
Loss of Individual Mitochondrial Ribonuclease P Complex Proteins Differentially Affects Mitochondrial tRNA Processing In Vivo


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
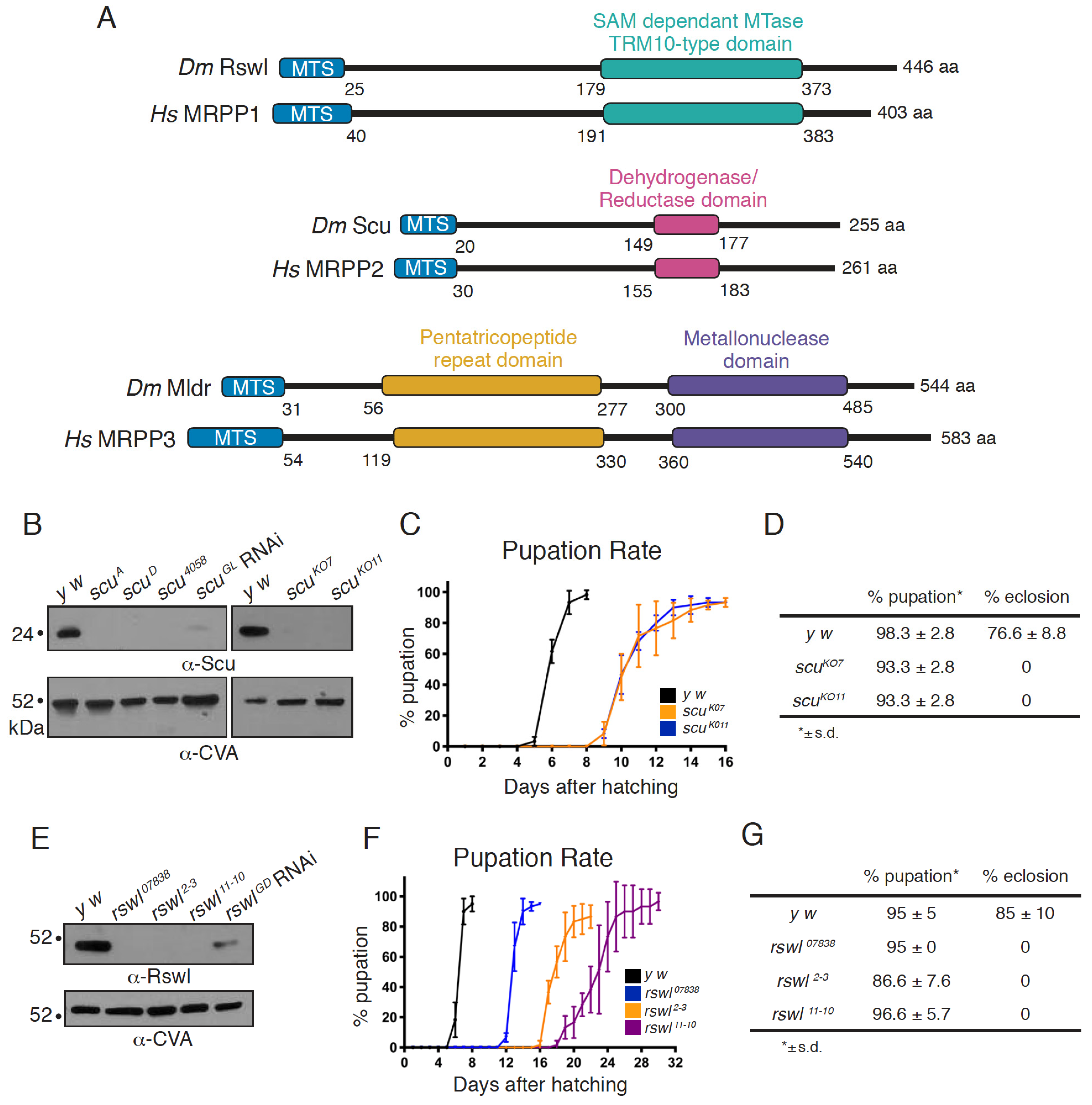
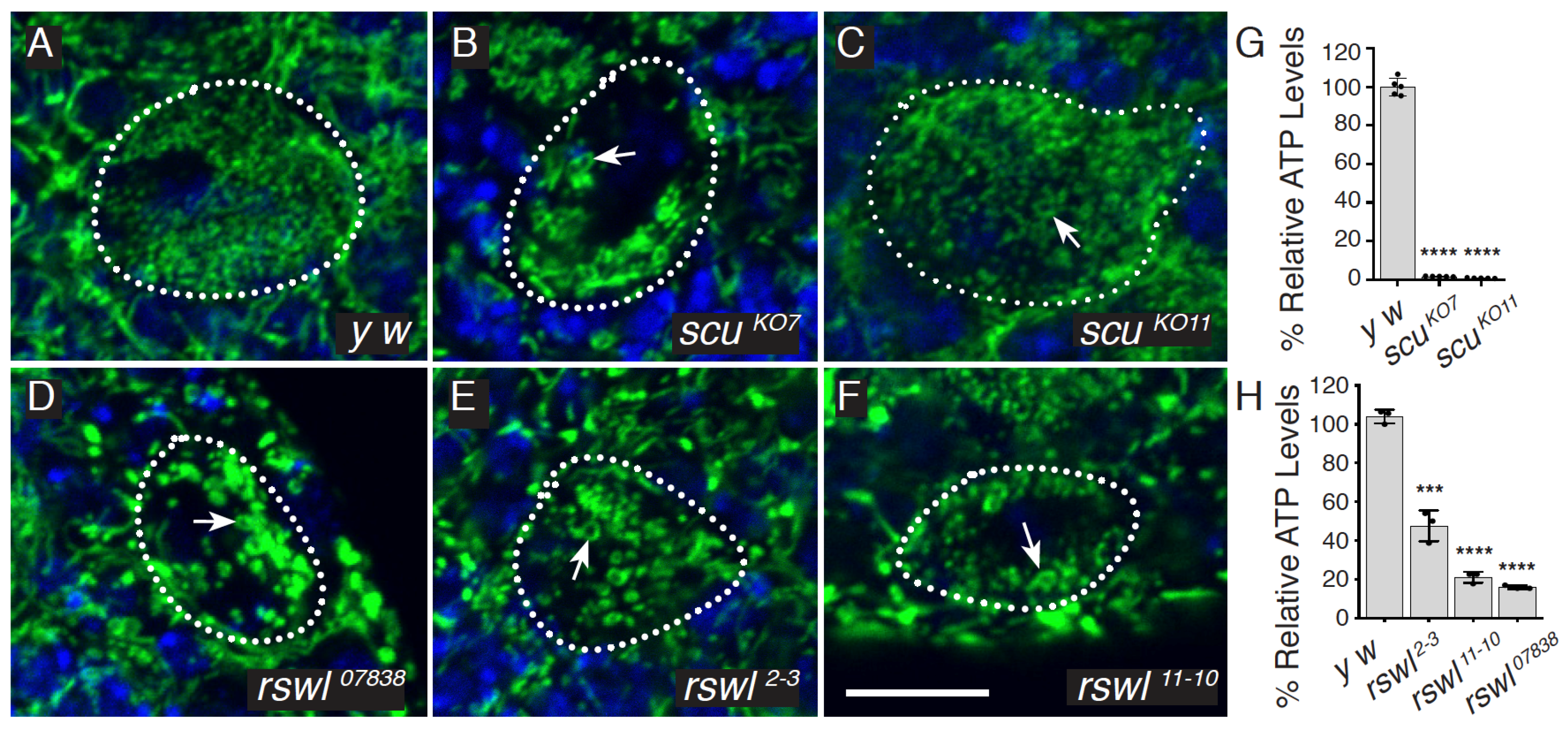
2.1. Protein Null Alleles of Scully and Roswell Confer Lethality and Mitochondrial Damage
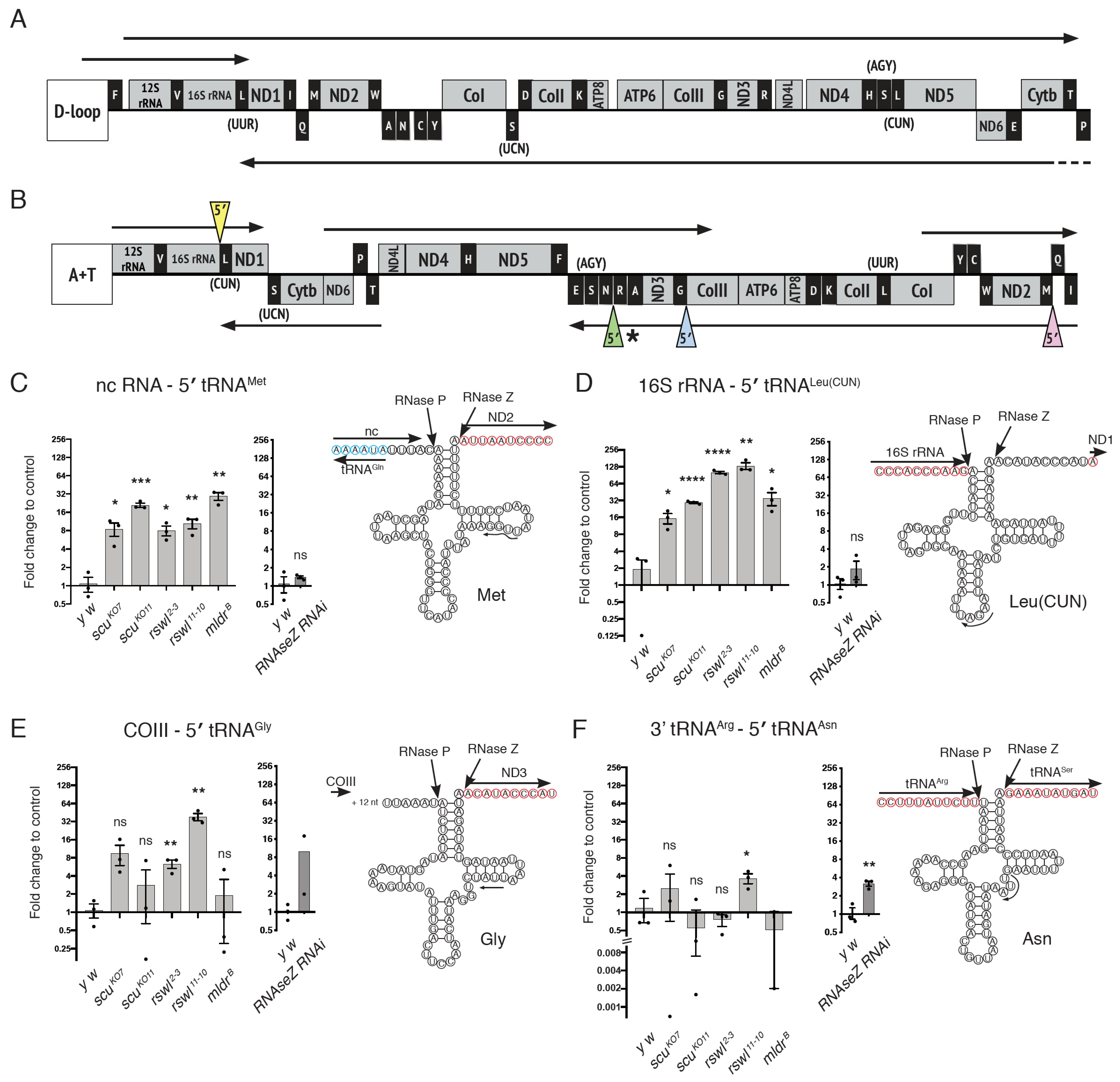
2.2. Loss of mtRNase P Complex Members Differentially Affects mt:tRNA 5′-End Processing In Vivo
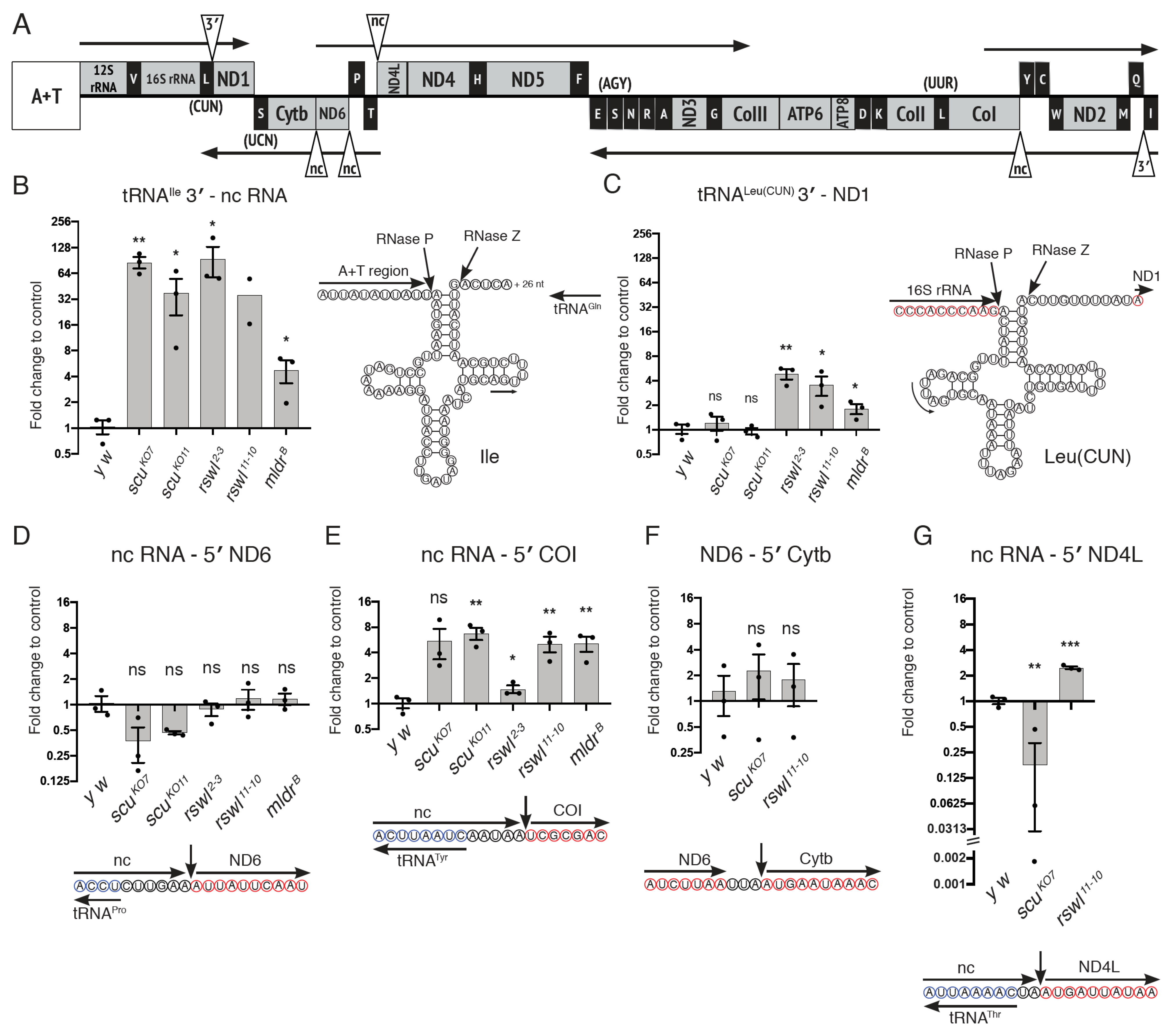
2.3. mt:tRNA 3′-End and Non-Canonical Junction Processing Is Affected Differentially by Loss of mtRNase P Complex Members
2.4. Loss of RNase P Differentially Affects the Abundance of Individual Transcripts Derived from the Same Polycistronic Transcript
3. Discussion
3.1. Protein Null Mutants for In Vivo Analysis of All Three RNase P Complex Members
3.2. Analyzing Specific mt:tRNA Junction Processing In Vivo
3.3. Canonical Junction Cleavage Is Most Affected by the Loss of Rswl
3.4. Loss of Rswl Affects Processing within a Cluster of mt:tRNAs in Drosophila
3.5. mt:tRNA Processing Is Not Always Dependent on Scu
3.6. mtRNase P Action In Vivo
4. Materials and Methods
4.1. Fly Stocks
4.2. Transgenic Flies and Crosses
4.3. Roswell and Scully Antibody Generation
4.4. Pupation and Eclosion Rates
4.5. Immunofluorescence
4.6. ATP Assay
4.7. Western Blots
4.8. qPCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, S.E.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Montoya, J.; Gaines, G.L.; Attardi, G. The pattern of transcription of the human mitochondrial rRNA genes reveals two overlapping transcription units. Cell 1983, 34, 151–159. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nat. Cell Biol. 