
Sub-3 Å Cryo-EM Structures of Necrosis Virus Particles via the Use of Multipurpose TEM with Electron Counting Camera

Abstract
1. Introduction
2. Results
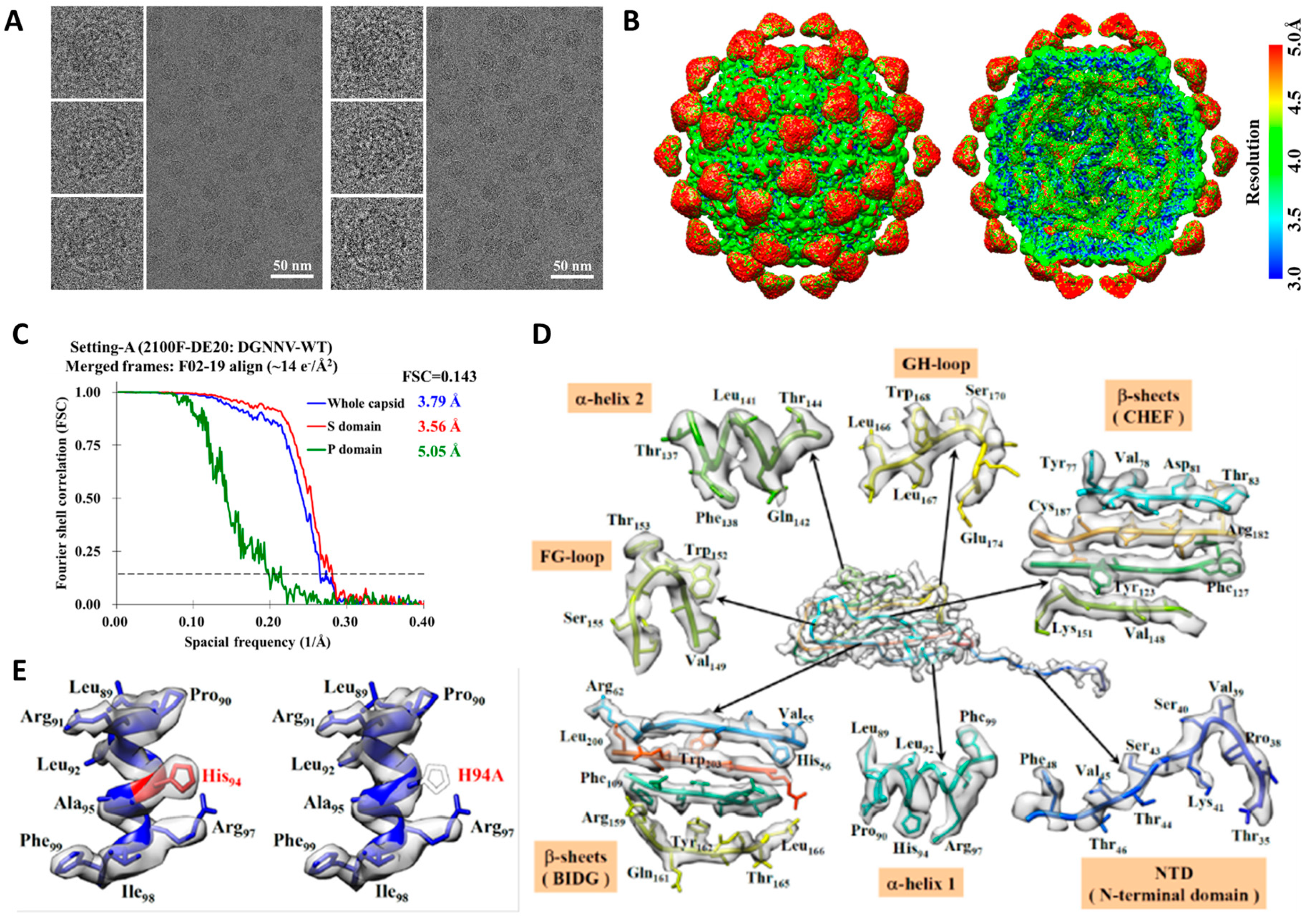
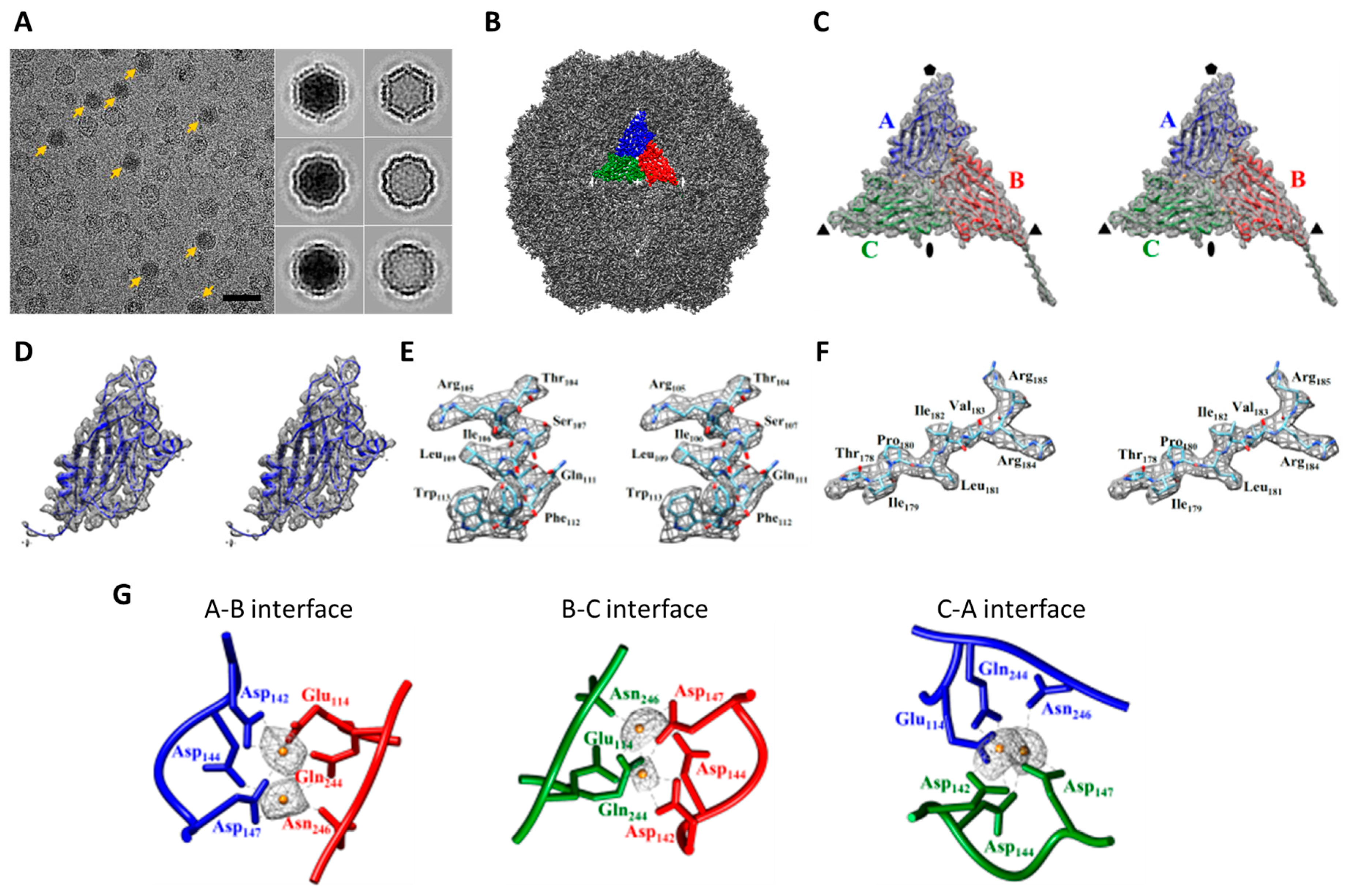
2.1. Cryo-EM Imaging of DGNNV with Setting A
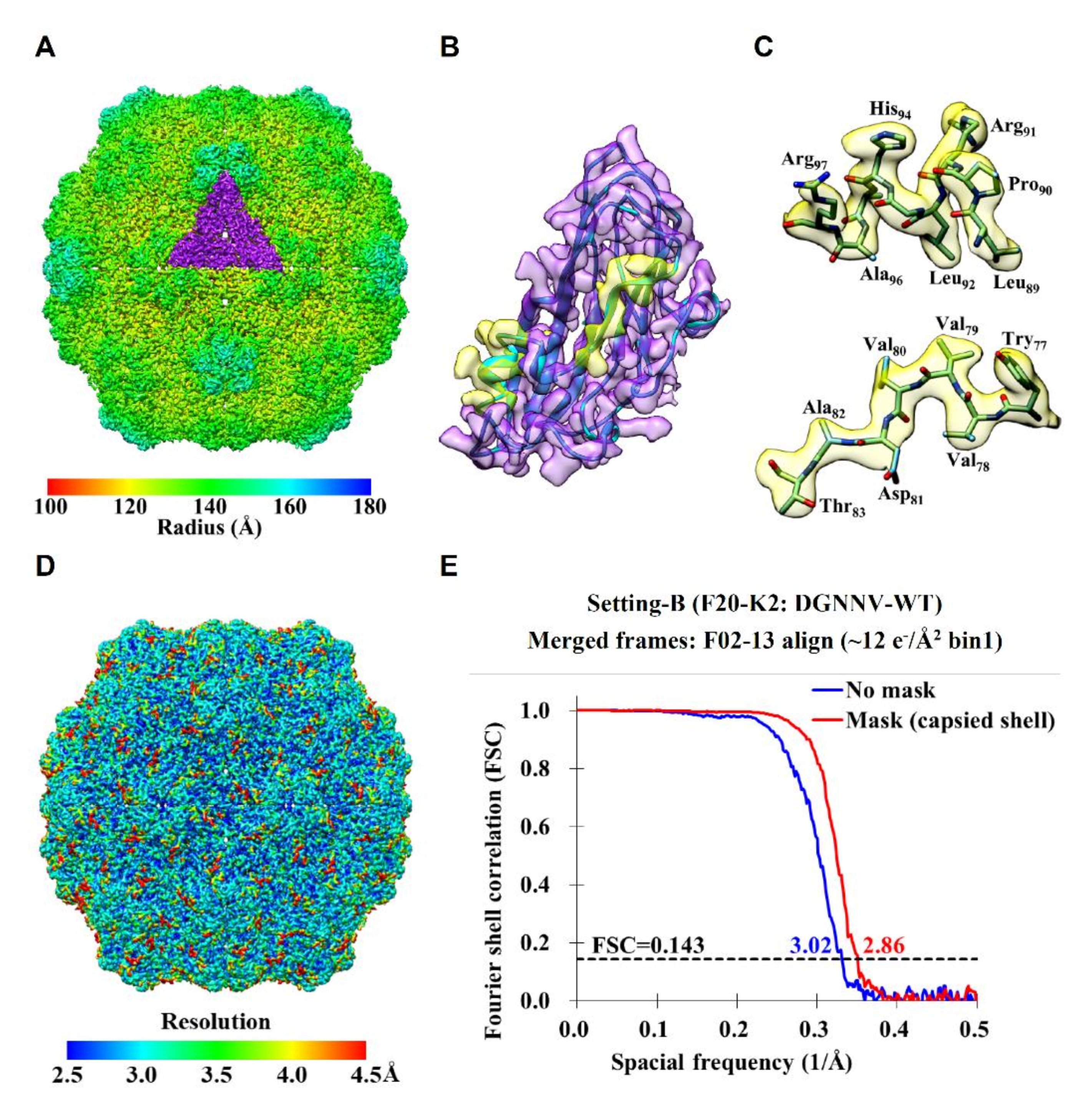
2.2. Cryo-EM Imaging of DGNNV with Setting B
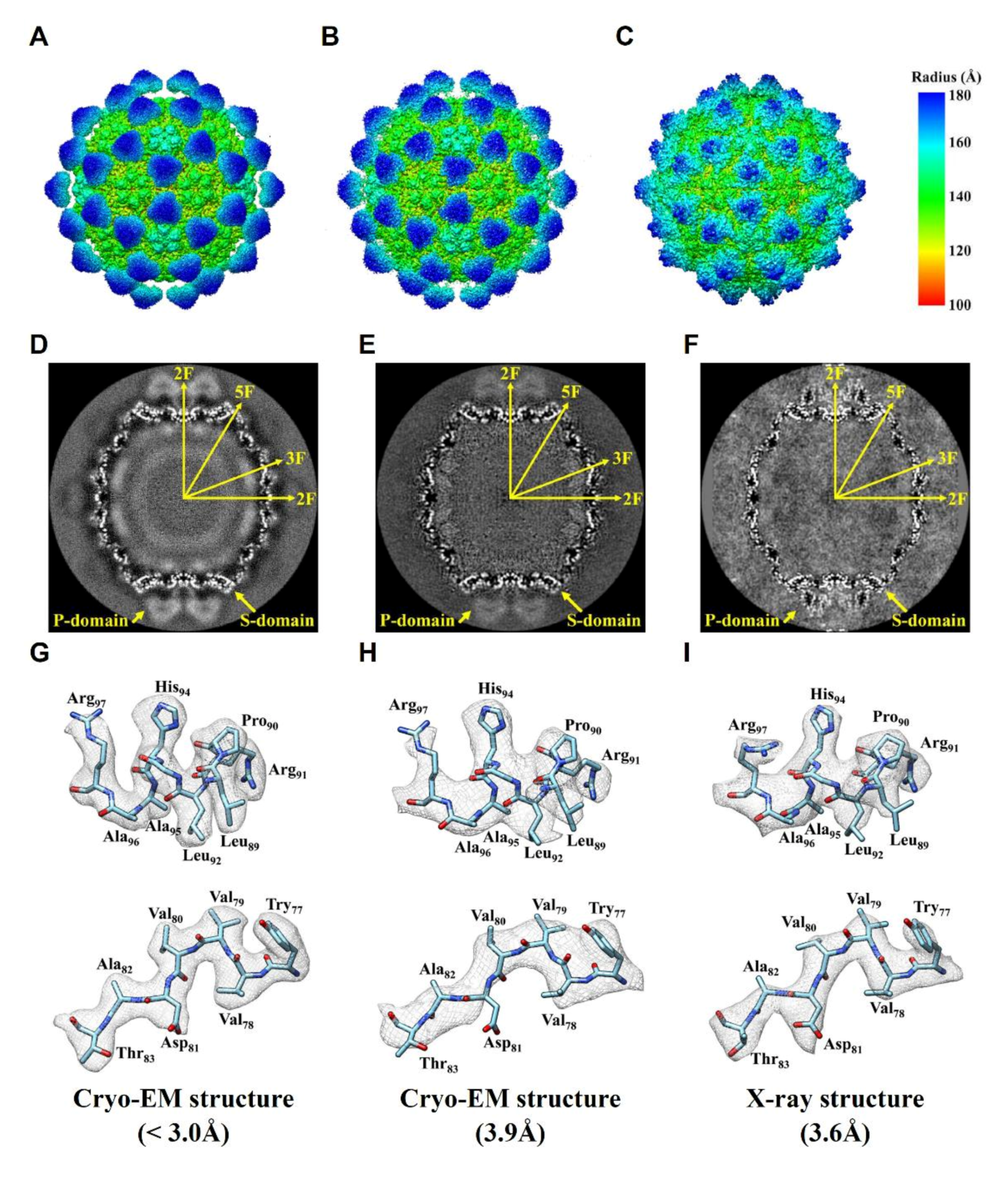
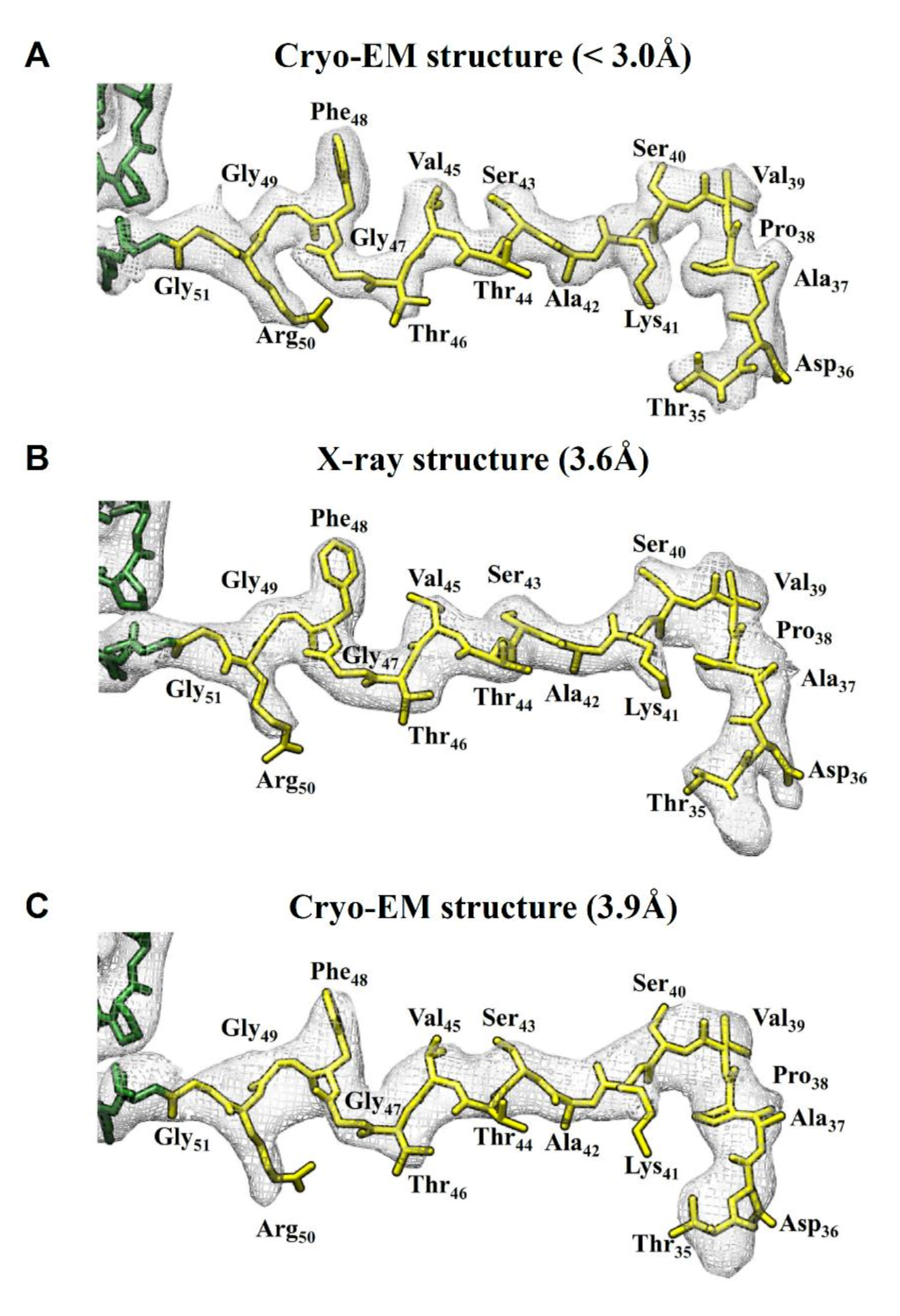
2.3. Comparision of DGNNV Maps from the Present and Earlier Studies
2.4. The Usage of the Automated Instrument Only Improves Efficiency
3. Discussion
4. Materials and Methods
4.1. Protein Preparation of DGNNV
4.2. Cryo-Electron Microscopy of DGNNV
4.3. Data Processing of DGNNV
4.4. Map Sharpening
4.5. Map Segmentation and Model Building
4.6. Protein Preparation of MrNV
4.7. Cryo-Electron Microscopy of MrNV
4.8. Data Processing of MrNV
4.9. Data Reprocessing with Modern Algorithms
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeRosier, D.J.; Klug, A. Reconstruction of three dimensional structures from electron micrographs. Nat. Cell Biol. 1968, 217, 130–134. [Google Scholar] [CrossRef]
- Crowther, R.A.; DeRosier, D.J.; Klug, A. The reconstruction of a three-dimensional structure from projections and its application to electron microscopy. Proc. R. Soc. Lond. A Math. Phys. Sci. 1970, 317, 319–340. [Google Scholar]
- Hoppe, W.; Grill, B. Prospects of three-dimensional high resolution electron microscopy of non-periodic structures. Ultramicroscopy 1976, 2, 153–168. [Google Scholar] [CrossRef]
- Frank, J. Advances in the field of single-particle cryo-electron microscopy over the last decade. Nat. Protoc. 2017, 12, 209–212. [Google Scholar] [CrossRef]
- Dubochet, J.; Adrian, M.; Chang, J.-J.; Homo, J.-C.; Lepault, J.; McDowall, A.W.; Schultz, P. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 1988, 21, 129–228. [Google Scholar] [CrossRef]
