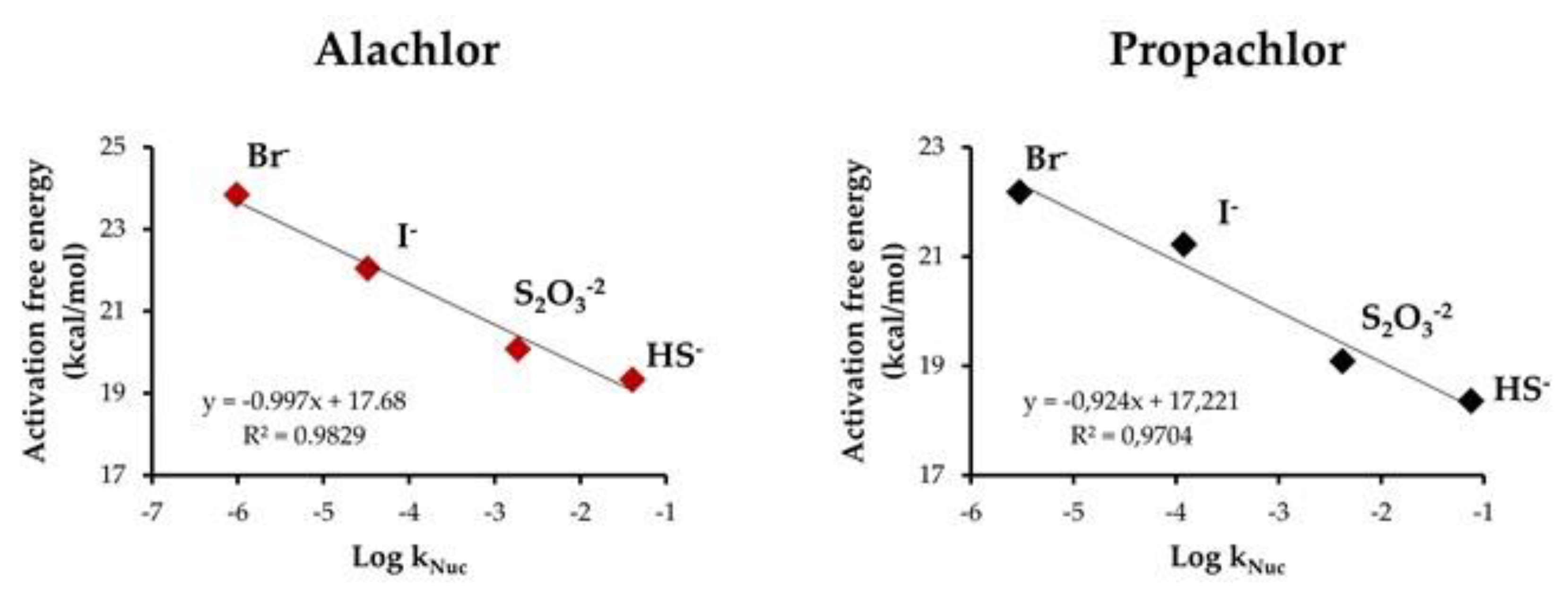
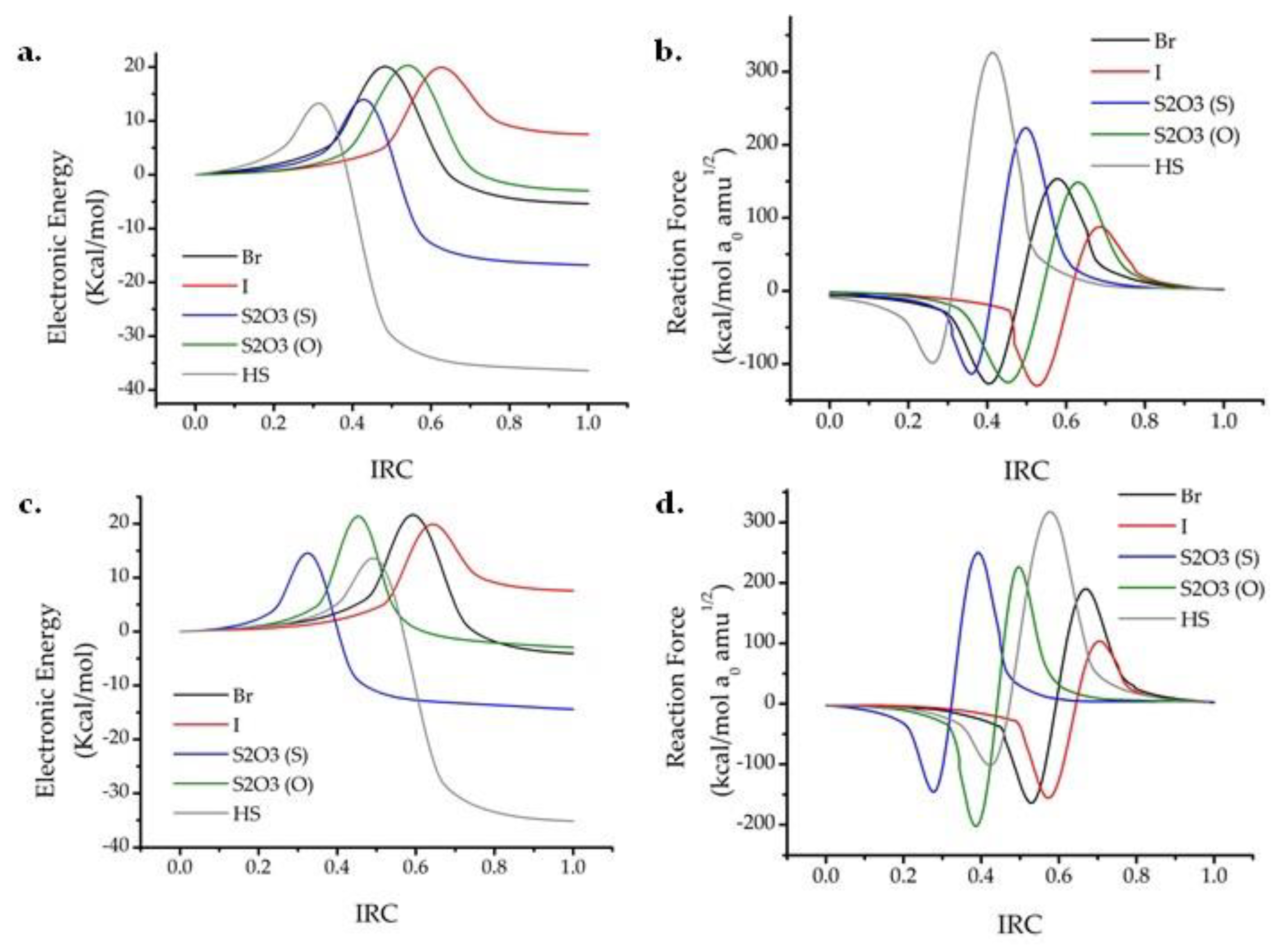
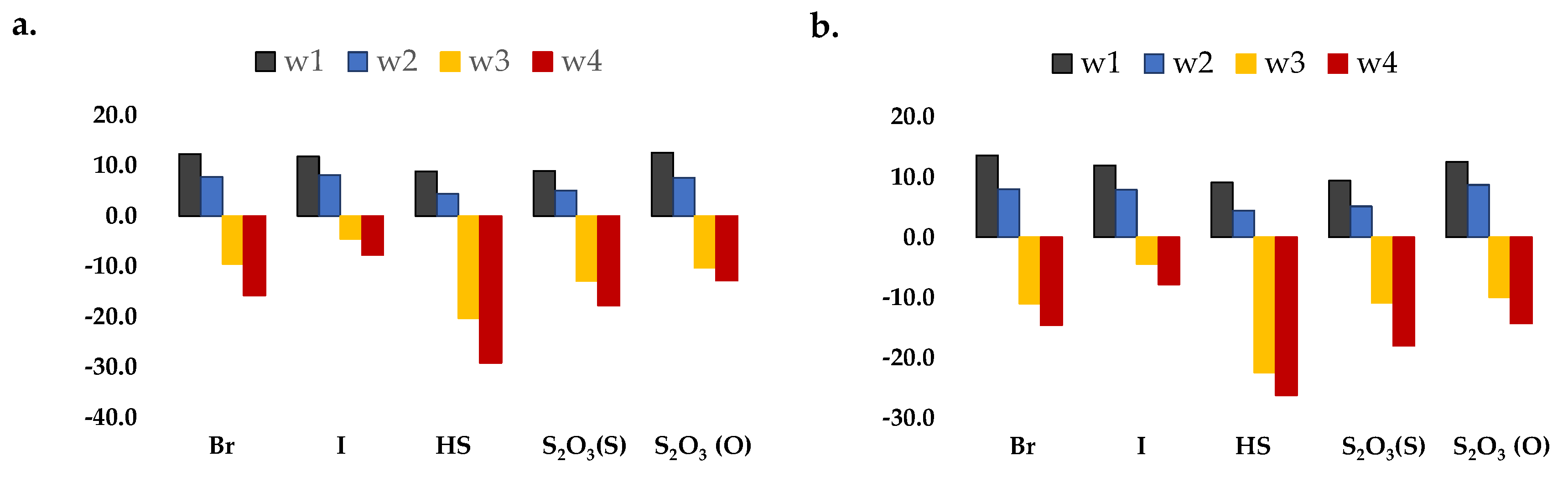
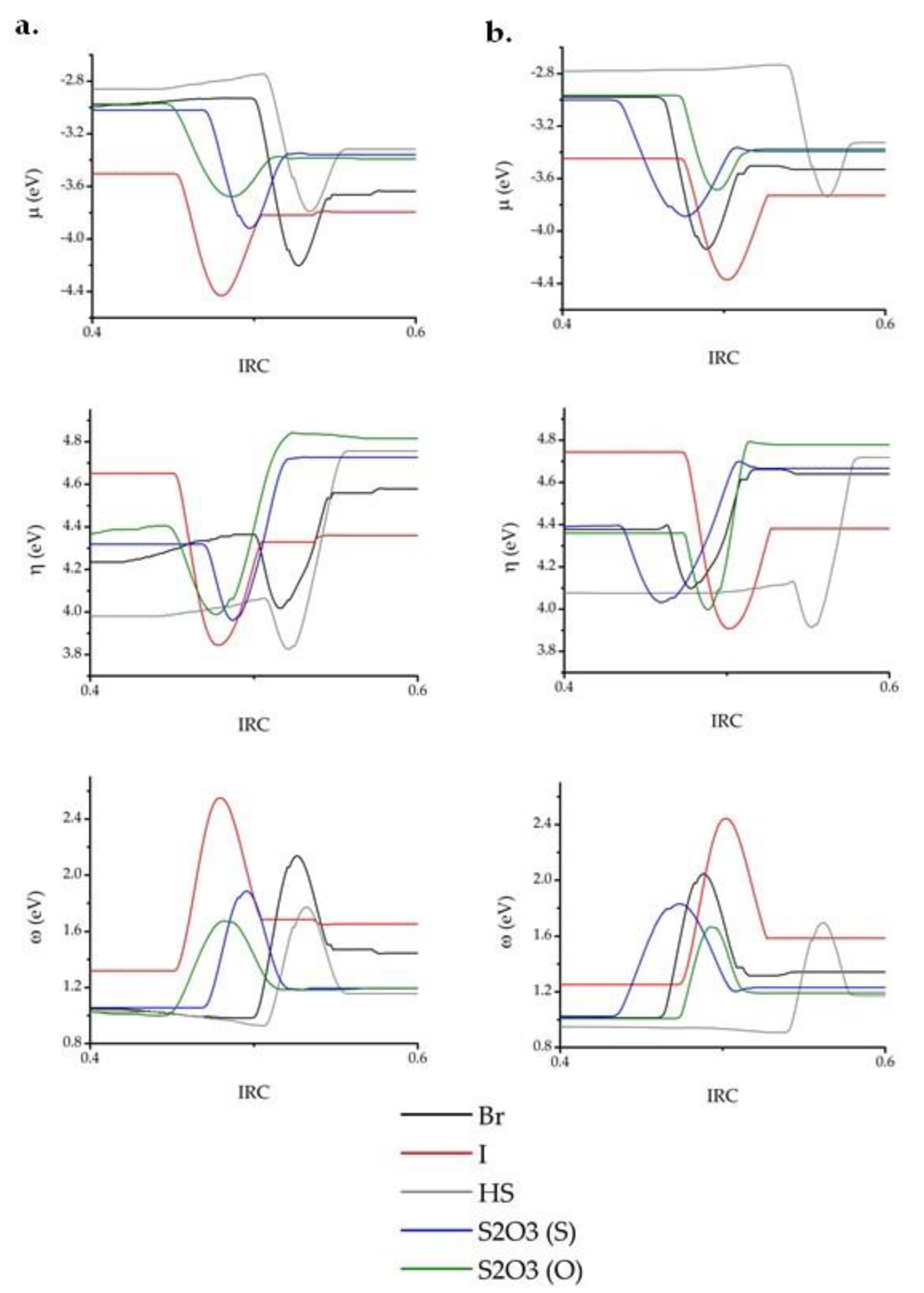
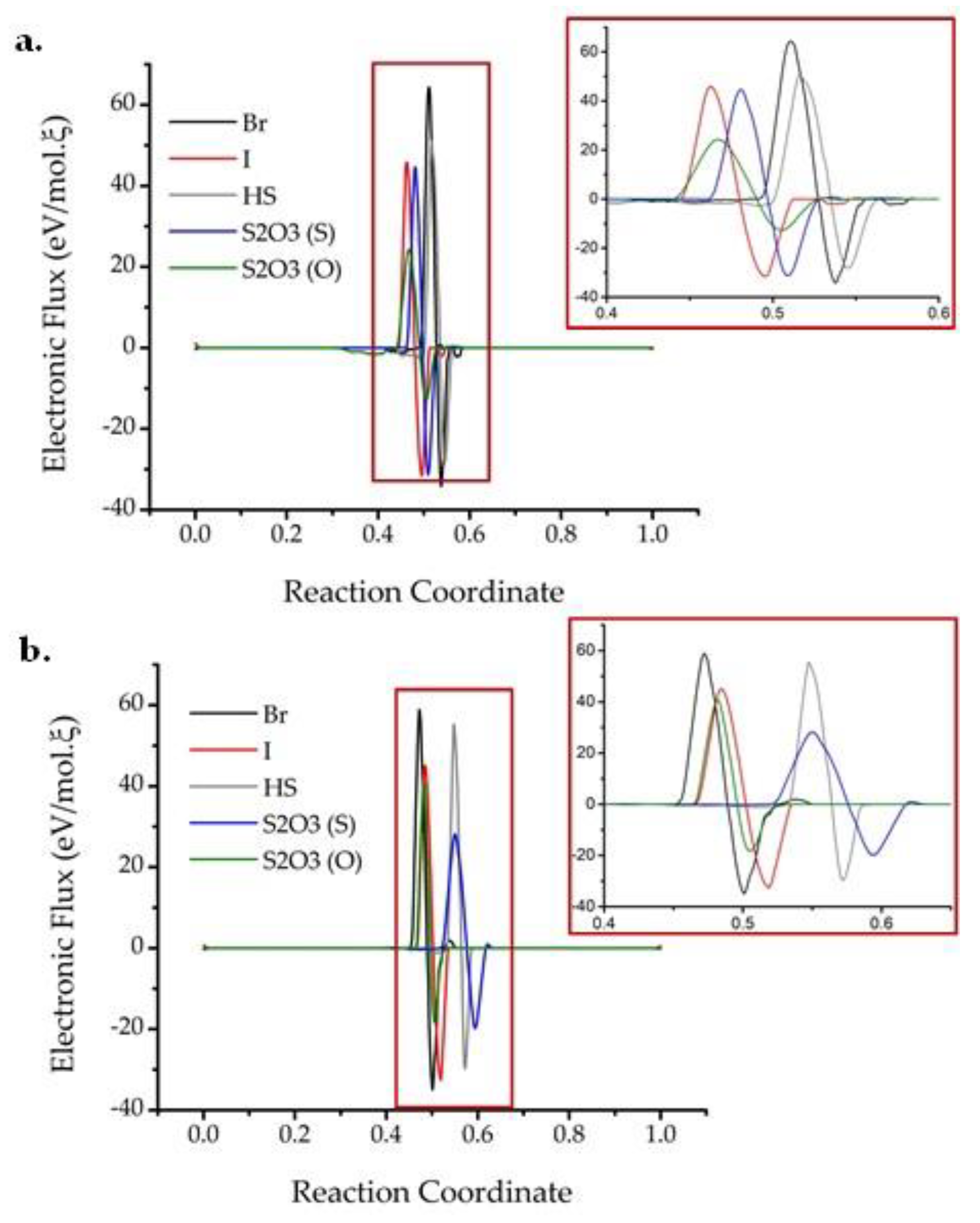
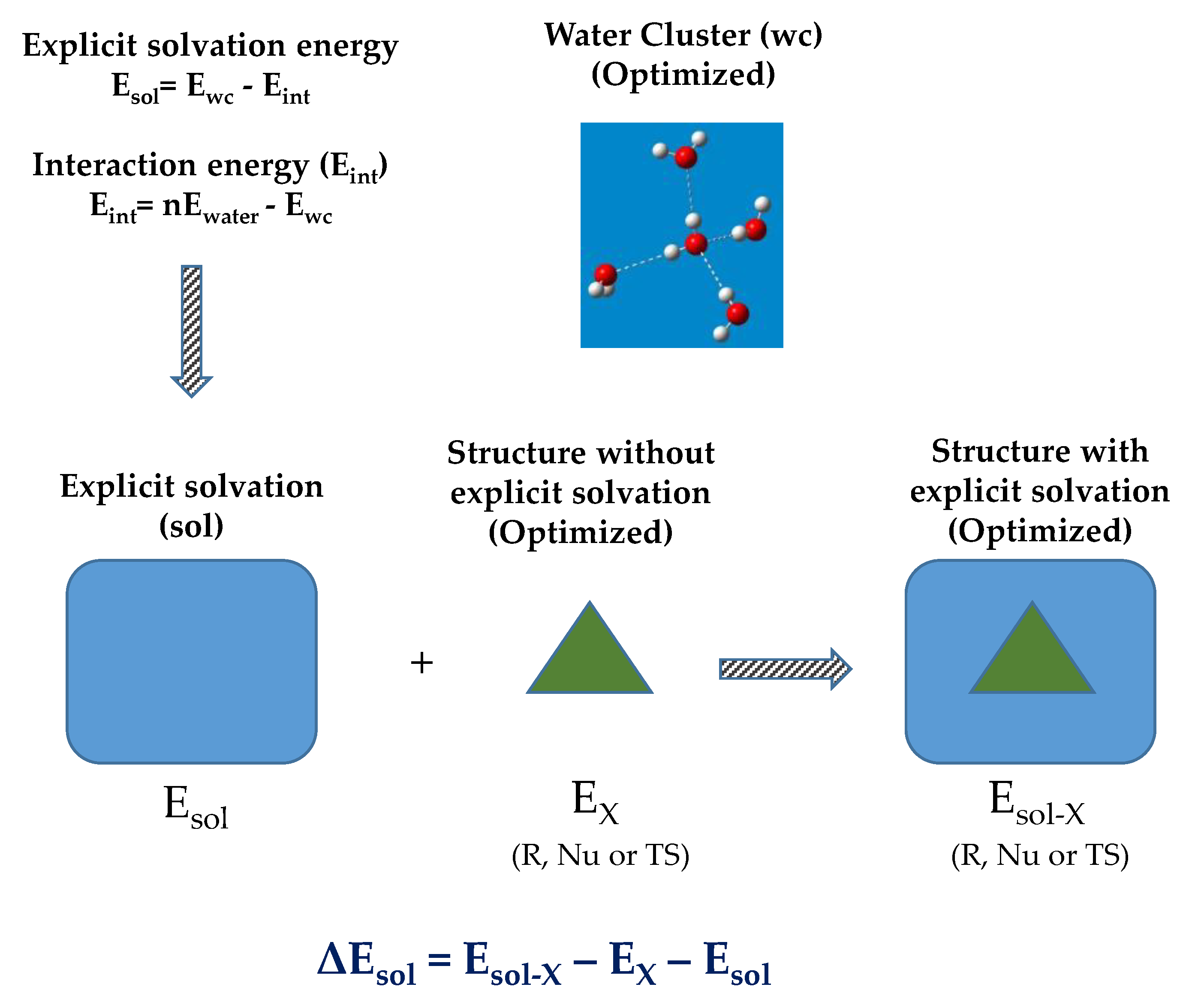
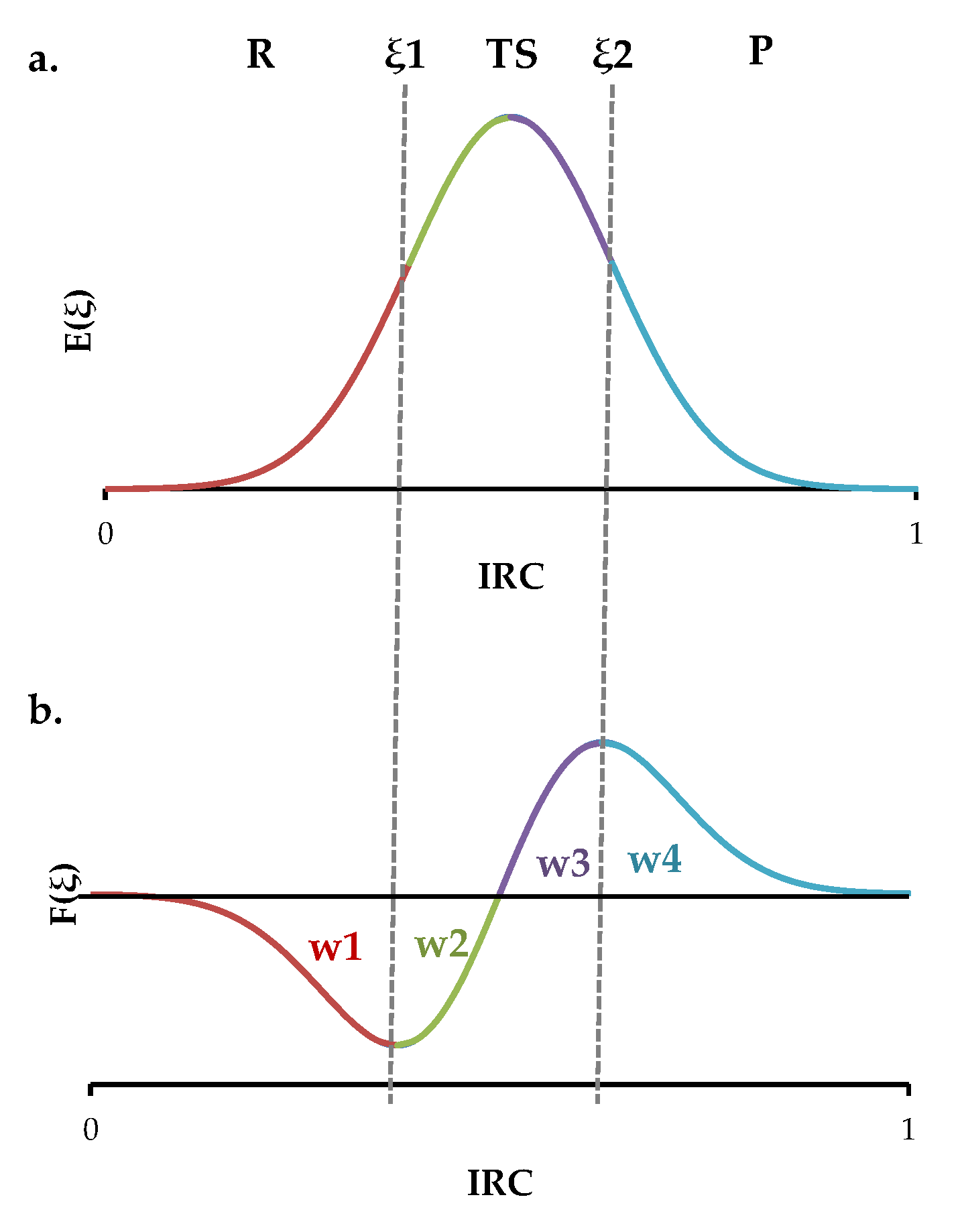
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Fartyal, D.; Agarwal, A.; James, D.; Borphukan, B.; Ram, B.; Sheri, V.; Agrawal, P.K.; Achary, V.M.M.; Reddy, M.K. Developing dual herbicide tolerant transgenic rice plants for sustainable weed management. Sci. Rep. 2018, 8, 11598. [Google Scholar] [CrossRef] [Green Version]
- Kraus, E.C.; Stout, M.J. Direct and indirect effects of herbicides on insect herbivores in rice, Oryza sativa. Sci. Rep. 2019, 9, 6998. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.S.; Jena, H.M. A systemic assessment of the environmental impacts and remediation strategies for chloroacetanilide herbicides. J. Water Process Eng. 2019, 31, 100860. [Google Scholar] [CrossRef]
- Bian, H.; Chen, J.; Cai, X.; Liu, P.; Wang, Y.; Huang, L.; Qiao, X.; Hao, C. Dechlorination of chloroacetanilide herbicides by plant growth regulator sodium bisulfite. Water Res. 2009, 43, 3566–3574. [Google Scholar] [CrossRef] [PubMed]
- Boparai, H.K.; Shea, P.J.; Comfort, S.D.; Snow, D.D. Dechlorinating chloroacetanilide herbicides by dithionite-treated aquifer sediment and surface soil. Environ. Sci. Technol. 2006, 40, 3043–3049. [Google Scholar] [CrossRef]
- Saha, S.; Dutta, D.; Karmakar, R.; Ray, D.P. Structure-toxicity relationship of chloroacetanilide herbicides: Relative impact on soil microorganisms. Environ. Toxicol. Pharmacol. 2012, 34, 307–314. [Google Scholar] [CrossRef]
- Barr, D.B.; Hines, C.J.; Olsson, A.O.; Deddens, J.A.; Bravo, R.; Striley, C.A.F.; Norrgran, J.; Needham, L.L. Identification of human urinary metabolites of acetochlor in exposed herbicide applicators by high-performance liquid chromatography-tandem mass spectrometry. J. Expo. Sci. Environ. Epidemiol. 2007, 17, 559–566. [Google Scholar] [CrossRef]