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, J.; Frank, P.; Löffler, E.; Bennett, K.L.; Gerner, C.; Rossmanith, W. RNase P without RNA: Identification and Functional Reconstitution of the Human Mitochondrial tRNA Processing Enzyme. Cell 2008, 135, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.I.L.; Mercer, T.R.; Davies, S.M.; Shearwood, A.-M.J.; Nygård, K.K.; Richman, T.R.; Mattick, J.S.; Rackham, O.; Filipovska, A. RNA processing in human mitochondria. Cell Cycle 2011, 10, 2904–2916. [Google Scholar] [CrossRef] [PubMed]
- Altman, S. Ribonuclease P: An Enzyme with a Catalytic RNA Subunit. Adv. Enzymol. Relat. Areas Mol. Biol. 2006, 62, 1–36. [Google Scholar] [CrossRef]
- Baer, M.F.; Wesolowski, D.; Altman, S. Characterization in vitro of the defect in a temperature-sensitive mutant of the protein subunit of RNase P from Escherichia coli. J. Bacteriol. 1989, 171, 6862–6866. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.E.; Montange, R.K.; Malik, H.S.; Phizicky, E.M. Identification of the yeast gene encoding the tRNA m1G methyltransferase responsible for modification at position 9. RNA 2003, 9, 574–585. [Google Scholar] [CrossRef]
- Vilardo, E.; Nachbagauer, C.; Buzet, A.; Taschner, A.; Holzmann, J.; Rossmanith, W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase--extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012, 40, 11583–11593. [Google Scholar] [CrossRef]
- Luo, M.J.; Mao, L.F.; Schulz, H. Short-chain 3-hydroxy-2-methylacyl-CoA dehydrogenase from rat liver: Purification and characterization of a novel enzyme of isoleucine metabolism. Arch. Biochem. Biophys. 1995, 321, 214–220. [Google Scholar] [CrossRef]
- Zschocke, J.; Ruiter, J.P.; Brand, J.; Lindner, M.; Hoffmann, G.F.; Wanders, R.J.; Mayatepek, E. Progressive infantile neurodegeneration caused by 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency: A novel inborn error of branched-chain fatty acid and isoleucine metabolism. Pediatr. Res. 2000, 48, 852–855. [Google Scholar] [CrossRef]
