
Methotrexate Neurotoxicity Is Related to Epigenetic Modification of the Myelination Process



Abstract
:1. Introduction
2. Results
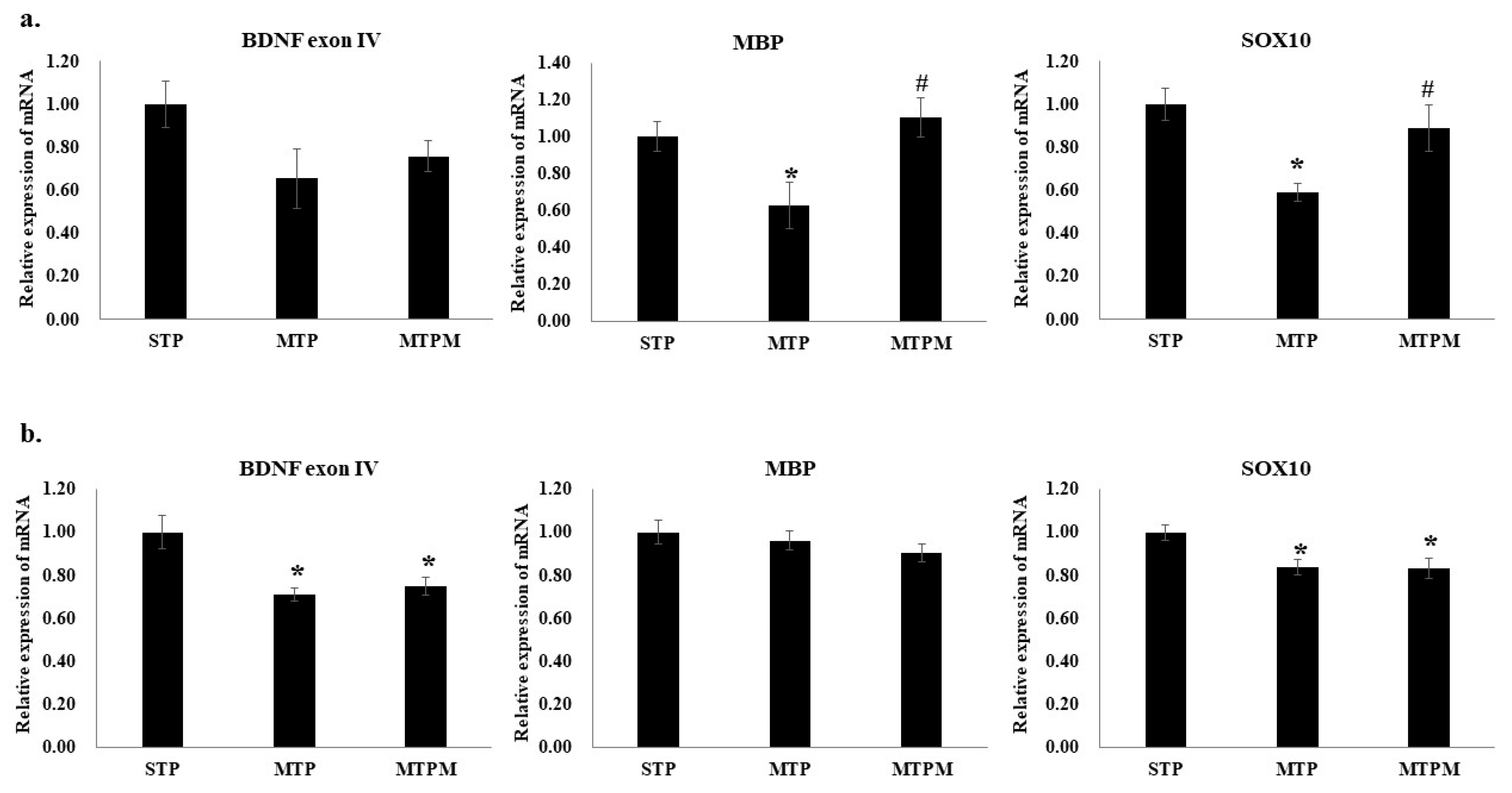
2.1. Methotrexate Treatment Resulted in Altered Myelination-Related Protein mRNA Expression
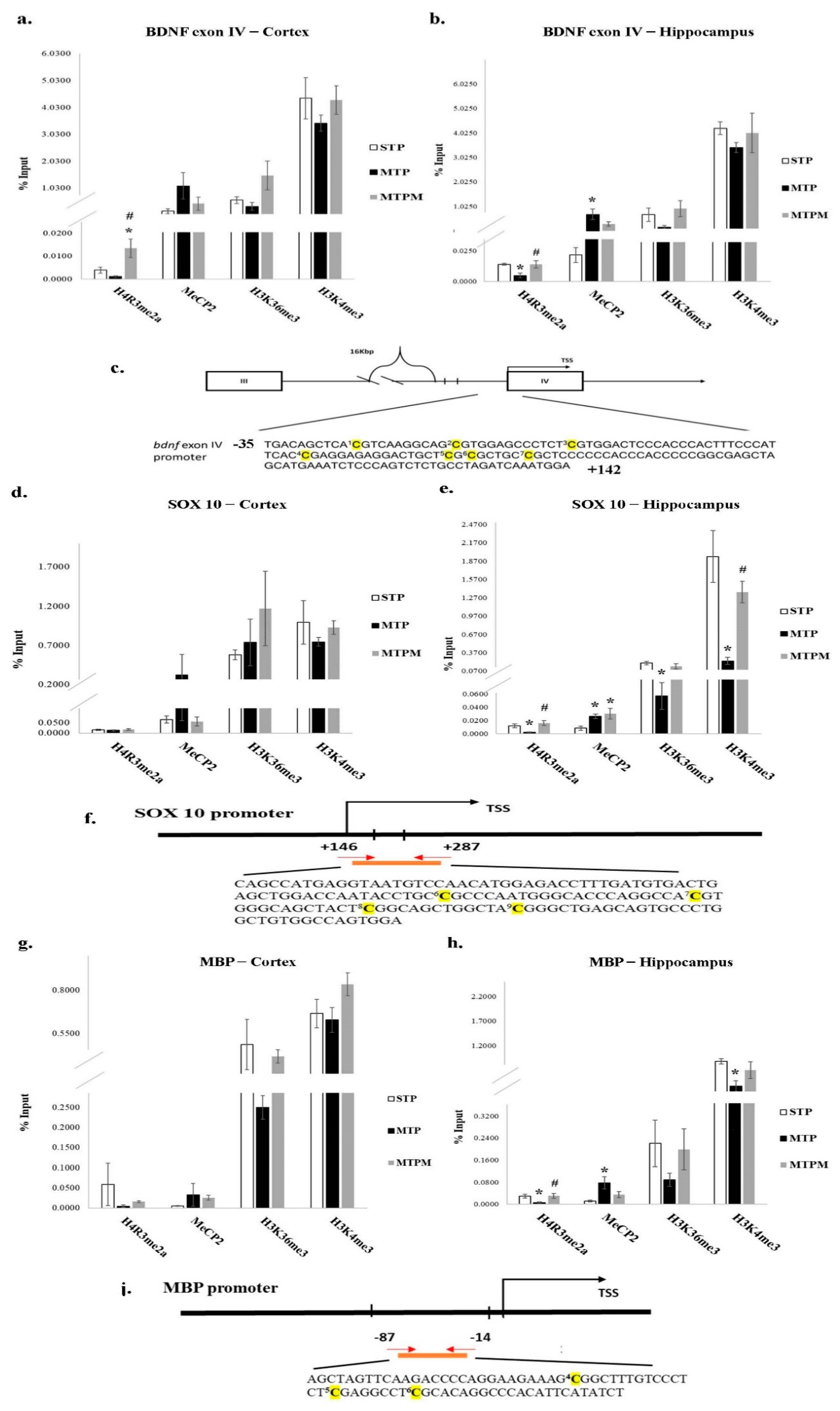
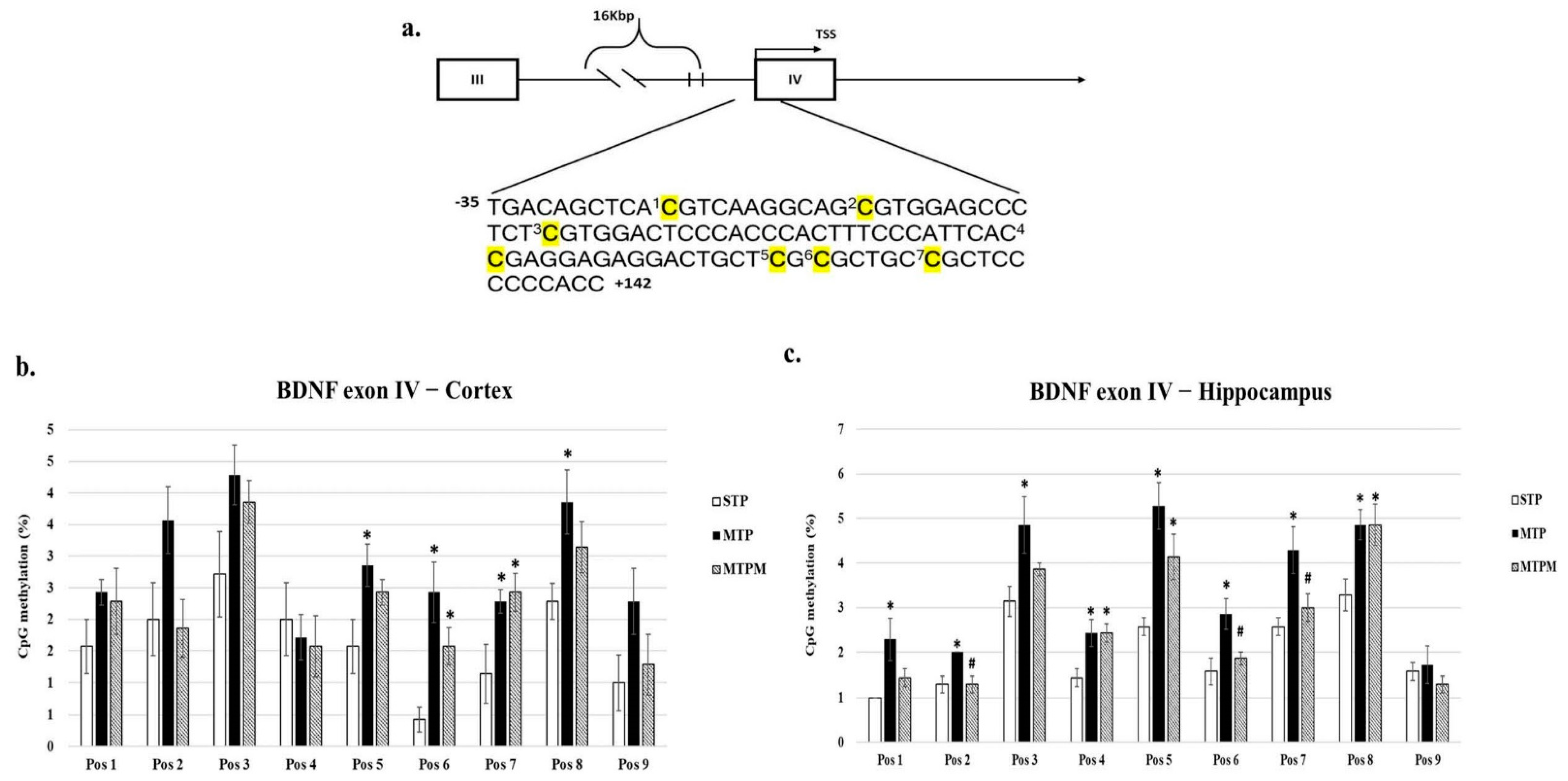
2.2. Combined Intrathecal and Intraperitoneal Methotrexate Treatment Resulted in an Altered CpG Methylation Status of the BDNF Exon IV Promoter in Both the Brain Cortex and Hippocampus
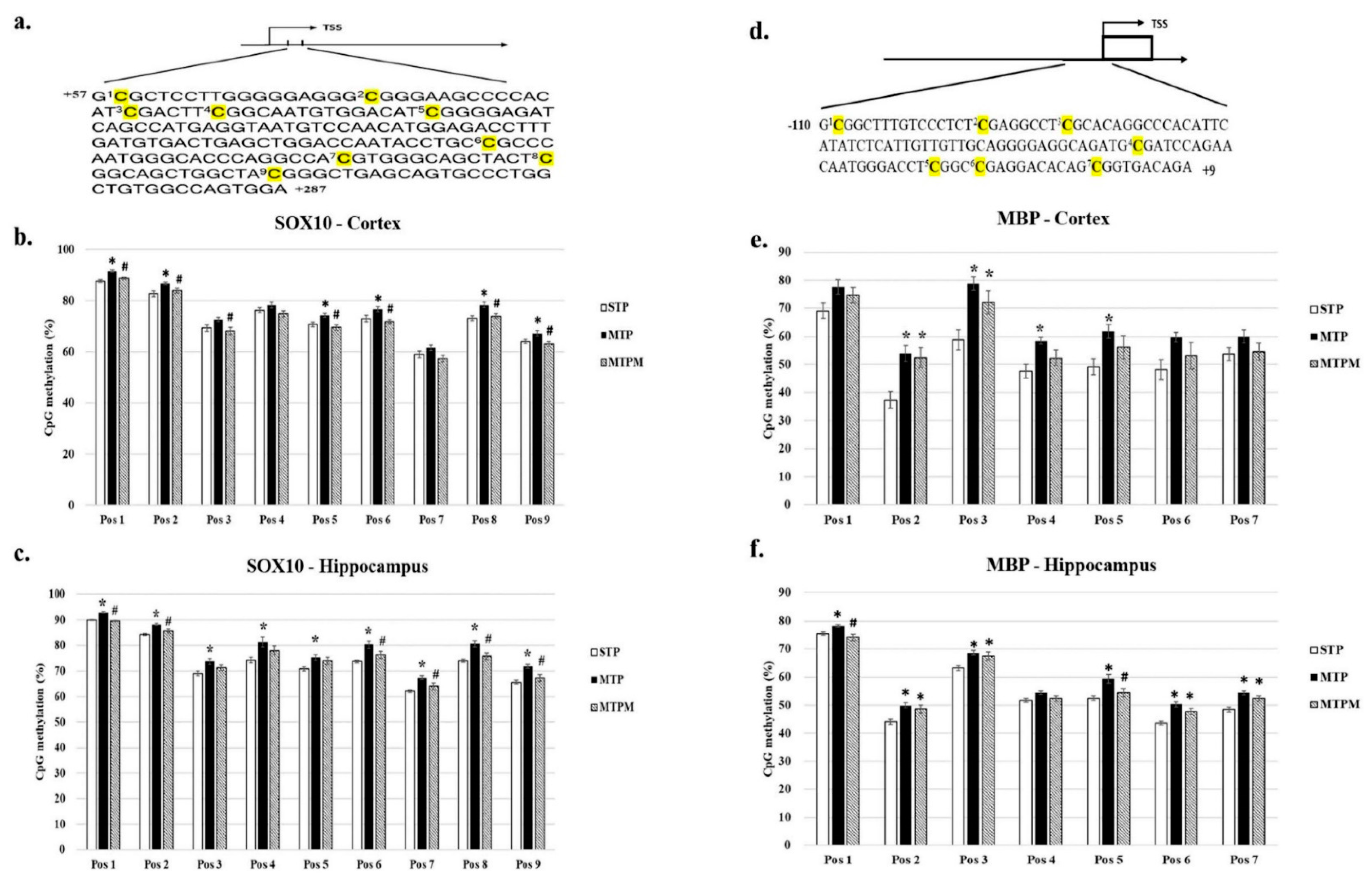
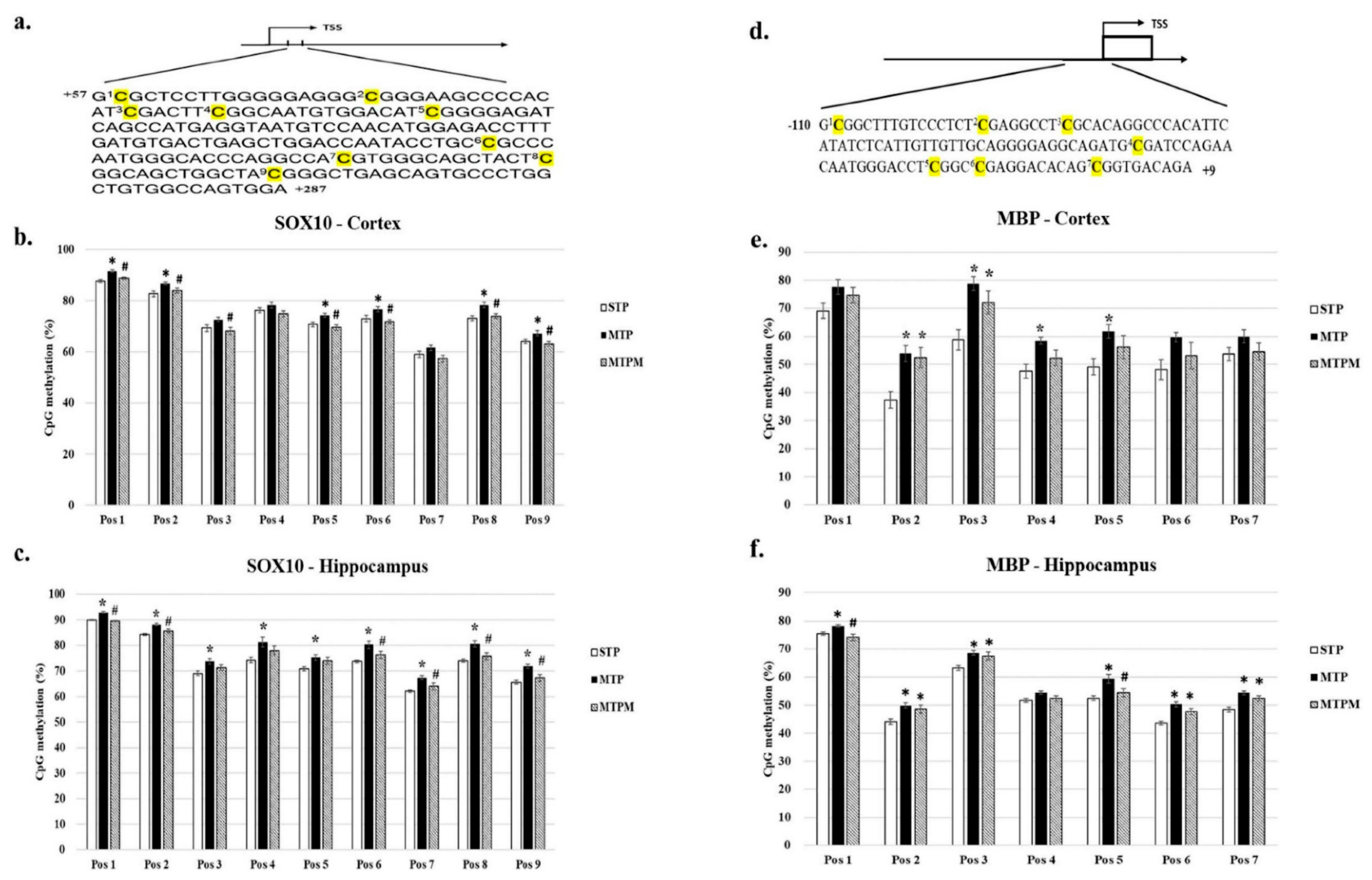
2.3. Combined Intrathecal and Intraperitoneal Methotrexate Treatment Resulted in Altered CpG Methylation Status of the SOX10 and MBP Promoter in Both Brain Cortex and Hippocampus
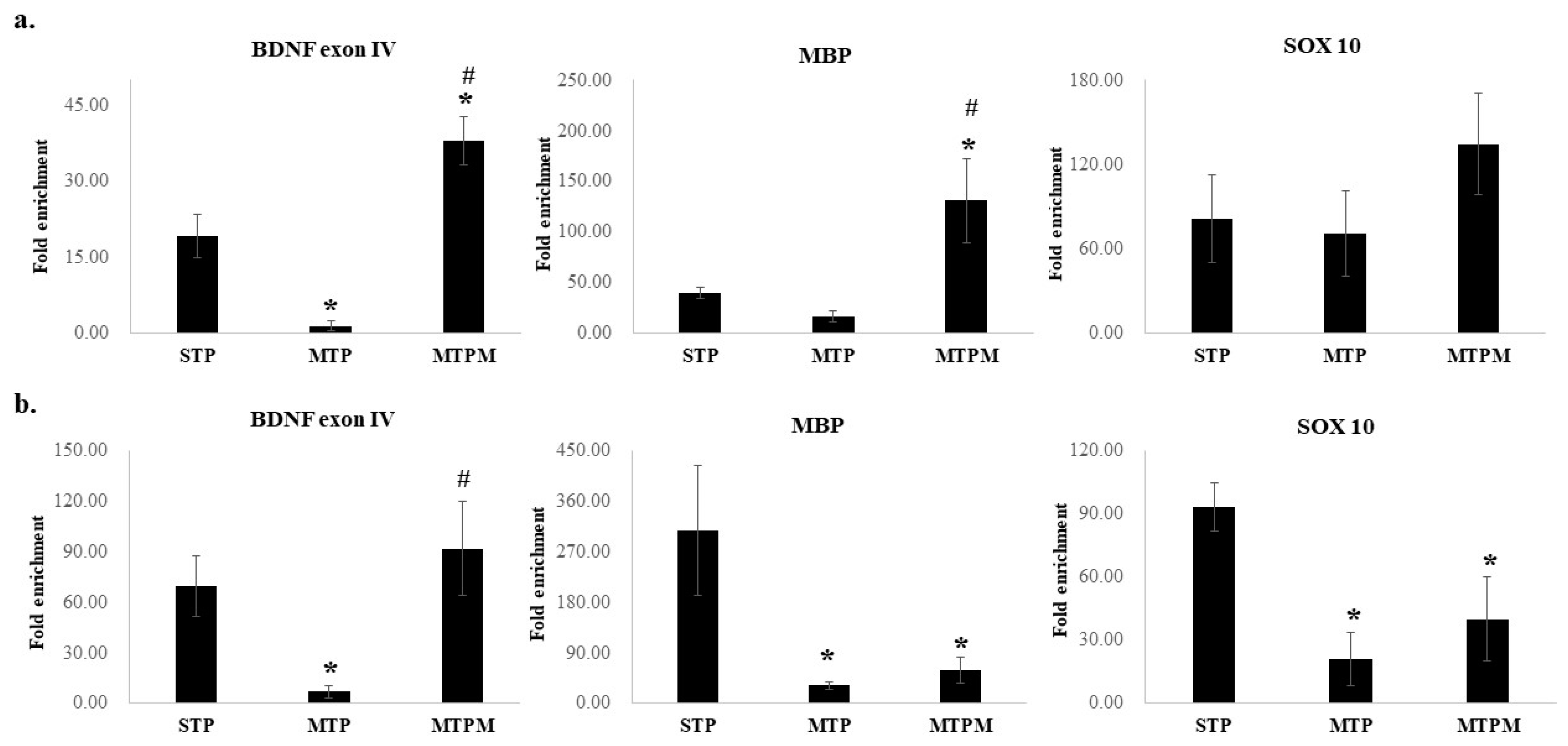
2.4. Combined Intrathecal and Intraperitoneal Methotrexate Treatment Resulted in Epigenetic Modification of BDNF Exon IV Promoter, Myelin Basic Protein (MBP) and SOX 10 Promoter
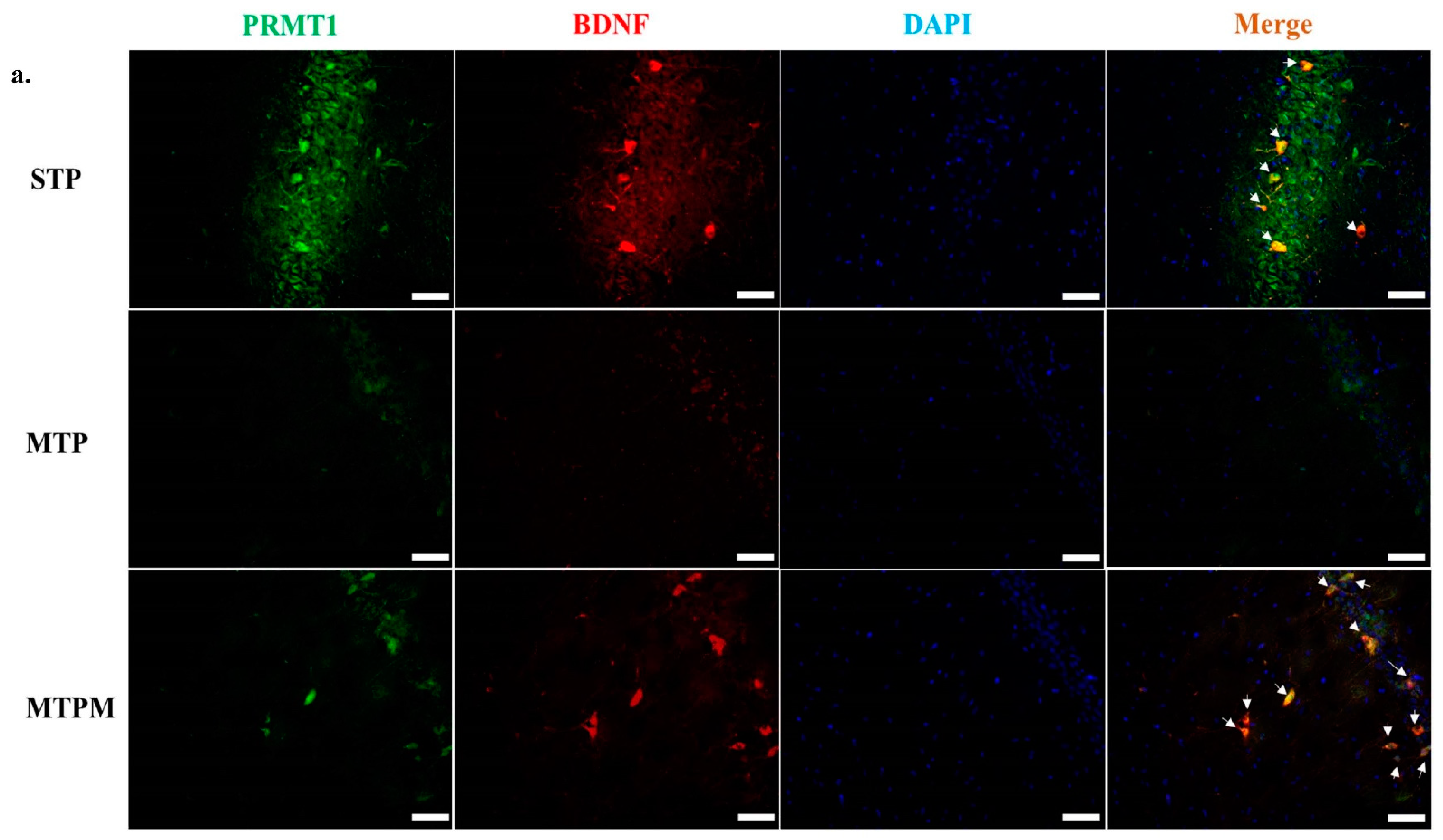
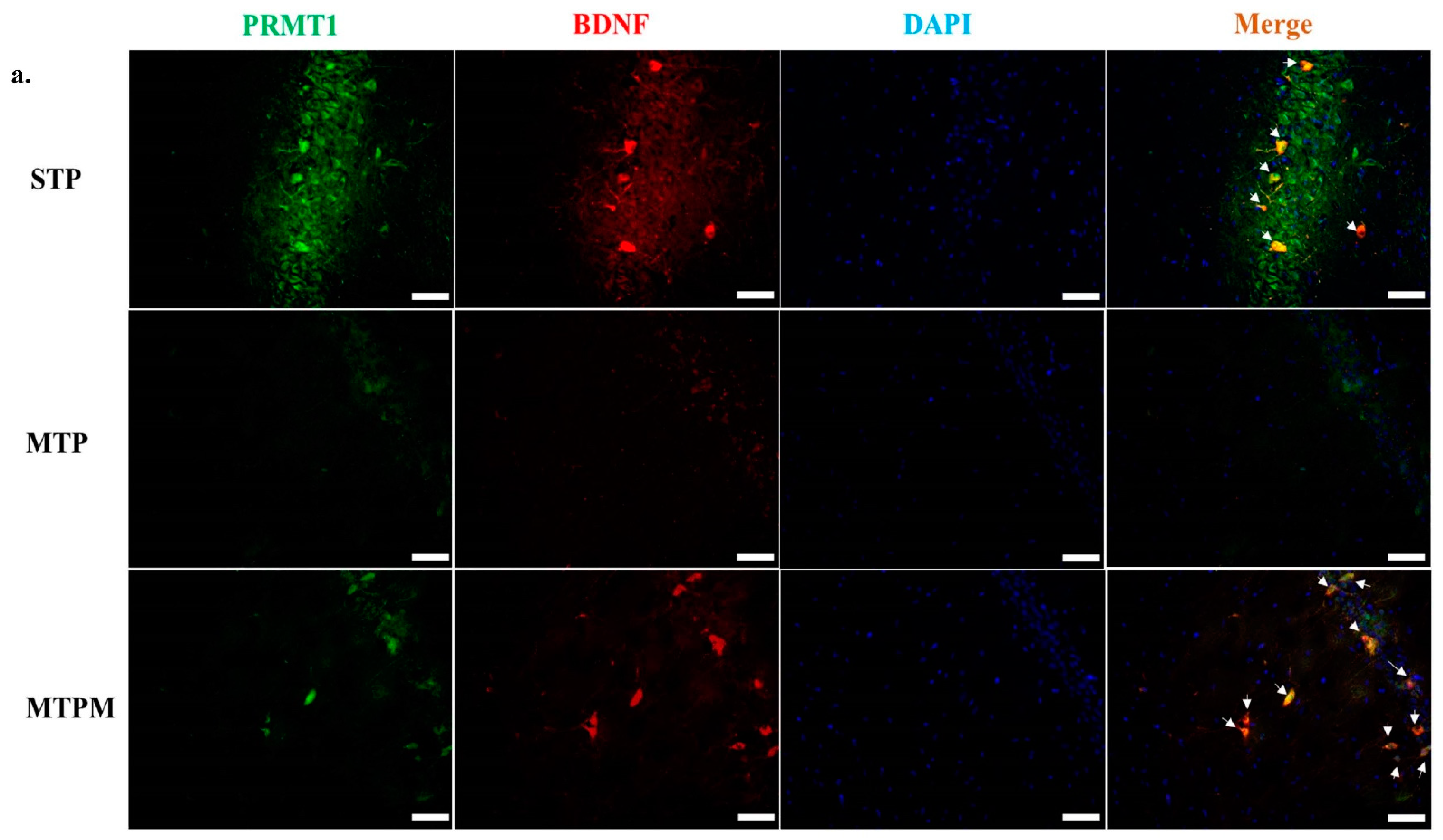
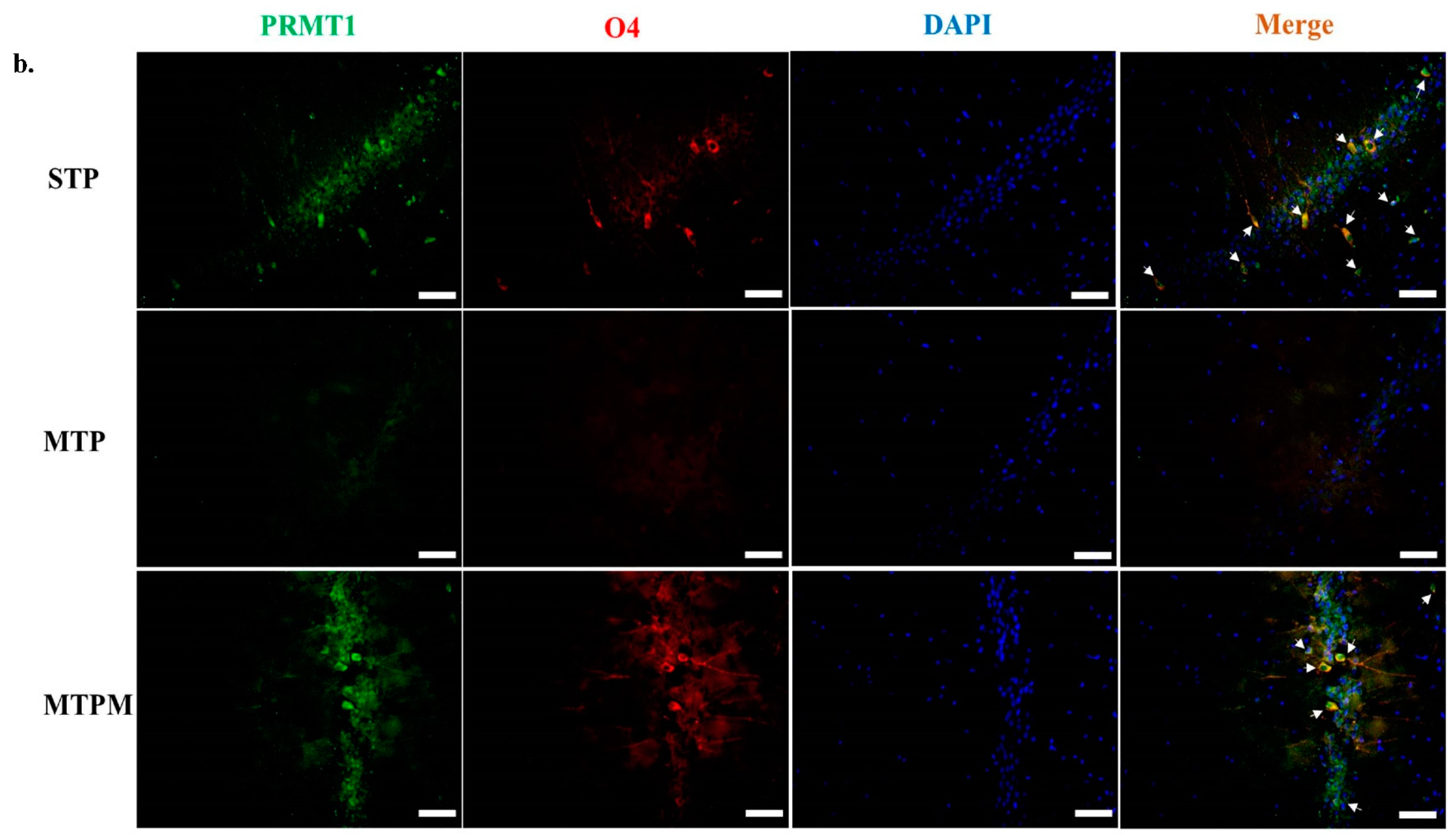
2.5. PRMT1 Is Involved in the Post-Translational Modification of BDNF and Myelination-Related Transcriptomes in MTX-Treated Developing Rats
3. Discussion
4. Material and Methods
4.1. Subjects
4.2. Experimental Procedures
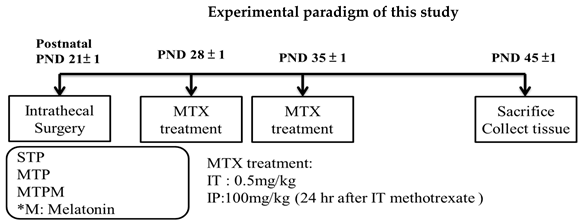
4.3. Brain Tissue Collection
4.4. DNA Bisulfite Modification and Pyrosequencing
4.5. Chromatin Immunoprecipitation (ChIP) Assay for BDNF and Myelination-Related Transcriptomes Modification
4.6. RNA-Protein Immunoprecipitation (RIP) Assay
4.7. Quantitative Real-Time PCR Analysis
4.8. Immunofluorescence Staining
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cascella, M.; Napoli, D.R.; Carbone, D.; Cuomo, G.F.; Bimonte, S.; Muzio, M.R. Chemotherapy-related cognitive impairment: Mechanisms, clinical features and research perspectives. Recent. Prog. Med. 2018, 109, 523–530. [Google Scholar]