- Vogel, R.H.; Provencher, S.W.; von Bonsdorff, C.-H.; Adrian, M.; Dubochet, J. Envelope structure of Semliki Forest virus reconstructed from cryo-electron micrographs. Nat. Cell Biol. 1986, 320, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Crowther, R.A.; Kiselev, N.A.; Böttcher, B.; Berriman, J.A.; Borisova, G.P.; Ose, V.; Pumpens, P. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 1994, 77, 943–950. [Google Scholar] [CrossRef]
- Zhou, Z.H.; Dougherty, M.; Jakana, J.; He, J.; Rixon, F.J.; Chiu, W. Seeing the Herpesvirus capsid at 8.5 Å. Science 2000, 288, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.; Rees, I.; Ludtke, S.J.; Chiu, W.; Baker, M.L. Constructing and validating initial Cα models from subnanometer resolution density maps with pathwalking. Structure 2012, 20, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jin, L.; Koh, S.B.S.; Atanasov, I.; Schein, S.; Wu, L.; Zhou, Z.H. Atomic Structure of Human Adenovirus by Cryo-EM Reveals Interactions Among Protein Networks. Science 2010, 329, 1038–1043. [Google Scholar] [CrossRef]
- Kuhlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jin, L.; Jih, J.; Shih, C.; Zhou, Z.H. 3.5 Å cryoEM Structure of Hepatitis B Virus Core Assembled from Full-Length Core Protein. PLoS ONE 2013, 8, e69729. [Google Scholar] [CrossRef]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 Å resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef]
- Kostyuchenko, V.A.; Lim, E.X.Y.; Zhang, S.; Fibriansah, G.; Ng, T.-S.; Ooi, J.; Shi, J.; Lok, S.-M. Structure of the thermally stable Zika virus. Nat. Cell Biol. 2016, 533, 425–428. [Google Scholar] [CrossRef]
- Miyazaki, N.; Murakami, K.; Oka, T.; Miki, M.; Iwasaki, K.; Katayama, K.; Murata, K. Atomic structure of human sapovirus capsid by single particle cryo-electron microscopy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Peukes, J.; Xiong, X.; Erlendsson, S.; Qu, K.; Wan, W.; Calder, L.J.; Schraidt, O.; Kummer, S.; Freund, S.M.V.; Kräusslich, H.