
Enhanced NAMPT-Mediated NAD Salvage Pathway Contributes to Psoriasis Pathogenesis by Amplifying Epithelial Auto-Inflammatory Circuits

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Cell Cultures and Treatments
2.3. Immunohistochemistry
2.4. NAD Measurement
2.5. RNA Isolation and RT-PCR
2.6. Immunoblotting and Densitometry
2.7. Proliferation Assays
2.8. Wounding Migration Assay
2.9. Flow Cytometry Analysis
2.10. Enzyme-Linked Immunosorbent Assays (ELISA)
2.11. Statistical Analysis
3. Results
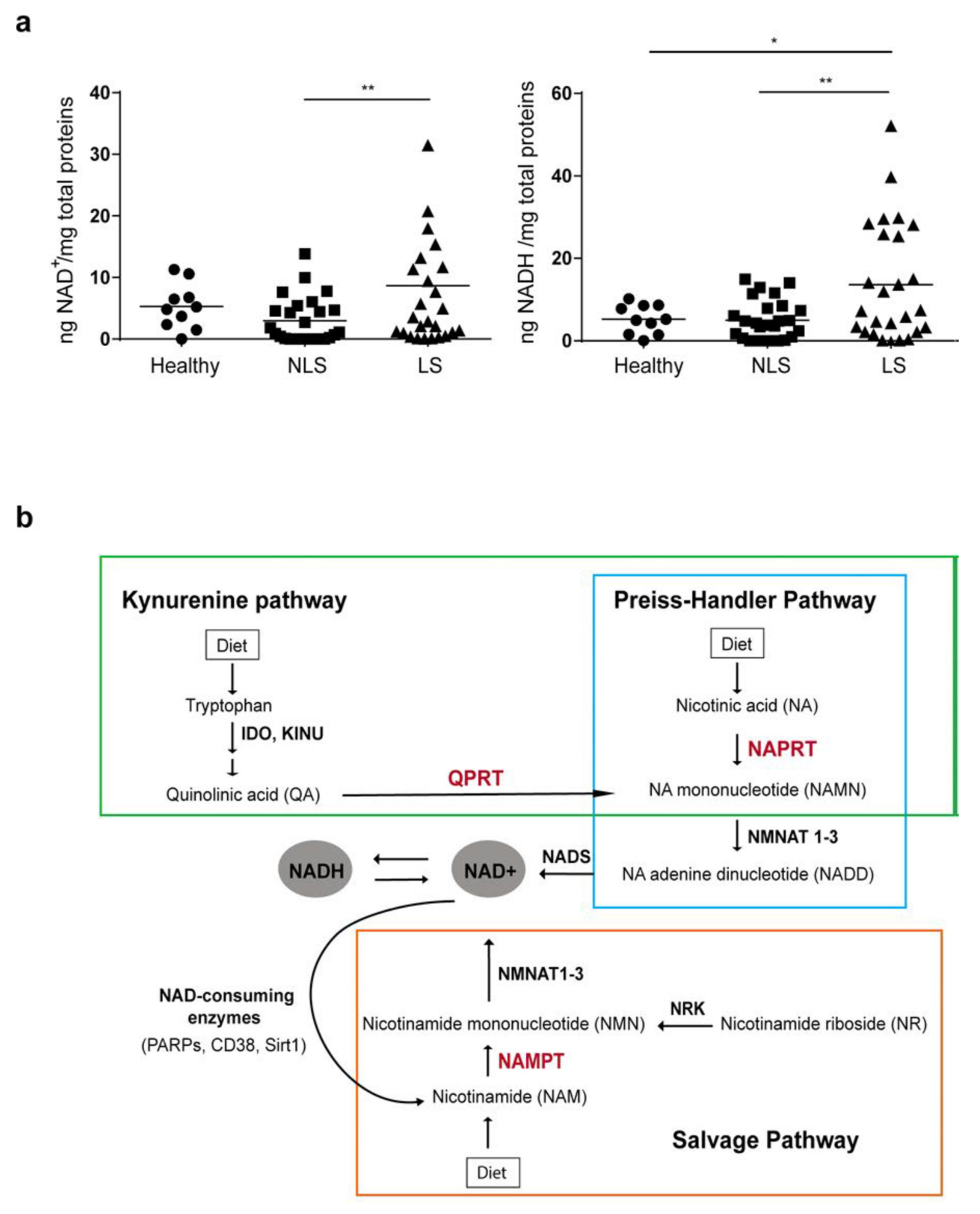
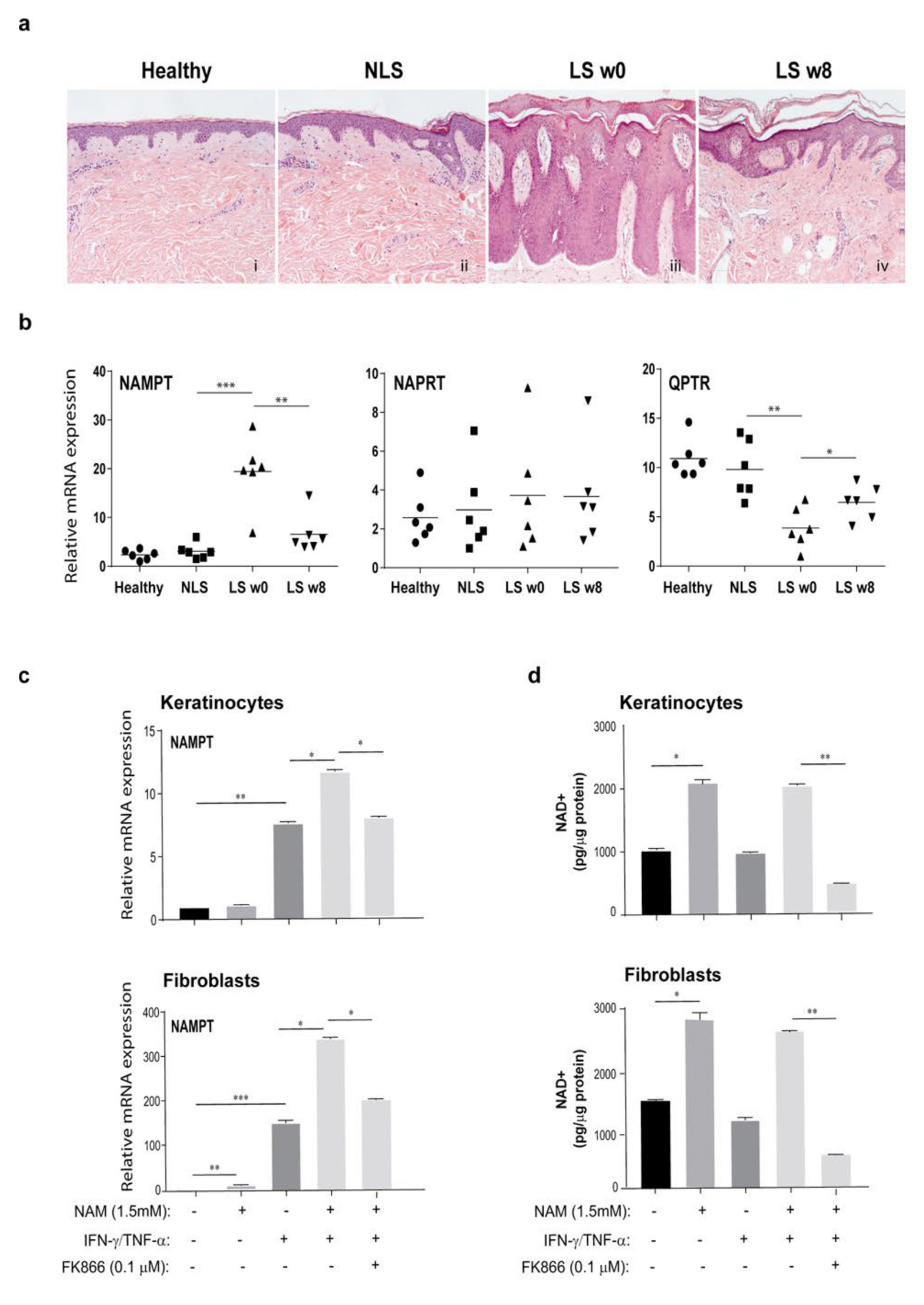
3.1. NAD Content Is Enhanced in LS Psoriatic Skin and Is Associated to High NAMPT Transcriptional Levels
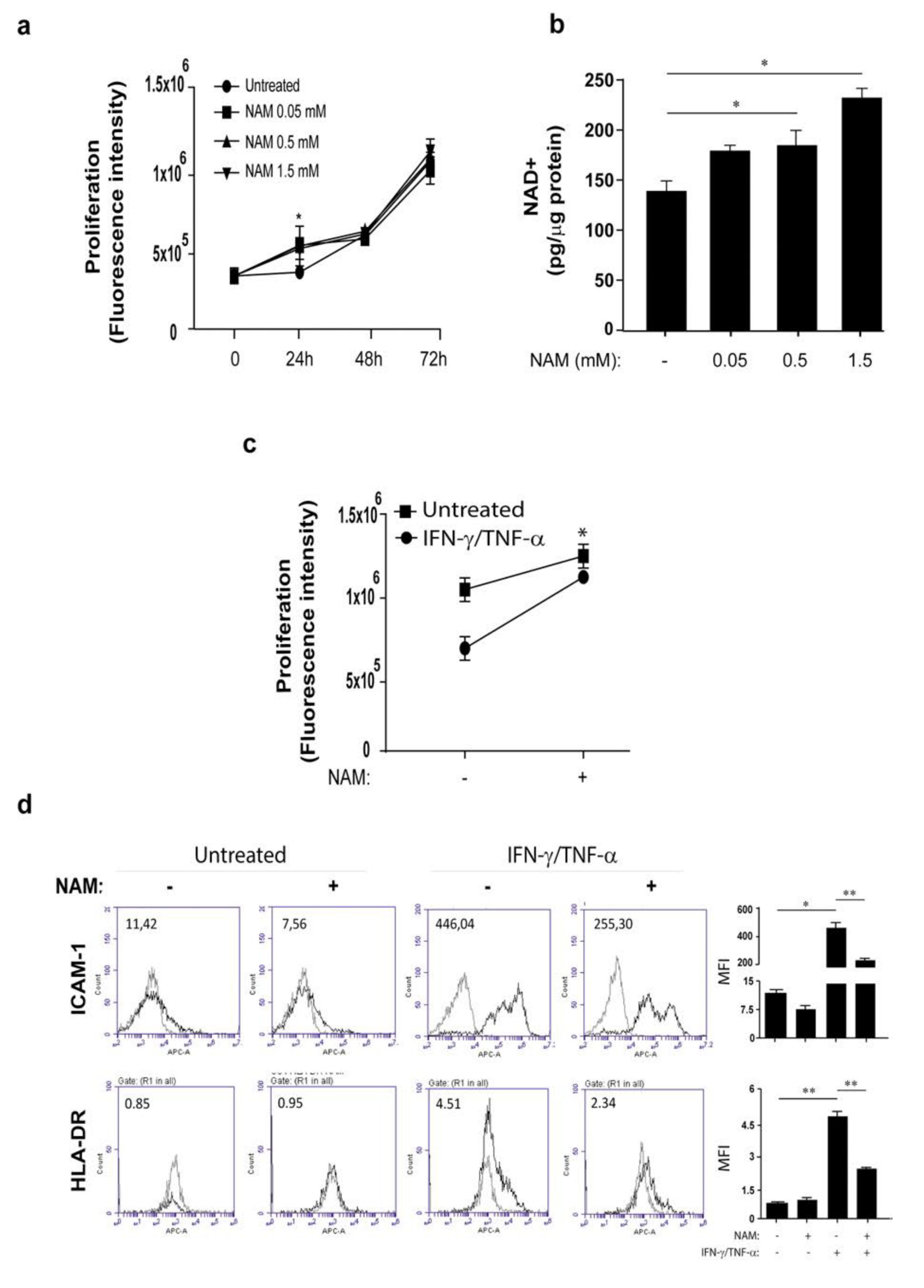
3.2. NAM Metabolism Synergizes with Psoriatic Cytokine Milieu in the Upregulation of NAMPT Expression
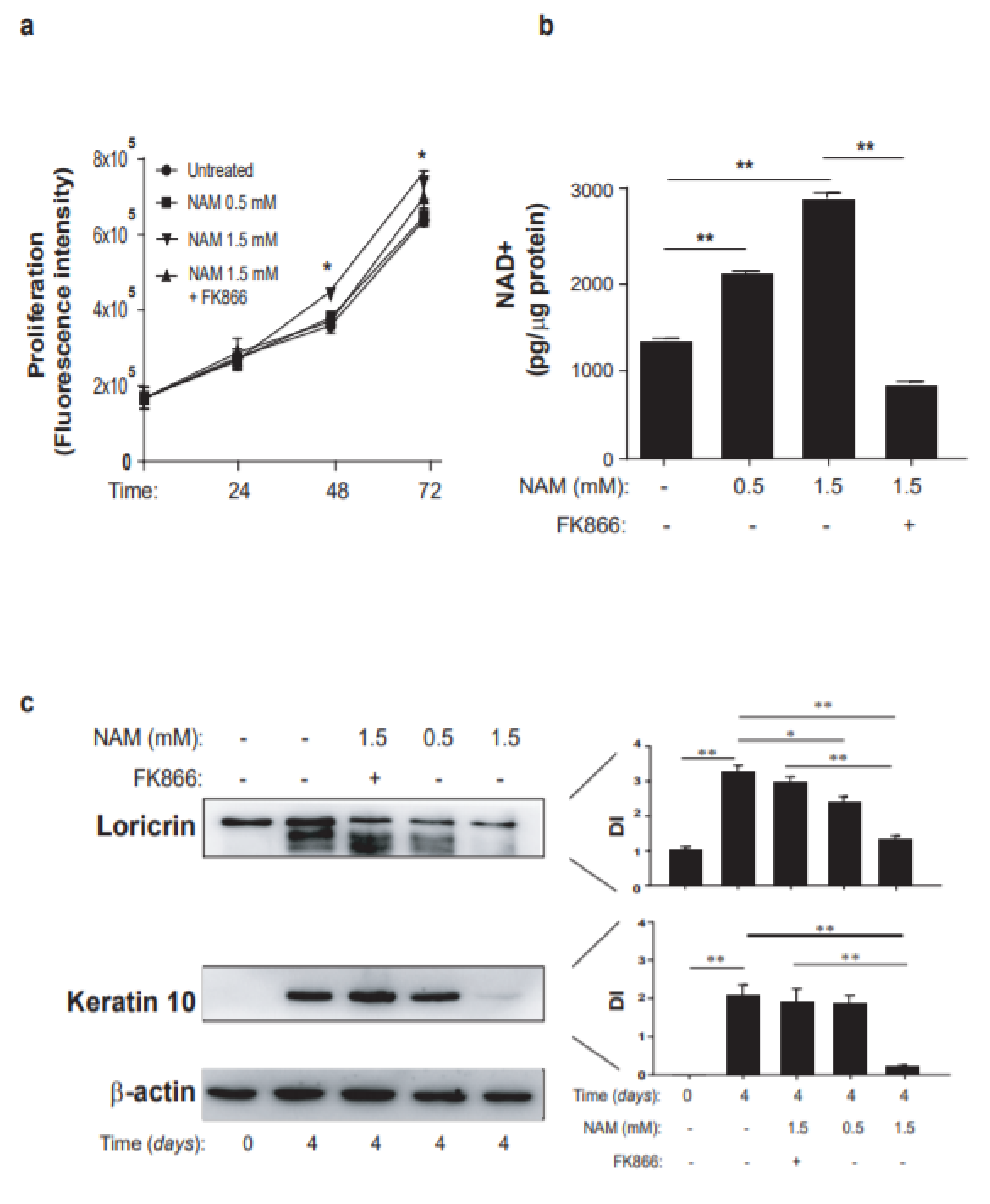
3.3. Opposite Effects of NAM on Cytokine-Activated Human Keratinocytes and Dermal Fibroblasts
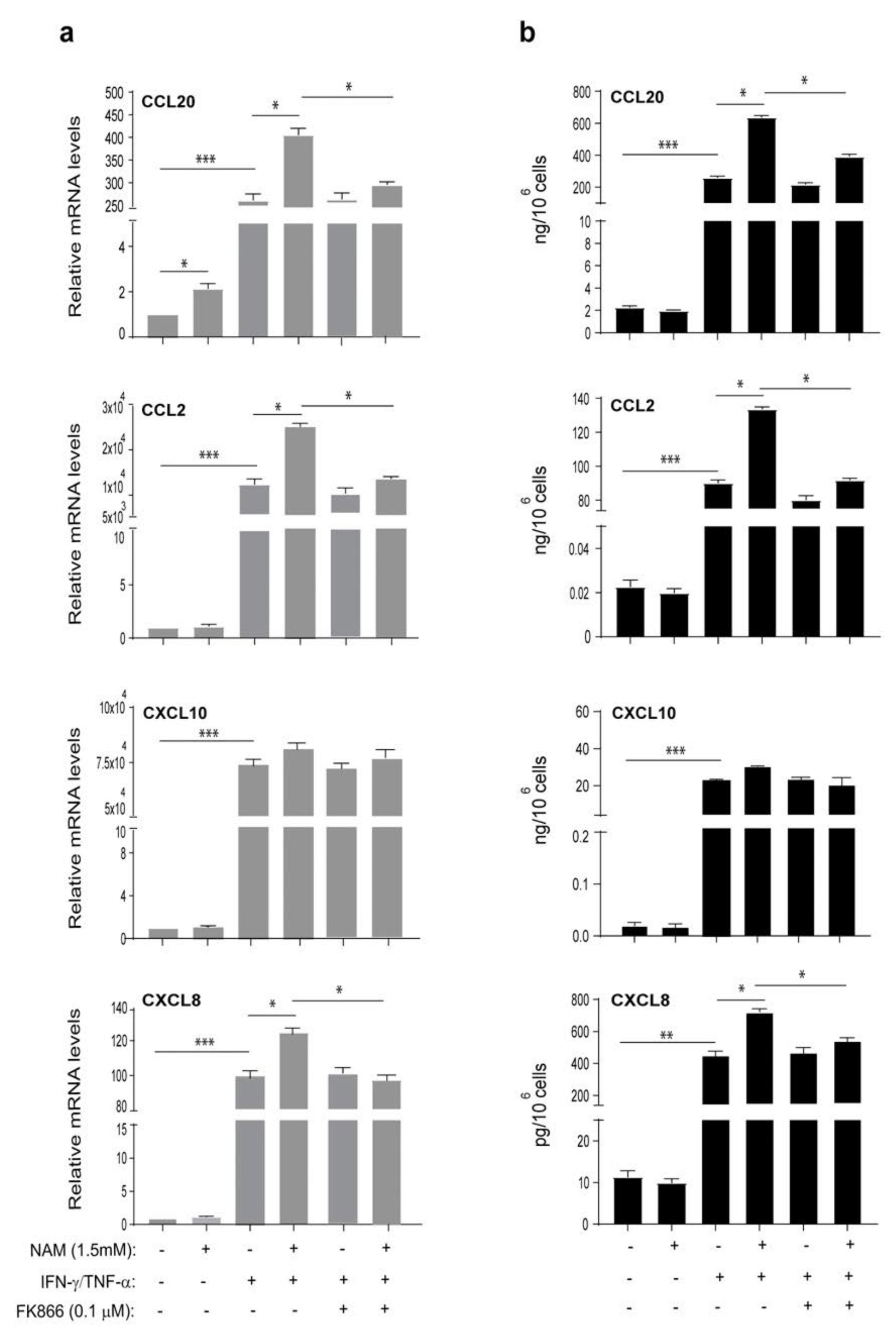
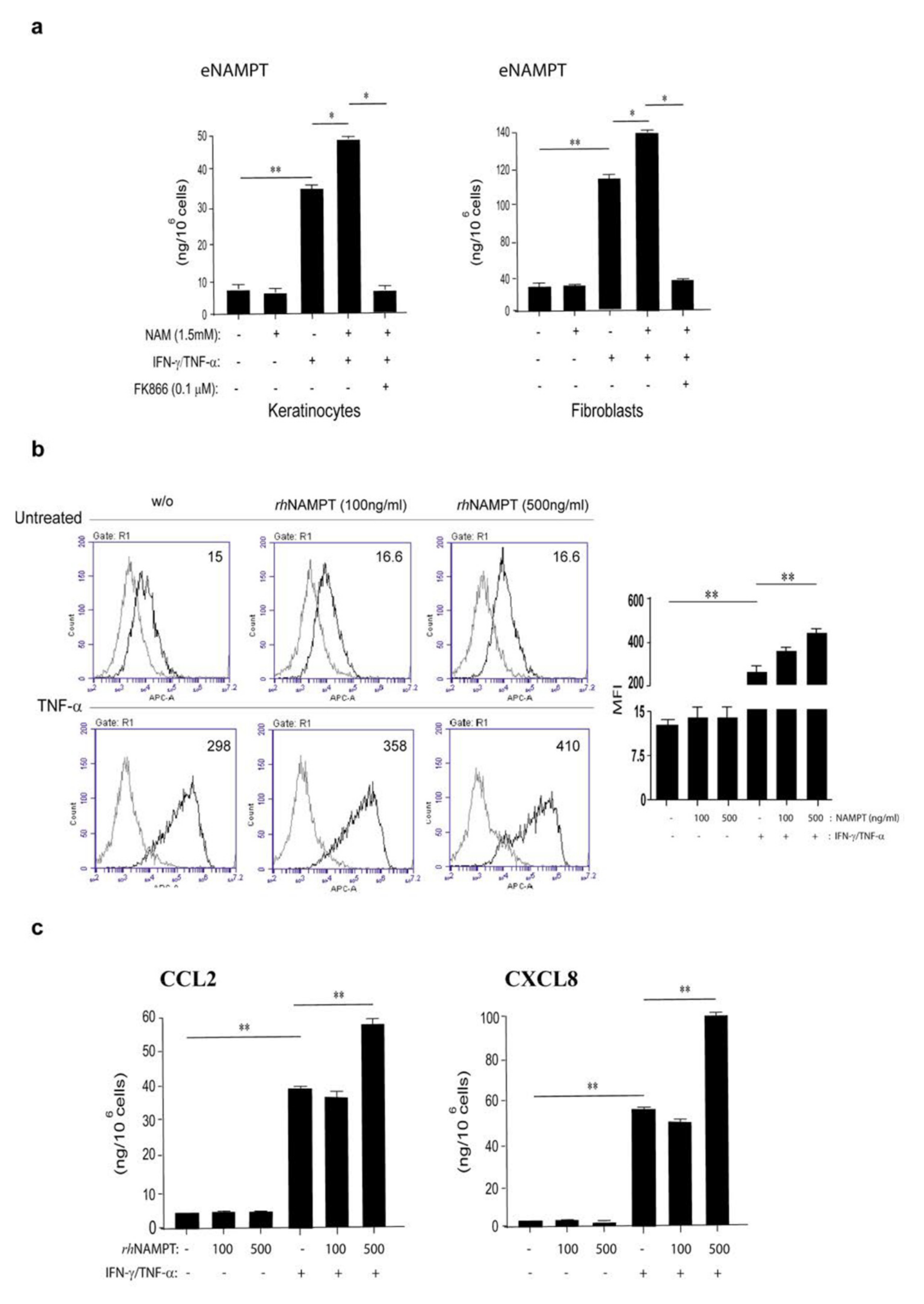
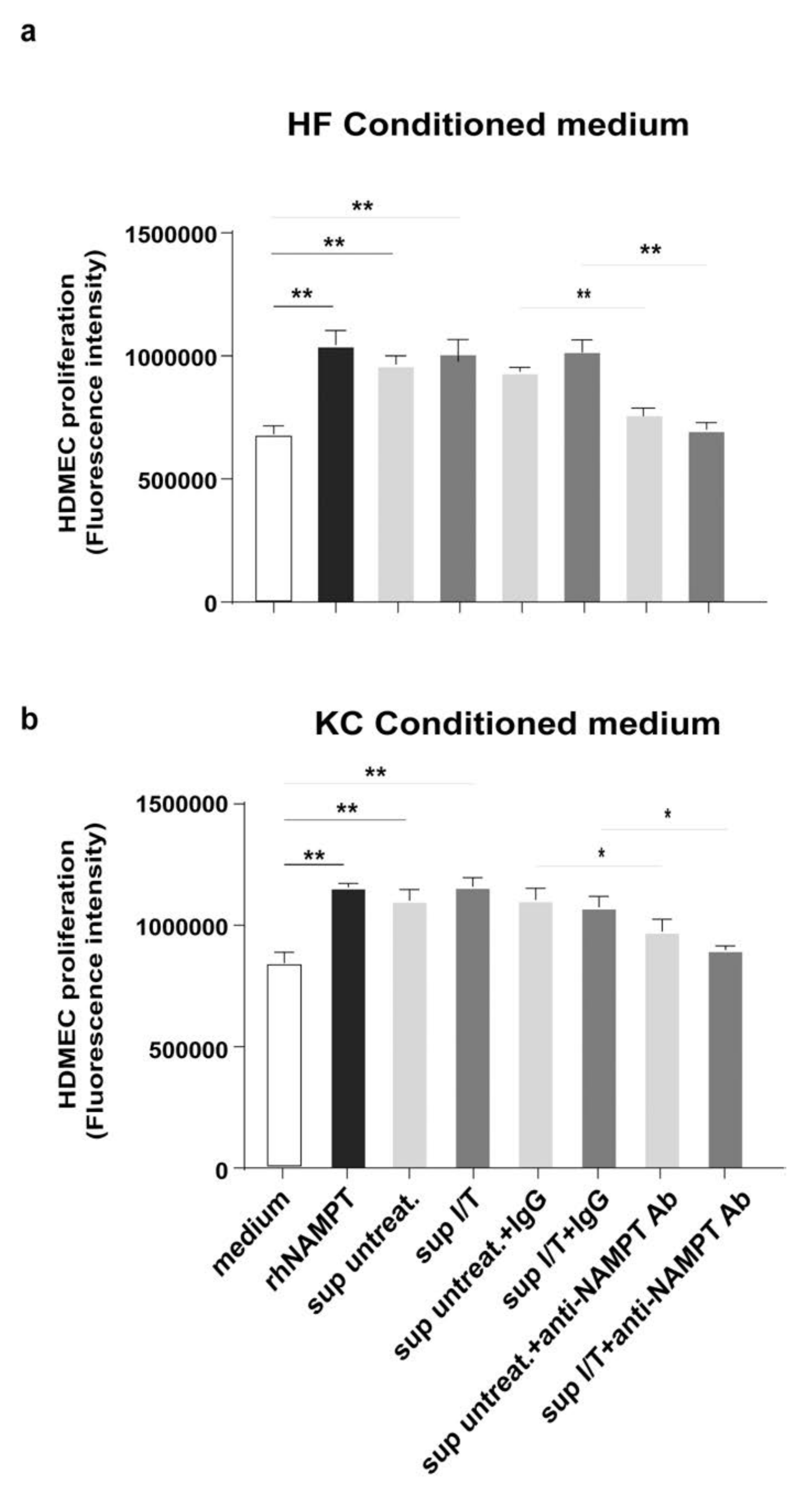
3.4. NAMPT Is Strongly Released by Activated Keratinocytes and Fibroblasts and Induces Proliferation and Inflammation in Dermal Endothelial Cells
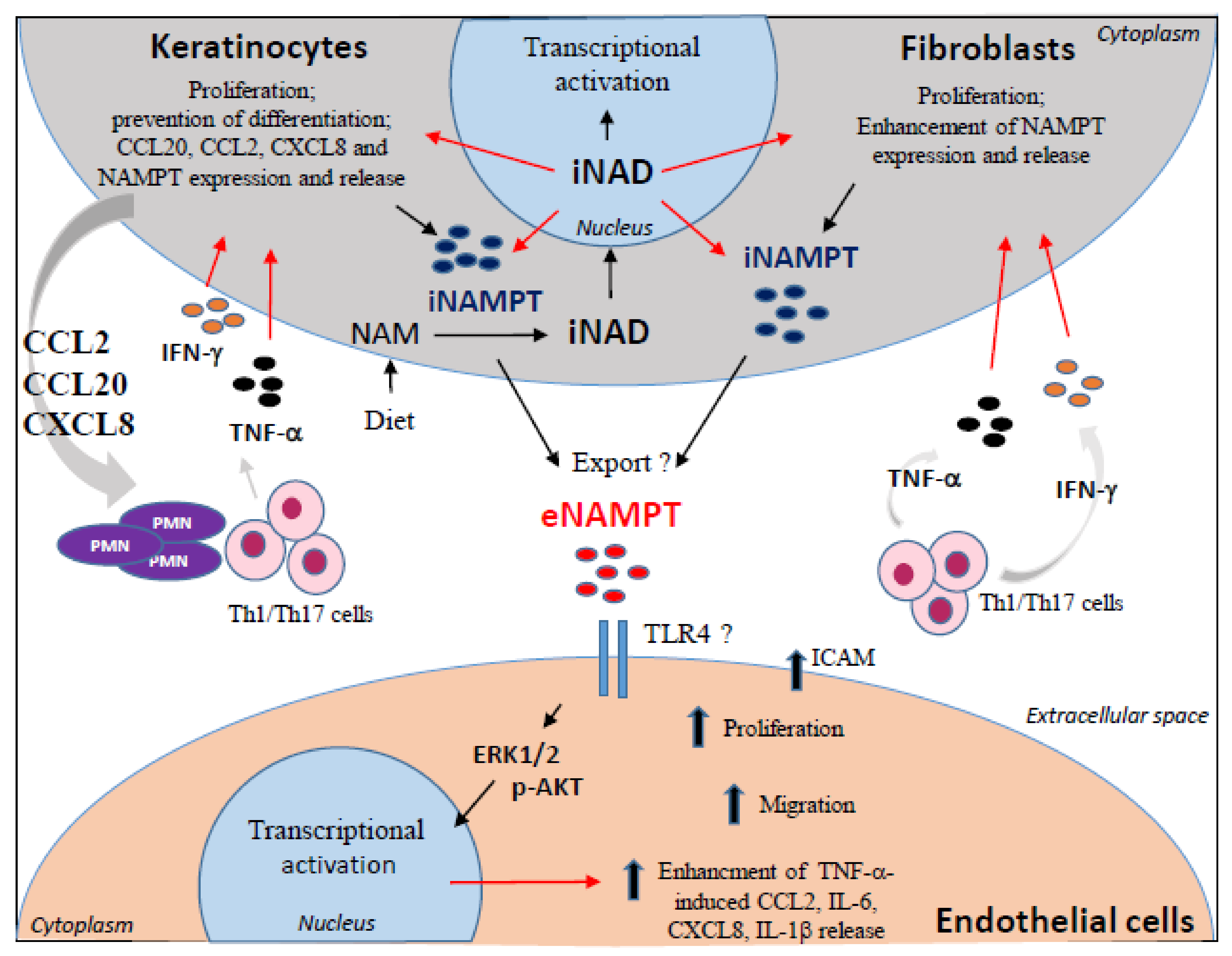
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Status
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Katsyuba, E.; Auwerx, J. Modulating NAD + metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef] [PubMed]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Burgos, E. NAMPT in Regulated NAD Biosynthesis and its Pivotal Role in Human Metabolism. Curr. Med. Chem. 2011, 18, 1947–1961. [Google Scholar] [CrossRef]
- Magni, G.; Amici, A.; Emanuelli, M.; Orsomando, G.; Raffaelli, N.; Ruggieri, S. Enzymology of NAD+ homeostasis in man. Cell. Mol. Life Sci. 2004, 61, 19–34. [Google Scholar] [CrossRef]
- Sauve, A.A. NAD+and Vitamin B3: From Metabolism to Therapies. J. Pharmacol. Exp. Ther. 2007, 324, 883–893. [Google Scholar] [CrossRef] [Green Version]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. Part B Clin. Cytom. 2013, 84, 207–217. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G. Age-Associated Changes In Oxidative Stress and NAD+ Metabolism In Human Tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S.-I. Nicotinamide Mononucleotide, a Key NAD+ Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Li, P.-H.; Zhang, R.; Cheng, L.-Q.; Liu, J.-J.; Chen, H.-Z. Metabolic regulation of immune cells in proinflammatory microenvironments and diseases during ageing. Ageing Res. Rev. 2020, 64, 101165. [Google Scholar] [CrossRef]