- Torrente, N.C.; Navarro Pastor, J.B.; dela Osa Chaparro, N. Systematic review of cognitive sequelae of non-central nervous system cancer and cancer therapy. J. Cancer Surviv. 2020, 14, 464–482. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Campana, D.; Pei, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Onciu, M.; et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N. Engl. J. Med. 2009, 360, 2730–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pui, C.H.; Mullighan, C.G.; Evans, W.E.; Relling, M.V. Pediatric acute lymphoblastic leukemia: Where are we going and how do we get there? Blood 2012, 120, 1165–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pui, C.H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood acute lymphoblastic leukemia: Progress through collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef]
- Iyer, N.S.; Balsamo, L.M.; Bracken, M.B.; Kadan-Lottick, N.S. Chemotherapy-only treatment effects on long-term neurocognitive functioning in childhood ALL survivors: A review and meta-analysis. Blood 2015, 126, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Millan, N.C.; Pastrana, A.; Guitter, M.R.; Zubizarreta, P.A.; Monges, M.S.; Felice, M.S. Acute and sub-acute neurological toxicity in children treated for acute lymphoblastic leukemia. Leuk. Res. 2018, 65, 86–93. [Google Scholar] [CrossRef]
- Cheung, Y.T.; Krull, K.R. Neurocognitive outcomes in long-term survivors of childhood acute lymphoblastic leukemia treated on contemporary treatment protocols: A systematic review. Neurosci. Biobehav. Rev. 2015, 53, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Moe, P.J.; Holen, A. High-dose methotrexate in childhood all. Pediatr. Hematol. Oncol. 2000, 17, 615–622. [Google Scholar] [CrossRef]
- Cohen, I.J. Neurotoxicity after high-dose methotrexate (MTX) is adequately explained by insufficient folinic acid rescue. Cancer Chemother. Pharmacol. 2017, 79, 1057–1065. [Google Scholar] [CrossRef]
- Vezmar, S.; Schüsseler, P.; Becker, A.; Bode, U.; Jaehde, U. Methotrexate-associated alterations of the folate and methyl-transfer pathway in the CSF of All patients with and without symptoms of neurotoxicity. Pediatr. Blood Cancer 2009, 52, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Sirichoat, A.; Krutsri, S.; Suwannakot, K.; Aranarochana, A.; Chaisawang, P.; Pannangrong, W.; Wigmore, P.; Welbat, J.U. Melatonin protects against methotrexate-induced memory deficit and hippocampal neurogenesis impairment in a rat model. Biochem. Pharmacol. 2019, 163, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Maxwell, R.R.; Wolf, A.J.; Spira, M.; Gulinello, M.E.; Cole, P.D. Methotrexate causes persistent deficits in memory and executive function in a juvenile animal model. Neuropharmacology 2018, 139, 76–84. [Google Scholar] [CrossRef]