- Gobert, A.; Gutmann, B.; Taschner, A.; Gössringer, M.; Holzmann, J.; Hartmann, R.K.; Rossmanith, W.; Giegé, P. A single Arabidopsis organellar protein has RNase P activity. Nat. Struct. Mol. Biol. 2010, 17, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.J.; Lim, W.H.; Fierke, C.A.; Koutmos, M. Mitochondrial ribonuclease P structure provides insight into the evolution of catalytic strategies for precursor-tRNA 5’ processing. Proc. Natl. Acad. Sci. USA 2012, 109, 16149–16154. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Davis, N.W.; Gegenheimer, P. Novel mechanisms for maturation of chloroplast transfer RNA precursors. EMBO J. 1988, 7, 1567–1574. [Google Scholar] [CrossRef]
- Deutschmann, A.J.; Amberger, A.; Zavadil, C.; Steinbeisser, H.; Mayr, J.A.; Feichtinger, R.G.; Oerum, S.; Yue, W.W.; Zschocke, J. Mutation or knock-down of 17beta-hydroxysteroid dehydrogenase type 10 cause loss of MRPP1 and impaired processing of mitochondrial heavy strand transcripts. Hum. Mol. Genet. 2014, 23, 3618–3628. [Google Scholar] [CrossRef]
- Reinhard, L.; Sridhara, S.; Hällberg, B.M. The MRPP1/MRPP2 complex is a tRNA-maturation platform in human mitochondria. Nucleic Acids Res. 2017, 45, 12469–12480. [Google Scholar] [CrossRef]
- Liu, X.; Wu, N.; Shanmuganathan, A.; Klemm, B.P.; Howard, M.J.; Lim, W.H.; Koutmos, M.; Fierke, C.A. Kinetic mechanism of human mitochondrial RNase P. bioRxiv 2019, 666792. [Google Scholar] [CrossRef]
- Karasik, A.; Fierke, C.A.; Koutmos, M. Interplay between substrate recognition, 5′ end tRNA processing and methylation activity of human mitochondrial RNase P. RNA 2019, 25, 1646–1660. [Google Scholar] [CrossRef] [PubMed]
- Rackham, O.; Busch, J.D.; Matic, S.; Siira, S.; Kuznetsova, I.; Atanassov, I.; Ermer, J.A.; Shearwood, A.-M.J.; Richman, T.; Stewart, J.B.; et al. Hierarchical RNA Processing Is Required for Mitochondrial Ribosome Assembly. Cell Rep. 2016, 16, 1874–1890. [Google Scholar] [CrossRef]