- Snaidr, V.A.; Damian, D.L.; Halliday, G.M. Nicotinamide for photoprotection and skin cancer chemoprevention: A review of efficacy and safety. Exp. Dermatol. 2019, 28, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Terakata, M.; Fukuwatari, T.; Sano, M.; Nakao, N.; Sasaki, R.; Fukuoka, S.-I.; Shibata, K. Establishment of True Niacin Deficiency in Quinolinic Acid Phosphoribosyltransferase Knockout Mice. J. Nutr. 2012, 142, 2148–2153. [Google Scholar] [CrossRef] [Green Version]
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; De Giorgis, T.; Kiess, W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015, 11, 535–546. [Google Scholar] [CrossRef]
- Revollo, J.R.; Körner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin Regulates Insulin Secretion in β Cells as a Systemic NAD Biosynthetic Enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Garten, A.; Petzold, S.; Barnikol-Oettler, A.; Körner, A.; Thasler, W.; Kratzsch, J.; Kiess, W.; Gebhardt, R. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is constitutively released from human hepatocytes. Biochem. Biophys. Res. Commun. 2010, 391, 376–381. [Google Scholar] [CrossRef]
- Kover, K.; Tong, P.Y.; Watkins, D.; Clements, M.; Stehno-Bittel, L.; Novikova, L.; Bittel, D.; Kibiryeva, N.; Stuhlsatz, J.; Yan, Y.; et al. Expression and Regulation of Nampt in Human Islets. PLoS ONE 2013, 8, e58767. [Google Scholar] [CrossRef]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an Adipocytokine with Proinflammatory and Immunomodulating Properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Nozaki, M.; Fukuhara, A.; Segawa, K.; Aoki, N.; Matsuda, M.; Komuro, R.; Shimomura, I. Visfatin is released from 3T3-L1 adipocytes via a non-classical pathway. Biochem. Biophys. Res. Commun. 2007, 359, 194–201. [Google Scholar] [CrossRef]
- Sierra-Honigmann, M.R.; Nath, A.K.; Murakami, C.; García-Cardeña, G.; Papapetropoulos, A.; Sessa, W.C.; Madge, L.A.; Schechner, J.S.; Schwabb, M.B.; Polverini, P.J.; et al. Biological Action of Leptin as an Angiogenic Factor. Science 1998, 281, 1683–1686. [Google Scholar] [CrossRef]