- Chen, Y.C.; Sheen, J.M.; Hsu, M.H.; Hsiao, C.C.; Wang, S.C.; Huang, L.T. Melatonin rescued methotrexate-induced spatial deficit and hyperhomocysteinemia and increased asymmetric dimethylarginine in plasma and dorsal hippocampus in developing rats. Life Sci. 2020, 242, 116931. [Google Scholar] [CrossRef] [PubMed]
- Aukema, E.J.; Caan, M.W.A.; Oudhuis, N.; Majoie, C.B.L.M.; Vos, F.M.; Reneman, L.; Last, B.F.; Grootenhuis, M.A.; Schouten-van Meeteren, A.Y.N. White matter fractional anisotropy correlates with speed of processing and motor speed in young childhood cancer survivors. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 837–843. [Google Scholar] [CrossRef]
- Kaiser, J.; Bledowski, C.; Dietrich, J. Neural correlates of chemotherapy-related cognitive impairment. Cortex 2014, 54, 33–50. [Google Scholar] [CrossRef]
- Pierson, C.; Waite, E.; Pyykkonen, B. A meta-analysis of the neuropsychological effects of chemotherapy in the treatment of childhood cancer. Pediatr. Blood Cancer 2016, 63, 1998–2003. [Google Scholar] [CrossRef]
- Gibson, E.M.; Nagaraja, S.; Ocampo, A.; Tam, L.T.; Wood, L.S.; Pallegar, P.N.; Greene, J.J.; Geraghty, A.C.; Goldstein, A.K.; Ni, L.; et al. Methotrexate chemotherapy induces persistent tri-glial dysregulation that underlies chemotherapy-related cognitive impairment. Cell 2019, 176, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Kolli, V.K.; Abraham, P.; Isaac, B.; Kasthuri, N. Preclinical efficacy of melatonin to reduce methotrexate-induced oxidative stress and small intestinal damage in rats. Dig. Dis. Sci. 2013, 58, 959–969. [Google Scholar] [CrossRef]
- Abraham, P.; Kolli, V.K.; Rabi, S. Melatonin attenuates methotrexate-induced oxidative stress and renal damage in rats. Cell Biochem. Funct. 2010, 28, 426–433. [Google Scholar] [CrossRef]
- Oguz, E.; Kocarslan, S.; Tabur, S.; Sezen, H.; Yilmaz, Z.; Aksoy, N. Effects of lycopene alone or combined with melatonin on methotrexate-induced nephrotoxicity in rats. Asian Pac. J. Cancer Prev. 2015, 16, 6061–6066. [Google Scholar] [CrossRef] [Green Version]