- Saoji, M.; Cox, R.T. Mitochondrial RNase P Complex in Animals: Mitochondrial tRNA Processing and Links to Disease. In Modified Nucleic Acids; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2018; pp. 47–71. [Google Scholar]
- Brandon, M.C.; Lott, M.T.; Nguyen, K.C.; Spolim, S.; Navathe, S.B.; Baldi, P.; Wallace, D.C. MITOMAP: A human mitochondrial genome database--2004 update. Nucleic Acids Res. 2004, 33, D611–D613. [Google Scholar] [CrossRef]
- Zschocke, J. HSD10 disease: Clinical consequences of mutations in the HSD17B10 gene. J. Inherit. Metab. Dis. 2011, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Chatfield, K.C.; Coughlin, C.R., 2nd; Friederich, M.W.; Gallagher, R.C.; Hesselberth, J.R.; Lovell, M.A.; Ofman, R.; Swanson, M.A.; Thomas, J.A.; Wanders, R.J.; et al. Mitochondrial energy failure in HSD10 disease is due to defective mtDNA transcript processing. Mitochondrion 2015, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Oerum, S.; Roovers, M.; Leichsenring, M.; Acquaviva-Bourdain, C.; Beermann, F.; Gemperle-Britschgi, C.; Fouilhoux, A.; Korwitz-Reichelt, A.; Bailey, H.J.; Droogmans, L.; et al. Novel patient missense mutations in the HSD17B10 gene affect dehydrogenase and mitochondrial tRNA modification functions of the encoded protein. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 3294–3302. [Google Scholar] [CrossRef]
- Vilardo, E.; Rossmanith, W. Molecular insights into HSD10 disease: Impact of SDR5C1 mutations on the human mitochondrial RNase P complex. Nucleic Acids Res. 2015, 43, 5112–5119. [Google Scholar] [CrossRef]
- Metodiev, M.D.; Thompson, K.; Alston, C.L.; Morris, A.A.; He, L.; Assouline, Z.; Rio, M.; Bahi-Buisson, N.; Pyle, A.; Griffin, H.; et al. Recessive Mutations in TRMT10C Cause Defects in Mitochondrial RNA Processing and Multiple Respiratory Chain Deficiencies. Am. J. Hum. Genet. 