- Mu, H.; Ohashi, R.; Yan, S.; Chai, H.; Yang, H.; Lin, P.; Yao, Q.; Chen, C. Adipokine resistin promotes in vitro angiogenesis of human endothelial cells. Cardiovasc. Res. 2006, 70, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Kobayashi, H.; Kihara, S.; Kumada, M.; Sato, K.; Inoue, T.; Funahashi, T.; Walsh, K. Adiponectin Stimulates Angiogenesis by Promoting Cross-talk between AMP-activated Protein Kinase and Akt Signaling in Endothelial Cells. J. Biol. Chem. 2004, 279, 1304–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samal, B.; Sun, Y.; Stearns, G.; Xie, C.; Suggs, S.; Mcniece, I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell. Biol. 1994, 14, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-R.; Bae, S.-K.; Choi, K.-S.; Park, S.-Y.; Jun, H.O.; Lee, J.-Y.; Jang, H.-O.; Yun, I.; Yoon, K.-H.; Kim, Y.-J.; et al. Visfatin promotes angiogenesis by activation of extracellular signal-regulated kinase 1/2. Biochem. Biophys. Res. Commun. 2007, 357, 150–156. [Google Scholar] [CrossRef]
- Bae, Y.-H.; Bae, M.-K.; Kim, S.-R.; Lee, J.H.; Wee, H.-J.; Bae, S.-K. Upregulation of fibroblast growth factor-2 by visfatin that promotes endothelial angiogenesis. Biochem. Biophys. Res. Commun. 2009, 379, 206–211. [Google Scholar] [CrossRef]
- Kanda, N.; Hau, C.S.; Tada, Y.; Tatsuta, A.; Sato, S.; Watanabe, S. Visfatin Enhances CXCL8, CXCL10, and CCL20 Production in Human Keratinocytes. Endocrinology 2011, 152, 3155–3164. [Google Scholar] [CrossRef] [Green Version]
- Gisondi, P.; Bellinato, F.; Girolomoni, G.; Albanesi, C. Pathogenesis of Chronic Plaque Psoriasis and Its Intersection With Cardio-Metabolic Comorbidities. Front. Pharmacol. 2020, 11, 117. [Google Scholar] [CrossRef]
- Boehncke, W.-H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Eyerich, S.; Eyerich, K.; Cavani, A.; Schmidt-Weber, C. IL-17 and IL-22: Siblings, not twins. Trends Immunol. 2010, 31, 354–361. [Google Scholar] [CrossRef]
- Albanesi, C.; Madonna, S.; Gisondi, P.; Girolomoni, G. The Interplay Between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front. Immunol. 2018, 9, 1549. [Google Scholar] [CrossRef] [Green Version]
- Wollina, U.; Schmidt, W.-D.; Koch, A.; Scheibe, A.; Erfurth, F.; Fassler, D. Fluorescence remission spectroscopy of psoriatic lesions and the effect of topical anthralin therapy. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 1409–1413. [Google Scholar] [CrossRef]
- Hammar, H. Epidermal nicotinamide adenine dinucleotides in psoriasis and neurodermatitis (Lichen Simplex Hypertrophicus). Arch. Dermatol. Res. 1975, 252, 217–227. [Google Scholar] [CrossRef]