- Wegner, M. All purpose sox: The many roles of sox proteins in gene expression. Int. J. Biochem. Cell Biol. 2010, 42, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weider, M.; Wegner, M. SoxE factors: Transcriptional regulators of neural differentiation and nervous system development. Semin. Cell Dev. Biol. 2017, 63, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Weider, M.; Reiprich, S.; Wegner, M. Sox appeal-Sox10 attracts epigenetic and transcriptional regulators in myelinating glia. Biol. Chem. 2013, 394, 1583–1593. [Google Scholar] [CrossRef]
- Fogarty, E.A.; Kitzman, J.O.; Antonellis, A. SOX10-regulated promoter use defines isoform-specific gene expression in Schwann cells. BMC Genom. 2020, 21, 549. [Google Scholar] [CrossRef]
- Saur, A.L.; Fröb, F.; Weider, M.; Wegner, M. Formation of the node of Ranvier by Schwann cells is under control of transcription factor Sox10. Glia 2021, 69, 1464–1477. [Google Scholar] [CrossRef]
- Frazzini, V.; Granzotto, A.; Bomba, M.; Massetti, N.; Castelli, V.; d’Aurora, M.; Punzi, M.; Iorio, M.; Mosca, A.; Delli Pizzi, S.; et al. The pharmacological perturbation of brain zinc impairs BDNF-related signaling and the cognitive performances of young mice. Sci. Rep. 2018, 8, 9768. [Google Scholar] [CrossRef] [Green Version]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Fletcher, J.L.; Murray, S.S.; Xiao, J. Brain-derived neurotrophic factor in central nervous system myelination: A new mechanism to promote myelin plasticity and repair. Int. J. Mol. Sci. 2018, 19, 4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, T.; Lee, Y.Y.; Chae, J.W.; Yeo, A.H.L.; Shwe, M.; Gan, Y.X.; Ng, R.C.H.; Chu, P.P.Y.; Khor, C.C.; Ho, H.K.; et al. Evaluation of plasma brain-derived neurotrophic factor levels and self-perceived cognitive impairment post-chemotherapy: A longitudinal study. BMC Cancer 2017, 17, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azoulay, D.; Leibovici, A.; Sharoni, R.; Shaoul, E.; Gross, B.; Braester, A.; Goldberg, H. Association between Met-BDNF allele and vulnerability to paclitaxel-induced peripheral neuropathy. Breast Cancer Res. Treat. 2015, 153, 703–704. [Google Scholar] [CrossRef]