- Friedman, C.L.; Lemley, A.T.; Hay, A. Degradation of chloroacetanilide herbicides by anodic Fenton treatment. J. Agric. Food Chem. 2006, 54, 2640–2651. [Google Scholar] [CrossRef] [PubMed]
- Foley, M.E.; Sigler, V.; Gruden, C.L. A multiphasic characterization of the impact of the herbicide acetochlor on freshwater bacterial communities. ISME J. 2008, 2, 56–66. [Google Scholar] [CrossRef]
- He, W.; Fontmorin, J.M.; Soutrel, I.; Floner, D.; Fourcade, F.; Amrane, A.; Geneste, F. Reductive dechlorination of a chloroacetanilide herbicide in water by a Co complex-supported catalyst. Mol. Catal. 2017, 432, 8–14. [Google Scholar] [CrossRef]
- Loch, A.R.; Lippa, K.A.; Carlson, D.L.; Chin, Y.P.; Traina, S.J.; Roberts, A.L. Nucleophilic aliphatic substitution reactions of propachlor, alachlor, and metolachlor with bisulfide (HS−) and polysulfides (Sn2−). Environ. Sci. Technol. 2002, 36, 4065–4073. [Google Scholar] [CrossRef]
- Liu, W.; Gan, J.; Papiernik, S.K.; Yates, S.R. Structural influences in relative sorptivity of chloroacetanilide herbicides on soil. J. Agric. Food Chem. 2000, 48, 4320–4325. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; Cervantes, C.; Marquez, E. New insight into the chloroacetanilide herbicide degradation mechanism through a nucleophilic attack of hydrogen sulfide. Int. J. Mol. Sci. 2018, 19, 2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippa, K.A.; Demel, S.; Lau, I.H.; Roberts, A.L. Kinetics and Mechanism of the Nucleophilic Displacement Reactions of Chloroacetanilide Herbicides: Investigation of α-Substituent Effects. J. Agric. Food Chem. 2004, 52, 3010–3021. [Google Scholar] [CrossRef] [PubMed]
- Roubeix, V.; Fauvelle, V.; Tison-Rosebery, J.; Mazzella, N.; Coste, M.; Delmas, F. Assessing the impact of chloroacetanilide herbicides and their metabolites on periphyton in the Leyre River (SW France) via short term growth inhibition tests on autochthonous diatoms. J. Environ. Monit. 2012, 14, 1655–1663. [Google Scholar] [CrossRef]
