
Single-Cell Transcriptome Analysis as a Promising Tool to Study Pluripotent Stem Cell Reprogramming


{kind=link}
Abstract
1. Introduction
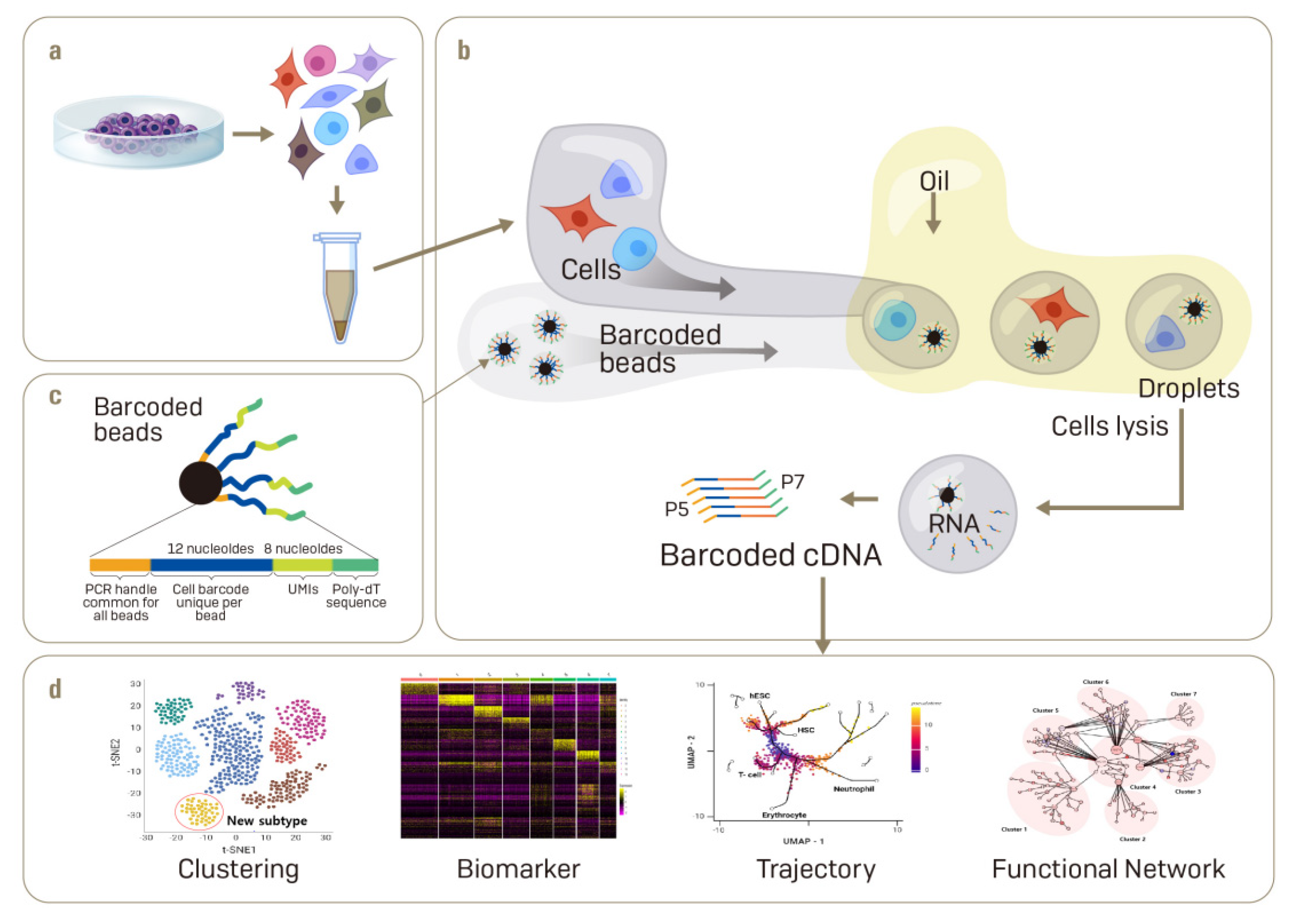
2. The Single-Cell mRNA Sequencing (scRNA-Seq) Technique
3. scRNA-Seq in PSCs
3.1. scRNA-Seq in Undifferentiated PSCs
3.2. scRNA-Seq in Somatic Cell Reprogramming
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Snyder, M.P.; Lin, S.; Posgai, A.; Atkinson, M.; Regev, A.; Rood, J.; Rozenblatt-Rosen, O.; Gaffney, L.; Hupalowska, A.; Satija, R.; et al. The human body at cellular resolution: The NIH Human Biomolecular Atlas Program. Nature 2019, 574, 187–192. [Google Scholar]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Spradling, A.C. Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.A.; Pogemiller, R.; Ogle, B.M. Heterogeneous differentiation of human mesenchymal stem cells in response to extended culture in extracellular matrices. Tissue Eng. Part A 2009, 15, 3911–3922. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Boudil, A.; Skhiri, L.; Candéias, S.; Pasqualetto, V.; Legrand, A.; Bedora-Faure, M.; Gautreau-Rolland, L.; Rocha, B.; Ezine, S. Single-cell analysis of thymocyte differentiation: Identification of transcription factor interactions and a major stochastic component in αβ-lineage commitment. PLoS ONE 2013, 8, e73098. [Google Scholar] [CrossRef]
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. Science forum: The Human Cell Atlas. Elife 2017, 6, e27041. [Google Scholar] [CrossRef]
- Aldridge, S.; Teichmann, S.A. Single cell transcriptomics comes of age. Nat. Commun. 2020, 11, 4307. [Google Scholar] [CrossRef]