- Geraghty, A.C.; Gibson, E.M.; Ghanem, R.A.; Greene, J.J.; Ocampo, A.; Goldstein, A.K.; Ni, L.; Yang, T.; Marton, R.M.; Paşca, S.P.; et al. Loss of adaptive myelination contributes to methotrexate chemotherapy-related cognitive impairment. Neuron 2019, 103, 250–265. [Google Scholar] [CrossRef]
- Hashimoto, M.; Fukamizu, A.; Nakagawa, T.; Kizuka, Y. Roles of protein arginine methyltransferase 1 (PRMT1) in brain development and disease. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129776. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Kumabe, A.; Kim, J.D.; Murata, K.; Sekizar, S.; Williams, A.; Lu, W.; Ishida, J.; Nakagawa, T.; Endo, M.; et al. Loss of PRMT1 in the central nervous system (CNS) induces reactive astrocytes and microglia during postnatal brain development. J. Neurochem. 2021, 156, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Forster, V.J.; McDonnell, A.; Theobald, R.; Mckay, J.A. Effect of methotrexate/vitamin B12 on DNA methylation as a potential factor in leukemia treatment-related neurotoxicity. Epigenomics 2017, 9, 1205–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Keeney, J.T.R.; Miriyala, S.; Noel, T.; Powell, D.K.; Chaiswing, L.; Bondada, S.; St Clair, D.K.; Butterfield, D.A. The triangle of death of neurons: Oxidative damage, mitochondrial dysfunction, and loss of choline-containing biomolecules in brains of mice treated with doxorubicin. Advanced insights into mechanisms of chemotherapy induced cognitive impairment (“chemobrain”) involving TNF-α. Free Radic. Biol. Med. 2019, 134, 1–8. [Google Scholar] [PubMed]
- Du, J.; Zhang, A.; Li, J.; Liu, X.; Wu, H.; Wang, B.; Wang, Y.; Jia, H. Doxorubicin-induced cognitive impairment: The mechanistic insights. Front. Oncol. 2021, 11, 673340. [Google Scholar] [CrossRef]
- Koh, Y.Q.; Tan, C.J.; Toh, Y.L.; Sze, S.K.; Ho, H.K.; Limoli, C.L.; Chan, A. role of exosomes in cancer-elated cognitive impairment. Int. J. Mol. Sci. 2020, 21, 2755. [Google Scholar] [CrossRef] [Green Version]
- Koh, Y.Q.; Ng, D.Q.; Ng, C.C.; Boey, A.; Wei, M.; Sze, S.K.; Ho, H.K.; Acharya, M.; Limoli, C.L.; Chan, A. Extracellular vesicle proteome of breast cancer patients with and without cognitive impairment following anthracycline-based chemotherapy: An exploratory study. Biomark. Insights 2021, 16, 1–12. [Google Scholar] [CrossRef]