- Cai, X.; Niu, L.; Zhang, Y.; Lang, X.; Yu, Y.; Chen, J. Discriminating multiple impacts of biogas residues amendment in selectively decontaminating chloroacetanilide herbicides. J. Agric. Food Chem. 2011, 59, 11177–11185. [Google Scholar] [CrossRef]
- Morton, M.D.; Walters, F.H.; Aga, D.S.; Thurman, E.M.; Larive, C.K. Nuclear Magnetic Resonance Identification of New Sulfonic Acid Metabolites of Chloroacetanilide Herbicides. J. Agric. Food Chem. 1997, 45, 1240–1243. [Google Scholar] [CrossRef]
- Gan, J.; Wang, Q.; Yates, S.R.; Koskinen, W.C.; Jury, W.A. Dechlorination of chloroacetanilide herbicides by thiosulfate salts. Proc. Natl. Acad. Sci. USA 2002, 99, 5189–5194. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Yates, S.R.; Papiernik, S.K.; Guo, M. Transformation of herbicide propachlor by an agrochemical thiourea. Environ. Sci. Technol. 2004, 38, 6855–6860. [Google Scholar] [CrossRef] [PubMed]
- Lippa, K.A.; Roberts, A.L. Correlation analyses for bimolecular nucleophilic substitution reactions of chloroacetanilide herbicides and their structural analogs with environmentally relevant nucleophiles. Environ. Toxicol. Chem. 2005, 24, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Vollhardt, P.; Schore, N. Organic Chemistry; Macmillan Learning: London, UK, 2014. [Google Scholar]
- Mann, D.J.; Hase, W.L. Trajectory studies of SN2 nucleophilic substitution. 6. Translational activation of the Cl− + CH3Cl reaction. J. Phys. Chem. A 1998, 102, 6208–6214. [Google Scholar] [CrossRef]
- Beg, H.; De, S.P.; Ash, S.; Das, D.; Misra, A. Polarizability, chemical hardness and ionization potential as descriptors to understand the mechanism of double proton transfer in acetamide dimer. Comput. Theor. Chem. 2013, 1005, 1–8. [Google Scholar] [CrossRef]
- Parr, R.G.; Chattaraj, P.K. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. [Google Scholar] [CrossRef]
- Toro-Labbe, A. Characterization of chemical reactions from the profiles of energy, chemical potential and hardness. J. Phys. Chem. A 1999, 103, 4398–4403. [Google Scholar] [CrossRef]