- Levine, D.; Even-Chen, Z.; Lipets, I.; Pritulo, O.A.; Svyatenko, T.V.; Andrashko, Y.; Lebwohl, M.; Gottlieb, A. Pilot, multicenter, double-blind, randomized placebo-controlled bilateral comparative study of a combination of calcipotriene and nicotinamide for the treatment of psoriasis. J. Am. Acad. Dermatol. 2010, 63, 775–781. [Google Scholar] [CrossRef]
- Khodadadi, M.; Siadat, A.H.; Iraji, F.; Jary, M.K. Topical nicotinamide in combination with calcipotriol for the treatment of mild to moderate psoriasis: A double-blind, randomized, comparative study. Adv. Biomed. Res. 2013, 2, 90. [Google Scholar] [CrossRef]
- Mercurio, L.; Morelli, M.; Scarponi, C.; Eisenmesser, E.Z.; Doti, N.; Pagnanelli, G.; Gubinelli, E.; Mazzanti, C.; Cavani, A.; Ruvo, M.; et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Morelli, M.; Scarponi, C.; Mercurio, L.; Facchiano, F.; Pallotta, S.; Madonna, S.; Girolomoni, G.; Albanesi, C. Selective Immunomodulation of Inflammatory Pathways in Keratinocytes by the Janus Kinase (JAK) Inhibitor Tofacitinib: Implications for the Employment of JAK-Targeting Drugs in Psoriasis. J. Immunol. Res. 2018, 2018, 1–18. [Google Scholar] [CrossRef]
- Scarponi, C.; Butturini, E.; Sestito, R.; Madonna, S.; Cavani, A.; Mariotto, S.; Albanesi, C. Inhibition of Inflammatory and Proliferative Responses of Human Keratinocytes Exposed to the Sesquiterpene Lactones Dehydrocostuslactone and Costunolide. PLoS ONE 2014, 9, e107904. [Google Scholar] [CrossRef] [Green Version]
- Albanesi, C.; Scarponi, C.; Pallotta, S.; Daniele, R.; Bosisio, D.; Madonna, S.; Fortugno, P.; Gonzalvo-Feo, S.; Franssen, J.-D.; Parmentier, M.; et al. Chemerin expression marks early psoriatic skin lesions and correlates with plasmacytoid dendritic cell recruitment. J. Exp. Med. 2008, 206, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, L.; Failla, C.M.; Capriotti, L.; Scarponi, C.; Facchiano, F.; Morelli, M.; Rossi, S.; Pagnanelli, G.; Albanesi, C.; Cavani, A.; et al. Interleukin (IL)-17/IL-36 axis participates to the crosstalk between endothelial cells and keratinocytes during inflammatory skin responses. PLoS ONE 2020, 15, e0222969. [Google Scholar] [CrossRef]
- Madonna, S.; Scarponi, C.; Sestito, R.; Pallotta, S.; Cavani, A.; Albanesi, C. The IFN-γ–Dependent Suppressor of Cytokine Signaling1 Promoter Activity Is Positively Regulated by IFN Regulatory Factor-1 and Sp1 but Repressed by Growth Factor Independence-1b and Krüppel-Like Factor-4, and It Is Dysregulated in Psoriatic Keratinocytes. J. Immunol. 2010, 185, 2467–2481. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, L.; Lulli, D.; Mascia, F.; Dellambra, E.; Scarponi, C.; Morelli, M.; Valente, C.; Carbone, M.L.; Pallotta, S.; Girolomoni, G.; et al. Intracellular Insulin-like growth factor binding protein 2 (IGFBP2) contributes to the senescence of keratinocytes in psoriasis by stabilizing cytoplasmic p21. Aging 2020, 12, 6823–6851. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, R.-S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef]
- Harden, J.L.; Lewis, S.M.; Lish, S.R.; Suárez-Fariñas, M.; Gareau, D.; Lentini, T.; Johnson-Huang, L.M.; Krueger, J.G.; Lowes, M.A. The tryptophan metabolism enzyme L-kynureninase is a novel inflammatory factor in psoriasis and other inflammatory diseases. J. Allergy Clin. Immunol. 2016, 137, 1830–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Yan, K.; Meng, Q.; Sun, R.; Yang, X.; Yuan, D.; Li, F.; Deng, H. Abnormal expression of SIRTs in psoriasis: Decreased expression of SIRT 1-5 and increased expression of SIRT 6 and 7. Int. J. Mol. Med. 2019, 44, 157–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sestito, R.; Madonna, S.; Scarponi, C.; Cianfarani, F.; Failla, C.M.; Cavani, A.; Girolomoni, G.; Albanesi, C. STAT3-dependent effects of IL-22 in human keratinocytes are counterregulated by sirtuin 1 through a direct inhibition of STAT3 acetylation. FASEB J. 2010, 25, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Hasmann, M.; Schemainda, I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar] [PubMed]
- Federici, M.; Giustizieri, M.L.; Scarponi, C.; Girolomoni, G.; Albanesi, C. Impaired IFN-γ-Dependent Inflammatory Responses in Human Keratinocytes Overexpressing the Suppressor of Cytokine Signaling 1. J. Immunol. 2002, 169, 434–442. [Google Scholar] [CrossRef]
- Madonna, S.; Scarponi, C.; Pallotta, S.; Cavani, A.; Albanesi, C. Anti-apoptotic effects of suppressor of cytokine signaling 3 and 1 in psoriasis. Cell Death Dis. 2012, 3, e334. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.L.; Chin, T.; Tan, C.Y.R.; Rovito, H.A.; Quek, L.S.; Oblong, J.E.; Bellanger, S. Nicotinamide Metabolism Modulates the Proliferation/Differentiation Balance and Senescence of Human Primary Keratinocytes. J. Investig. Dermatol. 2019, 139, 1638–1647. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Lei, H.; Zhang, Z. Pre-B cell colony enhancing factor (PBEF), a cytokine with multiple physiological functions. Cytokine Growth Factor Rev. 2013, 24, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, R.; Röcken, M.; Ghoreschi, K. Angiogenesis drives psoriasis pathogenesis. Int. J. Exp. Pathol. 2009, 90, 232–248. [Google Scholar] [CrossRef]