- Lyon, D.; Elmore, L.; Aboalela, N.; Merrill-Schools, J.; McCain, N.; Starkweather, A.; Elswick, R.K., Jr.; Jackson-Cook, C. Potential epigenetic mechanism(s) associated with the persistence of psychoneurological symptoms in women receiving chemotherapy for breast cancer: A hypothesis. Biol. Res. Nurs. 2014, 16, 160–174. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.M.; Walitt, B.; Saligan, L.; Tiwari, A.F.W.; Cheung, C.W.; Zhang, Z.-J. Chemobrain: A critical review and causal hypothesis of link between cytokines and epigenetic reprogramming associated with chemotherapy. Cytokine 2015, 72, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsythe, A.; Breland, T.; Majumdar, S.; Elkin, T.D.; Johnson, D.; Megason, G. Gender differences in incidence rates of childhood. B-precursor acute lymphocytic leukemia in Mississippi. J. Pediatr. Oncol. Nurs. 2010, 27, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.L.; Collins, G.J.; Ojima, Y.; Sullivan, R.D. A study of distribution of methotrexate in human tissues and tumors. Cancer Res. 1970, 30, 1344–1348. [Google Scholar] [PubMed]
- Balis, F.M.; Blaney, S.M.; McCully, C.L.; Bacher, J.D.; Murphy, R.F.; Poplack, D.G. Methotrexate distribution within the subarachnoid space after intraventricular and intravenous administration. Cancer Chemother. Pharmacol. 2000, 45, 259–264. [Google Scholar] [CrossRef]
- Westerhout, J.; van den Berg, D.-J.; Hartman, R.; Danhof, M.; de Lange, E.C.M. Prediction of methotrexate CNS distribution in different species-influence of disease conditions. Eur. J. Pharm. Sci. 2014, 16, 11–24. [Google Scholar] [CrossRef] [Green Version]
- del Río, B.; García Pedrero, J.M.; Martínez-Campa, C.; Zuazua, P.; Lazo, P.S.; Ramos, S. Melatonin, an endogenous-specific inhibitor of estrogen receptor alpha via calmodulin. J. Biol. Chem. 2004, 279, 38294–38302. [Google Scholar] [CrossRef] [Green Version]
- Cos, S.; González, A.; Martínez-Campa, C.; Mediavilla, M.D.; Alonso-González, C.; Sánchez-Barceló, E.J. Melatonin as a selective estrogen enzyme modulator. Curr. Cancer Drug Targets 2008, 8, 691–702. [Google Scholar] [CrossRef]