-G.; et al. The native structure of the assembled matrix protein 1 of influenza A virus. Nat. Cell Biol. 2020, 587, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, S.; Pintilie, G.; Chmielewski, D.; Schmid, M.F.; Simmons, G.; Jin, J.; Chiu, W. A 3.4-Å cryo-electron microscopy structure of the human coronavirus spike trimer computationally derived from vitrified NL63 virus particles. QRB Discov. 2020, 1, e11. [Google Scholar] [CrossRef]
- DeRosier, D.J. Correction of high-resolution data for curvature of the Ewald sphere. Ultramicroscopy 2000, 81, 83–98. [Google Scholar] [CrossRef]
- Glaeser, R.M.; Typke, D.; Tiemeijer, P.C.; Pulokas, J.; Cheng, A. Precise beam-tilt alignment and collimation are required to minimize the phase error associated with coma in high-resolution cryo-EM. J. Struct. Biol. 2011, 174, 1–10. [Google Scholar] [CrossRef]
- Brink, J.; Sherman, M.B.; Berriman, J.; Chiu, W. Evaluation of charging on macromolecules in electron cryomicroscopy. Ultramicroscopy 1998, 72, 41–52. [Google Scholar] [CrossRef]
- Alewijnse, B.; Ashton, A.W.; Chambers, M.G.; Chen, S.; Cheng, A.; Ebrahim, M.; Eng, E.; Hagen, W.J.; Koster, A.J.; López, C.S.; et al. Best practices for managing large CryoEM facilities. J. Struct. Biol. 2017, 199, 225–236. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, M.; Lander, G.C.; Herzik, M.A., Jr. Sub-2 Angstrom resolution structure determination using single-particle cryo-EM at 200 keV. J. Struct. Biol. X 2020, 4, 100020. [Google Scholar] [CrossRef] [PubMed]
- Danev, R.; Yanagisawa, H.; Kikkawa, M. Cryo-electron microscopy methodology: Current aspects and future directions. Trends Biochem. Sci. 2019, 44, 837–848. [Google Scholar] [CrossRef]
- Merk, A.; Fukumura, T.; Zhu, X.; Darling, J.E.; Grisshammer, R.; Ognjenovic, J.; Subramaniam, S. 1.8 Å resolution structure of β-galactosidase with a 200 kV CRYO ARM electron microscope. IUCrJ 2020, 7 Pt 4, 639–643. [Google Scholar] [CrossRef]
- Danev, R.; Baumeister, W. Cryo-EM single particle analysis with the Volta phase plate. eLife 2016, 5, e13046. [Google Scholar] [CrossRef]