- Adya, R.; Tan, B.K.; Punn, A.; Chen, J.; Randeva, H.S. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: Novel insights into visfatin-induced angiogenesis. Cardiovasc. Res. 2007, 78, 356–365. [Google Scholar] [CrossRef]
- Adya, R.; Tan, B.K.; Chen, J.; Randeva, H.S. Pre-B cell colony enhancing factor (PBEF)/visfatin induces secretion of MCP-1 in human endothelial cells: Role in visfatin-induced angiogenesis. Atherosclerosis 2009, 205, 113–119. [Google Scholar] [CrossRef]
- Colombo, G.; Clemente, N.; Zito, A.; Bracci, C.; Colombo, F.S.; Sangaletti, S.; Jachetti, E.; Ribaldone, D.G.; Caviglia, G.P.; Pastorelli, L.; et al. Neutralization of extracellular NAMPT (nicotinamide phosphoribosyltransferase) ameliorates experimental murine colitis. J. Mol. Med. 2020, 98, 595–612. [Google Scholar] [CrossRef]
- Guerard, S. The Role of Angiogenesis in the Pathogenesis of Psoriasis: Mechanisms and Clinical Implications. J. Clin. Exp. Dermatol. Res. 2014, 4, 4. [Google Scholar] [CrossRef]
- Morelli, M.; Galluzzo, M.; Madonna, S.; Scarponi, C.; Scaglione, G.L.; Galluccio, T.; Andreani, M.; Pallotta, S.; Girolomoni, G.; Bianchi, L.; et al. HLA-Cw6 and other HLA-C alleles, as well as MICB-DT, DDX58, and TYK2 genetic variants associate with optimal response to anti-IL-17A treatment in patients with psoriasis. Expert Opin. Biol. Ther. 2021, 21, 259–270. [Google Scholar] [CrossRef]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef]
- Iqbal, J.; Zaidi, M. TNF regulates cellular NAD+ metabolism in primary macrophages. Biochem. Biophys. Res. Commun. 2006, 342, 1312–1318. [Google Scholar] [CrossRef]
- Gosset, M.; Berenbaum, F.; Salvat, C.; Sautet, A.; Pigenet, A.; Tahiri, K.; Jacques, C. Crucial role of visfatin/pre–B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: Possible influence on osteoarthritis. Arthritis Rheum. 2008, 58, 1399–1409. [Google Scholar] [CrossRef]
- Nowell, M.A.; Richards, P.J.; Fielding, C.; Ognjanovic, S.; Topley, N.; Williams, A.S.; Bryant-Greenwood, G.; Jones, S.A. Regulation of pre–B cell colony-enhancing factor by STAT-3–dependent interleukin-6trans-signaling: Implications in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2084–2095. [Google Scholar] [CrossRef]