- Echegaray, E.; Toro-Labbé, A. Reaction electronic flux: A new concept to get insights into reaction mechanisms. Study of model symmetric nucleophilic substitutions. J. Phys. Chem. A 2008, 112, 11801–11807. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef] [Green Version]
- Sosa, C.; Andzelm, J.; Elkin, B.C.; Wimmer, E.; Dobbs, K.D.; Dixon, D.A. A local density functional study of the structure and vibrational frequencies of molecular transition-metal compounds. J. Phys. Chem. 1992, 96, 6630–6636. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nakajima, M.; Nemoto, T. Determination of the best functional and basis sets for optimization of the structure of hypervalent iodines and calculation of their first and second bond dissociation enthalpies. J. Phys. Org. Chem. 2019, 32, e3961. [Google Scholar] [CrossRef]
- Li, L.; Sun, R.; Zheng, R.; Huang, Y.; Chen, H. A Computational mechanistic study of the chemo- and enantioselectivity in the 1,4-addition reaction catalyzed by a rh complex of sulfinyl-phosphine. Eur. J. Org. Chem. 2018, 2018, 3426–3431. [Google Scholar] [CrossRef]
- Zara, Z.; Iqbal, J.; Ayub, K.; Irfan, M.; Mahmood, A.; Khera, R.A.; Eliasson, B. A comparative study of DFT calculated and experimental UV/Visible spectra for thirty carboline and carbazole based compounds. J. Mol. Struct. 2017, 1149, 282–298. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Performance of SM6, SM8, and SMD on the SAMPL1 test set for the prediction of small-molecule solvation free energies. J. Phys. Chem. B 2009, 113, 4538–4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Ouellette, R.J.; Rawn, J.D.; Ouellette, R.J.; Rawn, J.D. Introduction to Organic Reaction Mechanisms; Academic Press: London, UK, 2018; ISBN 978-0-12-812838-1. [Google Scholar]