2016, 98, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, I.; Demain, L.A.M.; Urquhart, J.E.; Amberger, A.; Deutschmann, A.J.; Demetz, S.; Thompson, K.; O’Sullivan, J.; Belyantseva, I.A.; Barzik, M.; et al. A homozygous variant in mitochondrial RNase P subunit PRORP is associated with Perrault syndrome characterized by hearing loss and primary ovarian insufficiency. BioRxiv 2017. [Google Scholar] [CrossRef]
- Rauschenberger, K.; Scholer, K.; Sass, J.O.; Sauer, S.; Djuric, Z.; Rumig, C.; Wolf, N.I.; Okun, J.G.; Kolker, S.; Schwarz, H.; et al. A non-enzymatic function of 17beta-hydroxysteroid dehydrogenase type 10 is required for mitochondrial integrity and cell survival. EMBO Mol. Med. 2010, 2, 51–62. [Google Scholar] [CrossRef]
- Sen, A.; Karasik, A.; Shanmuganathan, A.; Mirkovic, E.; Koutmos, M.; Cox, R.T. Loss of the mitochondrial protein-only ribonuclease P complex causes aberrant tRNA processing and lethality in Drosophila. Nucleic Acids Res. 2016, 44, 6409–6422. [Google Scholar] [CrossRef]
- Claros, M.G.; Vincens, P. Computational Method to Predict Mitochondrially Imported Proteins and their Targeting Sequences. JBIC J. Biol. Inorg. Chem. 1996, 241, 779–786. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; López, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Bassett, A.R.; Tibbit, C.; Ponting, C.P.; Liu, J.-L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013, 4, 220–228. [Google Scholar] [CrossRef]
- Ren, X.; Sun, J.; Housden, B.; Hu, Y.; Roesel, C.; Lin, S.; Liu, L.-P.; Yang, Z.; Mao, D.; Sun, L.; et al. Optimized gene editing technology for Drosophila melanogaster using germ line-specific Cas9. Proc. Natl. Acad. Sci. USA 2013, 110, 19012–19017. [Google Scholar] [CrossRef]
- Huser, J.; Blatter, L.A. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J. 1999, 343 Pt 2, 311–317. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Chandel, N.S.; Williamson, E.K.; Schumacker, P.T.; Thompson, C.B. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 1997, 91, 627–637. [Google Scholar] [CrossRef]