- Ruskin, R.S.; Yu, Z.; Grigorieff, N. Quantitative characterization of electron detectors for transmission electron microscopy. J. Struct. Biol. 2013, 184, 385–393. [Google Scholar] [CrossRef]
- Feist, A.; Bach, N.; da Silva, N.R.; Danz, T.; Möller, M.; Priebe, K.E.; Domröse, T.; Gatzmann, J.G.; Rost, S.; Schauss, J.; et al. Ultrafast transmission electron microscopy using a laser-driven field emitter: Femtosecond resolution with a high coherence electron beam. Ultramicroscopy 2017, 176, 63–73. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Kayama, Y.; Burton-Smith, R.N.; Song, C.; Terahara, N.; Kato, T.; Murata, K. Below 3 Å structure of apoferritin using a multipurpose TEM with a side entry cryoholder. Sci. Rep. 2021, 11, 8395. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Hsu, C.-H.; Wang, C.-H.; Liu, W.; Chang, W.-H.; Lin, C.-S. Role of the DxxDxD motif in the assembly and stability of betanodavirus particles. Arch. Virol. 2008, 153, 1633–1642. [Google Scholar] [CrossRef]
- Crane, M.; Hyatt, A. Viruses of Fish: An overview of significant pathogens. Viruses 2011, 3, 2025–2046. [Google Scholar] [CrossRef] [PubMed]
- Munday, B.; Kwang, J.; Moody, N. Betanodavirus infections of teleost fish: A review. J. Fish Dis. 2002, 25, 127–142. [Google Scholar] [CrossRef]
- Chen, N.-C.; Yoshimura, M.; Guan, H.-H.; Wang, T.-Y.; Misumi, Y.; Lin, C.-C.; Chuankhayan, P.; Nakagawa, A.; Chan, S.I.; Tsukihara, T.; et al. Crystal structures of a piscine betanodavirus: Mechanisms of capsid assembly and viral infection. PLoS Pathog. 2015, 11, e1005203. [Google Scholar] [CrossRef]
- Li, X.; Mooney, P.; Zheng, S.; Booth, C.R.; Braunfeld, M.B.; Gubbens, S.; Agard, D.A.; Cheng, Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 2013, 10, 584–590. [Google Scholar] [CrossRef]