- Rastelli, A.; Bagatti, M.; Gandolfi, R. Ab initio study of concerted cycloadditions of allene, monofluoroallene, and 1,1-difluoroallene with diazomethane, formonitrile oxide, cyclopentadiene, and furane. J. Am. Chem. Soc. 1995, 117, 4965–4975. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Accurate reaction paths using a Hessian based predictor-corrector integrator. J. Chem. Phys. 2004, 120, 9918–9924. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Using Hessian updating to increase the efficiency of a Hessian based predictor-corrector reaction path following method. J. Chem. Theory Comput. 2005, 1, 61–69. [Google Scholar] [CrossRef]
- Gonzalez, C.; Bernhard Schlegel, H. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Herrera, B.; Toro-Labbé, A. The role of reaction force and chemical potential in characterizing the mechanism of double proton transfer in the adenine-uracil complex. J. Phys. Chem. A 2007, 111, 5921–5926. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.; Toro-Labbé, A. The reaction force. A scalar property to characterize reaction mechanisms. J. Math. Chem. 2009, 45, 911–927. [Google Scholar] [CrossRef]
- Toro-Labbé, A.; Gutiérrez-Oliva, S.; Concha, M.C.; Murray, J.S.; Politzer, P. Analysis of two intramolecular proton transfer processes in terms of the reaction force. J. Chem. Phys. 2004, 121, 4570–4576. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Toro-Labbé, A.; Gutiérrez-Oliva, S.; Herrera, B.; Jaque, P.; Concha, M.C.; Murray, J.S. The reaction force: Three key points along an intrinsic reaction coordinate. J. Chem. Sci. 2005, 117, 467–472. [Google Scholar] [CrossRef]
- Jaque, P.; Toro-Labbé, A.; Politzer, P.; Geerlings, P. Reaction force constant and projected force constants of vibrational modes along the path of an intramolecular proton transfer reaction. Chem. Phys. Lett. 2008, 456, 135–140. [Google Scholar] [CrossRef]
- Pedraza-González, L.; Galindo, J.F.; González, R.; Reyes, A. Revisiting the dielectric constant effect on the nucleophile and leaving group of prototypical backside SN2 reactions: A reaction force and atomic contribution analysis. J. Phys. Chem. A 2016, 120, 8360–8368. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Arriagada, D.; Toro-Labbe, A.; Mora, J.R.; Rincón, L.; Mereau, R.; Torres, F.J. Theoretical analysis of C–F bond cleavage mediated by cob[I]alamin-based structures. J. Mol. Model. 