- Huang, L.T.; Chen, C.C.; Sheen, J.M.; Chen, Y.J.; Hsieh, C.S.; Tain, Y.L. The interaction between high ammonia diet and bileduct ligation in developing rats: Assessment by spatial memory and asymmetric dimethylarginine. Int. J. Dev. Neurosci. 2010, 28, 169–174. [Google Scholar] [CrossRef]
- Sheen, J.M.; Chen, Y.C.; Hsu, M.H.; Tain, Y.L.; Yu, H.R.; Huang, L.T. Combined intraperitoneal and intrathecal etanercept reduce increased brain tumor necrosis factor-alpha and asymmetric dimethylarginine levels and rescues spatial deficits in young rats after bile duct ligation. Front. Cell Neurosci. 2016, 10, 167. [Google Scholar] [CrossRef] [Green Version]
- Cole, P.D.; Vijayanathan, V.; Ali, N.F.; Wagshul, M.E.; Tanenbaum, E.J.; Price, J.; Dalal, V.; Gulinello, M.E. Memantine protects rats treated with intrathecal methotrexate from developing spatial memory deficits. Clin. Cancer Res. 2013, 19, 4446–4454. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, J.; Prust, M.; Kaiser, J. Chemotherapy, cognitive impairment and hippocampal toxicity. Neuroscience 2015, 309, 224–232. [Google Scholar] [CrossRef]
- Sloan, H.L.; Good, M.; Dunnett, S.B. Double dissociation between hippocampal and prefrontal lesions on an operant delayed matching task and a water maze reference memory task. Behav. Brain Res. 2006, 171, 116–126. [Google Scholar] [CrossRef]
- Parrish, R.R.; Buckingham, S.C.; Mascia, K.L.; Johnson, J.J.; Matyjasik, M.M.; Lockhart, R.M.; Lubin, F.D. Methionine increases BDNF DNA methylation and improves memory in epilepsy. Ann. Clin. Transl. Neurol. 2015, 2, 401–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demircan, B.; Dyer, L.M.; Gerace, M.; Lobenhofer, E.K.; Robertson, K.D.; Brown, K.D. Comparative epigenomics of human and mouse mammary tumors. Genes Chromosomes Cancer 2009, 48, 83–97. [Google Scholar] [CrossRef] [Green Version]
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Y.; Shen, X.M.; Yuan, F.F.; Guo, O.Y.; Zhong, Y.; Chen, J.G.; Zhu, L.Q.; Wu, J. The physiology of BDNF and its relationship with ADHD. Mol. Neurobiol. 2015, 52, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Ma, J.; Zheng, J.; Liu, X.; Liu, Y.; Ruan, X.; Shen, S.; Yang, C.; Wang, D.; Cai, H.; et al. PABPC1-induced stabilization of BDNF-AS inhibits malignant progression of glioblastoma cells through STAU1-mediated decay. Cell Death Dis. 2020, 11, 81. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′-3′) | Reverse (5′-3′) Biotinylated | Sequencing Primer |