- Yan, X.; Dryden, K.A.; Tang, J.; Baker, T.S. Ab initio random model method facilitates 3D reconstruction of icosahedral particles. J. Struct. Biol. 2007, 157, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Peng, L.; Baldwin, P.R.; Mann, D.S.; Jiang, W.; Rees, I.; Ludtke, S.J. EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 2007, 157, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.Q.; Palovcak, E.; Armache, J.-P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; O Forsberg, B.; Kimanius, D.; Hagen, W.J.; Lindahl, E.; Scheres, S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 2018, 7, e42166. [Google Scholar] [CrossRef] [PubMed]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; A Brubaker, M. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Ho, K.L.; Gabrielsen, M.; Beh, P.L.; Kueh, C.L.; Thong, Q.X.; Streetley, J.; Tan, W.S.; Bhella, D. Structure of the Macrobrachium rosenbergii nodavirus: A new genus within the Nodaviridae? PLoS Biol. 2018, 16, e3000038. [Google Scholar] [CrossRef]
- Ho, K.L.; Kueh, C.L.; Beh, P.L.; Tan, W.S.; Bhella, D. Cryo-electron microscopy structure of the Macrobrachium rosenbergii nodavirus capsid at 7 Angstroms resolution. Sci. Rep. 2017, 7, 2083. [Google Scholar] [CrossRef] [PubMed]
- Shiue, J.; Chang, C.-S.; Huang, S.-H.; Hsu, C.-H.; Tsai, J.-S.; Chang, W.-H.; Wu, Y.-M.; Lin, Y.-C.; Kuo, P.-C.; Huang, Y.-S.; et al. Phase TEM for biological imaging utilizing a Boersch electrostatic phase plate: Theory and practice. J. Electron Microscopy 2009, 58, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Li, K.; Gao, Y.; Huang, R.; Lai, Y.; Shi, Y.; Yang, S.; Zhu, G.; Zhang, Q.; He, J. Structural analysis and insertion study reveal the ideal sites for surface displaying foreign peptides on a betanodavirus-like particle. Vet. Res. 2016, 47, 16. [Google Scholar] [CrossRef]
- Brilot, A.; Chen, J.Z.; Cheng, A.; Pan, J.; Harrison, S.C.; Potter, C.S.; Carragher, B.; Henderson, R.; Grigorieff, N. Beam-induced motion of vitrified specimen on holey carbon film. J. Struct. Biol. 2012, 177, 630–637. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, Z.H. Limiting factors in atomic resolution cryo electron microscopy: No simple tricks. J. Struct. Biol. 2011, 175, 253–263. [Google Scholar] [CrossRef]