2017, 23, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Neelamraju, V.S.K.; Jaganade, T. A reaction force perspective of a model amide bond formation reaction. Comput. Theor. Chem. 2019, 1151, 91–98. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef]
- Flores-Morales, P.; Gutiérrez-Oliva, S.; Silva, E.; Toro-Labbé, A. The reaction electronic flux: A new descriptor of the electronic activity taking place during a chemical reaction. Application to the characterization of the mechanism of the Schiff’s base formation in the Maillard reaction. J. Mol. Struct. THEOCHEM 2010, 943, 121–126. [Google Scholar] [CrossRef]
- Giri, S.; Echegaray, E.; Ayers, P.W.; Nuñez, A.S.; Lund, F.; Toro-Labbé, A. Insights into the mechanism of an S N2 reaction from the reaction force and the reaction electronic flux. J. Phys. Chem. A 2012, 116, 10015–10026. [Google Scholar] [CrossRef]
- Koopmans, T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica 1934, 1, 104–113. (In Germany) [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Chandra, A.K.; Nguyen, M.T. Density functional approach to regiochemistry, activation energy, and hardness profile in 1,3-dipolar cycloadditions. J. Phys. Chem. A 1998, 102, 6181–6185. [Google Scholar] [CrossRef]
- De Luca, G.; Sicilia, E.; Russo, N.; Mineva, T. On the hardness evaluation in solvent for neutral and charged systems. J. Am. Chem. Soc. 2002, 124, 1494–1499. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aurell, M.J.; Contreras, R. Quantitative characterization of the global electrophilicity pattern of some reagents involved in 1,3-dipolar cycloaddition reactions. Tetrahedron 2003, 59, 3117–3125. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Meneses, L.; Fuentealba, P.; Contreras, R. Relationship between the electrophilicity of substituting agents and substrate selectivity in Friedel-Crafts reactions. Tetrahedron 2005, 61, 831–836. [Google Scholar] [CrossRef]