|---|---|---|---|
| BDNF exon IV | AGGTAGAGGAGGTATTATATGATAGT AGGTAGAGGAGGTATTATATGATAGT | ACTATATATTTCCCCTTCTCTTCAATT AACTCTTACTATATATTTCCCCTTCT | AGGAGGTATTATATGATAGTT ATTTATAGAGGAGAGGATTGT |
| MBP | GAGATAGTTAGTTTAAGATTTTAGGAAGAA | ACCCTACCAATTATTCTTTAAATCTACT | GTTAGTTTAAGATTTTAGGAAGAAA |
| SOX10 | GTTAGGTAAGGTAGATTTTAAAAGGGATG | TAACCACAACCAAAACACTACTCAA | GTAGATTTTAAAAGGGATGG ATGTGATTGAGTTGGAT |
| Forward 5′-3′ | Reverse 5′-3′ | |
|---|---|---|
| BDNF exon IV promoter | TGACAGCTCACGTCAAGGCA | ATCTAGGCAGAGACTGGGAGAT |
| MBP promoter | AGCTAGTTCAAGACCCCAGG | AGATATGAATGTGGGCCTGTGC |
| SOX10 promoter | CAGCCATGAGGTAATGTCCA | GTAGCCAGCTGCCGAGTAG |
| Forward 5′-3′ | Reverse 5′-3′ | |
|---|---|---|
| BDNF exon IV | ATTCACCGAGGAGAGGACTG | AGTCTTTGGTGGCCGATATG |
| MBP | CTCTGGCAAGG CTCACACAC | TCTGCTGAGGGACAGGCCTCTC |
| SOX10 | TACAAGTACCAACCTCGGCG | CATGGGGGAGCCTTCTTCTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-C.; Sheen, J.-M.; Wang, S.-C.; Hsu, M.-H.; Hsiao, C.-C.; Chang, K.-A.; Huang, L.-T. Methotrexate Neurotoxicity Is Related to Epigenetic Modification of the Myelination Process. Int. J. Mol. Sci. 2021, 22, 6718. https://doi.org/10.3390/ijms22136718
Chen Y-C, Sheen J-M, Wang S-C, Hsu M-H, Hsiao C-C, Chang K-A, Huang L-T. Methotrexate Neurotoxicity Is Related to Epigenetic Modification of the Myelination Process. International Journal of Molecular Sciences. 2021; 22(13):6718. https://doi.org/10.3390/ijms22136718
Chicago/Turabian StyleChen, Yu-Chieh, Jiunn-Ming Sheen, Su-Chen Wang, Mei-Hsin Hsu, Chih-Cheng Hsiao, Kow-Aung Chang, and Li-Tung Huang. 2021. "Methotrexate Neurotoxicity Is Related to Epigenetic Modification of the Myelination Process" International Journal of Molecular Sciences 22, no. 13: 6718. https://doi.org/10.3390/ijms22136718
APA StyleChen, Y.-C., Sheen, J.-M., Wang, S.-C., Hsu, M.-H., Hsiao, C.-C., Chang, K.-A., & Huang, L.-T. (2021). Methotrexate Neurotoxicity Is Related to Epigenetic Modification of the Myelination Process. International Journal of Molecular Sciences, 22(13), 6718. https://doi.org/10.3390/ijms22136718