- Russo, C.J.; Henderson, R. Ewald sphere correction using a single side-band image processing algorithm. Ultramicroscopy 2018, 187, 26–33. [Google Scholar] [CrossRef]
- Tan, Y.Z.; Aiyer, S.; Mietzsch, M.; Hull, J.A.; McKenna, R.; Grieger, J.; Samulski, R.J.; Baker, T.S.; Agbandje-McKenna, M.; Lyumkis, D. Sub-2 Å Ewald curvature corrected structure of an AAV2 capsid variant. Nat. Commun. 2018, 9, 362. [Google Scholar] [CrossRef]
- Li, X.; Zhou, N.; Chen, W.; Zhu, B.; Wang, X.; Xu, B.; Wang, J.; Liu, H.; Cheng, L. Near-atomic resolution structure determination of a Cypovirus capsid and polymerase complex using cryo-EM at 200 kV. J. Mol. Biol. 2017, 429, 79–87. [Google Scholar] [CrossRef]
- Naydenova, K.; McMullan, G.; Peet, M.J.; Lee, Y.; Edwards, P.C.; Chen, S.; Leahy, E.; Scotcher, S.; Henderson, R.; Russo, C.J. CryoEM at 100 keV: A demonstration and prospects. IUCrJ 2019, 6, 1086–1098. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Eng, E.; Alink, L.; Rice, W.J.; Jordan, K.D.; Kim, L.Y.; Potter, C.S.; Carragher, B. High resolution single particle cryo-electron microscopy using beam-image shift. J. Struct. Biol. 2018, 204, 270–275. [Google Scholar] [CrossRef]
- Wu, C.; Huang, X.; Cheng, J.; Zhu, D.; Zhang, X. High-quality, high-throughput cryo-electron microscopy data collection via beam tilt and astigmatism-free beam-image shift. J. Struct. Biol. 2019, 208, 107396. [Google Scholar] [CrossRef]
- Cash, J.N.; Kearns, S.; Li, Y.; Cianfrocco, M.A. High-resolution cryo-EM using beam-image shift at 200 keV. IUCrJ 2020, 7, 1179–1187. [Google Scholar] [CrossRef]
- Yan, X.; Sinkovits, R.S.; Baker, T.S. AUTO3DEM—An automated and high throughput program for image reconstruction od icosahedral particles. J. Struct. Biol. 2007, 157, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Mindell, J.A.; Grigorieff, N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 2003, 142, 334–347. [Google Scholar] [CrossRef]