- Pérez, P.; Toro-Labbé, A.; Contreras, R. Solvent effects on electrophilicity. J. Am. Chem. Soc. 2001, 123, 5527–5531. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.; Parida, R.; Jana, M.; Gutiérrez-Oliva, S.; Toro-Labbe, A. Insights into the Mechanism of Ground and Excited State Double Proton Transfer Reaction in Formic Acid Dimer. J. Phys. Chem. A 2017, 121, 9531–9543. [Google Scholar] [CrossRef]
- Duarte, F.; Toro-Labbé, A. The mechanism of H2 activation by (amino)carbenes. J. Phys. Chem. A 2011, 115, 3050–3059. [Google Scholar] [CrossRef]
- Lendvay, G. Characterization of the progress of chemical reactions by ab initio bond orders. J. Phys. Chem. 1994, 98, 6098–6104. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Brea, O.; Loroño, M.; Marquez, E.; Mora, J.R.; Cordova, T.; Chuchani, G. Theoretical study of methoxy group influence in the gas-phase elimination kinetics of methoxyalkyl chlorides. Int. J. Quantum Chem. 2012, 112, 2504–2514. [Google Scholar] [CrossRef]
- Marquez, E.; Domínguez, R.M.; Mora, J.R.; Córdova, T.; Chuchani, G. Experimental and theoretical studies of the homogeneous, unimolecular gas-phase elimination kinetics of trimethyl orthovalerate and trimethyl orthochloroacetate. J. Phys. Chem. A 2010, 114, 4203–4209. [Google Scholar] [CrossRef] [PubMed]
- Lezama, J.; Márquez, E.; Mora, J.R.; Córdova, T.; Chuchani, G. Theoretical calculations on the mechanisms of the gas phase elimination kinetics of chlorocyclohexane, 3-chlorocyclohexene and 4-chlorocyclohexene. J. Mol. Struct. THEOCHEM 2009, 916, 17–22. [Google Scholar] [CrossRef]
- Bhuvaneswari, R.; Divya Bharathi, M.; Anbalagan, G.; Chakkaravarthi, G.; Murugesan, K.S. Molecular structure, vibrational spectroscopic (FT-IR, FT-Raman), NBO, HOMO and LUMO analysis of morpholinium oxalate by density functional method. J. Mol. Struct. 2018, 1173, 188–195. [Google Scholar] [CrossRef]
- Halim, S.A.; Khalil, A.K. TD-DFT calculations, NBO analysis and electronic absorption spectra of some thiazolo[3,2-a] pyridine derivatives. J. Mol. Struct. 2017, 1147, 651–667. [Google Scholar] [CrossRef]