- Berthier, F.; Renaud, M.; Alziari, S.; Durand, R. RNA mapping on Drosophila mitochondrial DNA: Precursors and template strands. Nucleic Acids Res. 1986, 14, 4519–4533. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Et Biophys. Acta (BBA) Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Torres, T.T.; Dolezal, M.; Schlötterer, C.; Ottenwälder, B. Expression profiling of Drosophila mitochondrial genes via deep mRNA sequencing. Nucleic Acids Res. 2009, 37, 7509–7518. [Google Scholar] [CrossRef]
- Xie, X.; Dubrovskaya, V.A.; Dubrovsky, E.B. RNAi knockdown of dRNaseZ, the Drosophila homolog of ELAC2, impairs growth of mitotic and endoreplicating tissues. Insect Biochem. Mol. Biol. 2011, 41, 167–177. [Google Scholar] [CrossRef]
- Brzezniak, L.; Bijata, M.; Szczesny, R.J.; Stepien, P.P. Involvement of human ELAC2 gene product in 3’ end processing of mitochondrial tRNAs. RNA Biol. 2011, 8, 616–626. [Google Scholar] [CrossRef]
- Szczesny, R.J.; Borowski, L.S.; Brzezniak, L.; Dmochowska, A.; Gewartowski, K.; Bartnik, E.; Stepien, P.P. Human mitochondrial RNA turnover caught in flagranti: Involvement of hSuv3p helicase in RNA surveillance. Nucleic Acids Res. 2009, 38, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Neph, S.; Dinger, M.; Crawford, J.; Smith, M.A.; Shearwood, A.-M.J.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The Human Mitochondrial Transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Ruzzenente, B.; Metodiev, M.D.; Wredenberg, A.; Bratic, A.; Park, C.B.; Cámara, Y.; Milenkovic, D.; Zickermann, V.; Wibom, R.; Hultenby, K.; et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J. 2011, 31, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Karasik, A.; Wilhelm, C.A.; Fierke, C.A.; Koutmos, M. Disease-associated mutations in mitochondrial precursor tRNAs affect binding, m1R9 methylation, and tRNA processing by mtRNase P. RNA 2021, 27, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, X.; Zhou, W.; Yang, X.; Shen, Y. Auto-inhibitory Mechanism of the Human Mitochondrial RNase P Protein Complex. Sci. Rep. 2015, 5, 9878. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.J.; Gai, X.; Shigematsu, M.; Vilardo, E.; Takase, R.; McCormick, E.; Christian, T.; Place, E.; Pierce, E.; Consugar, M.; et al. A novelHSD17B10mutation impairing the activities of the mitochondrial RNase P complex causes X-linked intractable epilepsy and neurodevelopmental regression. RNA Biol. 2015, 13, 477–485. [Google Scholar] [CrossRef]
- Oerum, S.; Roovers, M.; Rambo, R.P.; Kopec, J.; Bailey, H.J.; Fitzpatrick, F.; Newman, J.A.; Newman, W.G.; Amberger, A.; Zschocke, J.; et al. Structural insight into the human mitochondrial tRNA purine N1-methyltransferase and ribonuclease P complexes. J. Biol. Chem. 2018, 293, 12862–12876. [Google Scholar] [CrossRef]
- Pan, T.; Sosnick, T. RNA FOLDING DURING TRANSCRIPTION. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, H.W.; Oktay, Y.; Zhang, J.; Allen, E.L.; Smith, G.M.; Fan, K.C.; Hong, J.S.; French, S.W.; McCaffery, J.M.; et al. PNPASE regulates RNA import into mitochondria. Cell 2010, 142, 456–467. [Google Scholar] [CrossRef]
- Puranam, R.S.; Attardi, G. The RNase P Associated with HeLa Cell Mitochondria Contains an Essential RNA Component Identical in Sequence to That of the Nuclear RNase P. Mol. Cell. Biol. 2001, 21, 548–561. [Google Scholar] [CrossRef]
- Noh, J.H.; Kim, K.M.; Abdelmohsen, K.; Yoon, J.H.; Panda, A.C.; Munk, R.; Kim, J.; Curtis, J.; Moad, C.A.; Wohler, C.M.; et al. HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes Dev. 2016, 30, 1224–1239. [Google Scholar]