- Henderson, R.; Sali, A.; Baker, M.L.; Carragher, B.; Devkota, B.; Downing, K.H.; Egelman, E.; Feng, Z.; Frank, J.; Grigorieff, N.; et al. Outcome of the First Electron Microscopy Validation Task Force Meeting. Structure 2012, 20, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, P.B.; Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 2003, 333, 721–745. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.-J.; Luque, D.; Caston, J.; Carrascosa, J. Sharpening high resolution information in single particle electron cryomicroscopy. J. Struct. Biol. 2008, 164, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K.D. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Afonine, P.V.; Poon, B.K.; Read, R.J.; Sobolev, O.V.; Terwilliger, T.C.; Urzhumtsev, A.; Adams, P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 531–544. [Google Scholar] [CrossRef]
- Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016, 193, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kimanius, D.; Forsberg, B.; Scheres, S.H.; Lindahl, E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 2016, 5, e18722. [Google Scholar] [CrossRef] [PubMed]
- Kucukelbir, A.; Sigworth, F.J.; Tagare, H.D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 2014, 11, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Rohou, A.; Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015, 192, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Grant, T.; Rohou, A.; Grigorieff, N. cisTEM, user-friendly software for single-particle image processing. eLife 2018, 7, e35383. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | Setting A (2100F-DE20) (DGNNV-WT) | Setting B (F20-K2) (DGNNV-WT) | Setting C (Talos-Falcon3 EC) (MrNV) | |||
|---|---|---|---|---|---|---|
| Software | Auto3dem | Relion v3 and cryoSPARC v2 | Auto3dem | Relion v3 and cryoSPARC v2 | cryoSPARC v1 and Relion v2 | Relion v3 and cryoSPARC v2 |