- Ognjanovic, S.; Bao, S.; Yamamoto, S.Y.; Garibay-Tupas, J.; Samal, B.; Bryant-Greenwood, G.D. Genomic organization of the gene coding for human pre-B-cell colony enhancing factor and expression in human fetal membranes. J. Mol. Endocrinol. 2001, 26, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audrito, V.; Messana, V.G.; Deaglio, S. NAMPT and NAPRT: Two Metabolic Enzymes With Key Roles in Inflammation. Front. Oncol. 2020, 10, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skokowa, J.; Lan, D.; Thakur, B.K.; Wang, F.; Gupta, K.; Cario, G.; Brechlin, A.M.; Schambach, A.; Hinrichsen, L.; Meyer, G.; et al. NAMPT is essential for the G-CSF–induced myeloid differentiation via a NAD+–sirtuin-1–dependent pathway. Nat. Med. 2009, 15, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Travelli, C.; Colombo, G.; Mola, S.; Genazzani, A.A.; Porta, C. NAMPT: A pleiotropic modulator of monocytes and macrophages. Pharmacol. Res. 2018, 135, 25–36. [Google Scholar] [CrossRef]
- Grolla, A.A.; Torretta, S.; Gnemmi, I.; Amoruso, A.; Orsomando, G.; Gatti, M.; Caldarelli, A.; Lim, D.; Penengo, L.; Brunelleschi, S.; et al. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is a tumoural cytokine released from melanoma. Pigment. Cell Melanoma Res. 2015, 28, 718–729. [Google Scholar] [CrossRef]
- Koczan, D.; Guthke, R.; Thiesen, H.-J.; Ibrahim, S.M.; Kundt, G.; Krentz, H.; Gross, G.; Kunz, M. Gene expression profiling of peripheral blood mononuclear leukocytes from psoriasis patients identifies new immune regulatory molecules. Eur. J. Dermatol. EJD 2005, 15, 251–257. [Google Scholar]
- Bao, J.; Chen, W.; Wu, L. Visfatin: A Potential Therapeutic Target for Rheumatoid Arthritis. J. Int. Med Res. 2009, 37, 1655–1661. [Google Scholar] [CrossRef]
- Brentano, F.; Schorr, O.; Ospelt, C.; Stanczyk, J.; Gay, R.E.; Gay, S.; Kyburz, D. Pre–B cell colony-enhancing factor/visfatin, a new marker of inflammation in rheumatoid arthritis with proinflammatory and matrix-degrading activities. Arthritis Rheum. 2007, 56, 2829–2839. [Google Scholar] [CrossRef]
- Gerner, R.R.; Klepsch, V.; Macheiner, S.; Arnhard, K.; E Adolph, T.; Grander, C.; Wieser, V.; Pfister, A.; Moser, P.; Hermann-Kleiter, N.; et al. NAD metabolism fuels human and mouse intestinal inflammation. Gut 2017, 67, 1813–1823. [Google Scholar] [CrossRef]
- Busso, N.; Karababa, M.; Nobile, M.; Rolaz, A.; Van Gool, F.; Galli, M.; Leo, O.; So, A.; De Smedt, T. Pharmacological Inhibition of Nicotinamide Phosphoribosyltransferase/Visfatin Enzymatic Activity Identifies a New Inflammatory Pathway Linked to NAD. PLoS ONE 2008, 3, e2267. [Google Scholar] [CrossRef] [Green Version]