- Foriel, S.; Willems, P.; Smeitink, J.; Schenck, A.; Beyrath, J. Mitochondrial diseases: Drosophila melanogaster as a model to evaluate potential therapeutics. Int. J. Biochem. Cell Biol. 2015, 63, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Cox, R.T. Fly Models of Human Diseases: Drosophila as a Model for Understanding Human Mitochondrial Mutations and Disease. Curr. Top Dev. Biol. 2017, 121, 1–27. [Google Scholar]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. Dis. Model. Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef]
- Gratz, S.J.; Cummings, A.M.; Nguyen, J.N.; Hamm, D.C.; Donohue, L.K.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. Genome Engineering of Drosophila with the CRISPR RNA-Guided Cas9 Nuclease. Genetics 2013, 194, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Clemente, P.; Pajak, A.; Laine, I.; Wibom, R.; Wedell, A.; Freyer, C.; Wredenberg, A. SUV3 helicase is required for correct processing of mitochondrial transcripts. Nucleic Acids Res. 2015, 43, 7398–7413. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saoji, M.; Sen, A.; Cox, R.T. Loss of Individual Mitochondrial Ribonuclease P Complex Proteins Differentially Affects Mitochondrial tRNA Processing In Vivo. Int. J. Mol. Sci. 2021, 22, 6066. https://doi.org/10.3390/ijms22116066
Saoji M, Sen A, Cox RT. Loss of Individual Mitochondrial Ribonuclease P Complex Proteins Differentially Affects Mitochondrial tRNA Processing In Vivo. International Journal of Molecular Sciences. 2021; 22(11):6066. https://doi.org/10.3390/ijms22116066
Chicago/Turabian StyleSaoji, Maithili, Aditya Sen, and Rachel T. Cox. 2021. "Loss of Individual Mitochondrial Ribonuclease P Complex Proteins Differentially Affects Mitochondrial tRNA Processing In Vivo" International Journal of Molecular Sciences 22, no. 11: 6066. https://doi.org/10.3390/ijms22116066
APA StyleSaoji, M., Sen, A., & Cox, R. T. (2021). Loss of Individual Mitochondrial Ribonuclease P Complex Proteins Differentially Affects Mitochondrial tRNA Processing In Vivo. International Journal of Molecular Sciences, 22(11), 6066. https://doi.org/10.3390/ijms22116066