| Selected frames (no.) | 18 | 50 | 12 | 50 | 19 | 30 |
| Electron exposure (e−/Å2) | ~14 | ~40 | ~12 | ~50 | ~23 | ~37 |
| Dose weighting | No | Yes | No | Yes | No | Yes |
| Micrographs stacks (no.) | 588 | 588 | 1009 | 1009 | 1353 | 1353 |
| Final particle images (no.) | 41,318 | 46,796 | 29,575 | 41,152 | 19,049 | 27,733 |
| Symmetry imposed | I (532) | I | I (532) | I | I (I 1) | I |
| Pixel size (Å/pixel) | 1.16 | 1.16 | 1.24 | 1.24 | 0.86 | 0.86 |
| Nyquist limit (Å) | 2.32 | 2.32 | 2.48 | 2.48 | 1.72 | 1.72 |
| Map resolution of shell (Å) 1 | 3.56 | 3.24 | 3.18 | 2.72 | 2.92 | 2.70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-H.; Chen, D.-H.; Huang, S.-H.; Wu, Y.-M.; Chen, Y.-Y.; Hwu, Y.; Bushnell, D.; Kornberg, R.; Chang, W.-H. Sub-3 Å Cryo-EM Structures of Necrosis Virus Particles via the Use of Multipurpose TEM with Electron Counting Camera. Int. J. Mol. Sci. 2021, 22, 6859. https://doi.org/10.3390/ijms22136859
Wang C-H, Chen D-H, Huang S-H, Wu Y-M, Chen Y-Y, Hwu Y, Bushnell D, Kornberg R, Chang W-H. Sub-3 Å Cryo-EM Structures of Necrosis Virus Particles via the Use of Multipurpose TEM with Electron Counting Camera. International Journal of Molecular Sciences. 2021; 22(13):6859. https://doi.org/10.3390/ijms22136859
Chicago/Turabian StyleWang, Chun-Hsiung, Dong-Hua Chen, Shih-Hsin Huang, Yi-Min Wu, Yi-Yun Chen, Yeukuang Hwu, David Bushnell, Roger Kornberg, and Wei-Hau Chang. 2021. "Sub-3 Å Cryo-EM Structures of Necrosis Virus Particles via the Use of Multipurpose TEM with Electron Counting Camera" International Journal of Molecular Sciences 22, no. 13: 6859. https://doi.org/10.3390/ijms22136859
APA StyleWang, C.-H., Chen, D.-H., Huang, S.-H., Wu, Y.-M., Chen, Y.-Y., Hwu, Y., Bushnell, D., Kornberg, R., & Chang, W.-H. (2021). Sub-3 Å Cryo-EM Structures of Necrosis Virus Particles via the Use of Multipurpose TEM with Electron Counting Camera. International Journal of Molecular Sciences, 22(13), 6859. https://doi.org/10.3390/ijms22136859