- Mora, J.R.; Nuñez, O.; Rincón, L.; Torres, F.J. Understanding the role of Zn2+ in the hydrolysis of glycylserine: A mechanistic study by using density functional theory. Mol. Phys. 2017, 115, 403–412. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleophile | Acetochlor | Alachlor | Metolachlor | Propachlor |
|---|---|---|---|---|
| Br− | 23.81 | 23.84 | 22.98 | 22.18 |
| I− | 21.80 | 22.04 | 22.53 | 21.22 |
| HS− | 20.45 | 19.33 | 20.25 | 18.36 |
| S2O3−2 (S) | 18.73 | 20.08 | 20.29 | 19.09 |
| S2O3−2 (O) | 26.28 | 26.37 | 26.77 | 26.68 |
| System | Br− | I− | HS− | S2O3−2 (S) | S2O3−2 (S) | |
|---|---|---|---|---|---|---|
| Alachlor | %Evav | 49.1 | 56.8 | 40.8 | 45.0 | 45.8 |
| Sy | 0.904 | 0.928 | 0.899 | 0.892 | 0.839 | |
| Propachlor | %Evav | 49.2 | 56.4 | 41.2 | 46.4 | 46.5 |
| Sy | 0.903 | 0.917 | 0.895 | 0.892 | 0.833 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuesta, S.A.; Torres, F.J.; Rincón, L.; Paz, J.L.; Márquez, E.A.; Mora, J.R. Effect of the Nucleophile’s Nature on Chloroacetanilide Herbicides Cleavage Reaction Mechanism. A DFT Study. Int. J. Mol. Sci. 2021, 22, 6876. https://doi.org/10.3390/ijms22136876
Cuesta SA, Torres FJ, Rincón L, Paz JL, Márquez EA, Mora JR. Effect of the Nucleophile’s Nature on Chloroacetanilide Herbicides Cleavage Reaction Mechanism. A DFT Study. International Journal of Molecular Sciences. 2021; 22(13):6876. https://doi.org/10.3390/ijms22136876
Chicago/Turabian StyleCuesta, Sebastián A., F. Javier Torres, Luis Rincón, José Luis Paz, Edgar A. Márquez, and José R. Mora. 2021. "Effect of the Nucleophile’s Nature on Chloroacetanilide Herbicides Cleavage Reaction Mechanism. A DFT Study" International Journal of Molecular Sciences 22, no. 13: 6876. https://doi.org/10.3390/ijms22136876
APA StyleCuesta, S. A., Torres, F. J., Rincón, L., Paz, J. L., Márquez, E. A., & Mora, J. R. (2021). Effect of the Nucleophile’s Nature on Chloroacetanilide Herbicides Cleavage Reaction Mechanism. A DFT Study. International Journal of Molecular Sciences, 22(13), 6876. https://doi.org/10.3390/ijms22136876