- Fivenson, D.P.; Breneman, D.L.; Rosen, G.B.; Hersh, C.S.; Cardone, S.; Mutasim, D. Nicotinamide and Tetracycline Therapy of Bullous Pemphigoid. Arch. Dermatol. 1994, 130, 753–758. [Google Scholar] [CrossRef]
- Navarrete-Solís, J.; Castanedo-Cázares, J.P.; Torres-Álvarez, B.; Oros-Ovalle, C.; Fuentes-Ahumada, C.; González, F.J.; Martínez-Ramírez, J.D.; Moncada, B. A Double-Blind, Randomized Clinical Trial of Niacinamide 4% versus Hydroquinone 4% in the Treatment of Melasma. Dermatol. Res. Pract. 2011, 2011, 1–5. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
| NAMPT | TGA ATG CCG TGA AAA GAA GA | AAT TTG TTG CCA CTG TGA TT |
| NAPRT | GTG AGG TGA ATG TCA TTGG | GGC CAC CAG CTT ATA GAC |
| CCL20 | GTG CTG CTA CTC CAC CTC TG | TGT ATC CAA GAC AGC AGT CAAA |
| CCL2 | CAC CAG CAG CAA GTG TCCC | CCA TGG AAT CCT GAA CCC AC |
| CXCL10 | TGG CAT TCA AGG AGT ACC TCT CT | CTG ATG CAG GTA CAG CGT ACG |
| CXCL8 | CTC TGT GTG AAG GTG CAG TTTT | GGG TGG AAA GGT TTG GAG TAT |
| HPRT1 | ATG GAC AGG ACT GAA CGT CTT | CTT GAG CAC ACA GAG GGC TA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercurio, L.; Morelli, M.; Scarponi, C.; Scaglione, G.L.; Pallotta, S.; Avitabile, D.; Albanesi, C.; Madonna, S. Enhanced NAMPT-Mediated NAD Salvage Pathway Contributes to Psoriasis Pathogenesis by Amplifying Epithelial Auto-Inflammatory Circuits. Int. J. Mol. Sci. 2021, 22, 6860. https://doi.org/10.3390/ijms22136860
Mercurio L, Morelli M, Scarponi C, Scaglione GL, Pallotta S, Avitabile D, Albanesi C, Madonna S. Enhanced NAMPT-Mediated NAD Salvage Pathway Contributes to Psoriasis Pathogenesis by Amplifying Epithelial Auto-Inflammatory Circuits. International Journal of Molecular Sciences. 2021; 22(13):6860. https://doi.org/10.3390/ijms22136860
Chicago/Turabian StyleMercurio, Laura, Martina Morelli, Claudia Scarponi, Giovanni Luca Scaglione, Sabatino Pallotta, Daniele Avitabile, Cristina Albanesi, and Stefania Madonna. 2021. "Enhanced NAMPT-Mediated NAD Salvage Pathway Contributes to Psoriasis Pathogenesis by Amplifying Epithelial Auto-Inflammatory Circuits" International Journal of Molecular Sciences 22, no. 13: 6860. https://doi.org/10.3390/ijms22136860
APA StyleMercurio, L., Morelli, M., Scarponi, C., Scaglione, G. L., Pallotta, S., Avitabile, D., Albanesi, C., & Madonna, S. (2021). Enhanced NAMPT-Mediated NAD Salvage Pathway Contributes to Psoriasis Pathogenesis by Amplifying Epithelial Auto-Inflammatory Circuits. International Journal of Molecular Sciences, 22(13), 6860. https://doi.org/10.3390/ijms22136860