- Park, J.E.; Botting, R.A.; Domínguez Conde, C.; Popescu, D.M.; Lavaert, M.; Kunz, D.J.; Goh, I.; Stephenson, E.; Ragazzini, R.; Tuck, E.; et al. A cell atlas of human thymic development defines T cell repertoire formation. Science 2020, 367. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.-E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef]
- Xing, Q.R.; El Farran, C.A.; Gautam, P.; Chuah, Y.S.; Warrier, T.; Toh, C.X.D.; Kang, N.Y.; Sugii, S.; Chang, Y.T.; Xu, J.; et al. Diversification of reprogramming trajectories revealed by parallel single-cell transcriptome and chromatin accessibility sequencing. Sci. Adv. 2020, 6, eaba1190. [Google Scholar] [CrossRef]
- Schiebinger, G.; Shu, J.; Tabaka, M.; Cleary, B.; Subramanian, V.; Solomon, A.; Gould, J.; Liu, S.; Lin, S.; Berube, P.; et al. Optimal-Transport Analysis of Single-Cell Gene Expression Identifies Developmental Trajectories in Reprogramming. Cell 2019, 176, 928–943.e22. [Google Scholar] [CrossRef]
- Tran, K.A.; Pietrzak, S.J.; Zaidan, N.Z.; Siahpirani, A.F.; McCalla, S.G.; Zhou, A.S.; Iyer, G.; Roy, S.; Sridharan, R. Defining Reprogramming Checkpoints from Single-Cell Analyses of Induced Pluripotency. Cell Rep. 2019, 27, 1726–1741.e5. [Google Scholar] [CrossRef]
- Yu, J.; Thomson, J.A. Pluripotent stem cell lines. Genes Dev. 2008, 22, 1987–1997. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Amato, P.; Sparman, M.; Gutierrez, N.M.; Tippner-Hedges, R.; Ma, H.; Kang, E.; Fulati, A.; Lee, H.S.; Sritanaudomchai, H.; et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell 2013, 153, 1228–1238. [Google Scholar] [CrossRef]
- Stewart, M.H.; Bossé, M.; Chadwick, K.; Menendez, P.; Bendall, S.C.; Bhatia, M. Clonal isolation of hESCs reveals heterogeneity within the pluripotent stem cell compartment. Nat. Methods 2006, 3, 807–815. [Google Scholar] [CrossRef]
- de Souza, N. Taming stem cell heterogeneity. Nat. Methods 2012, 9, 645. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the safety of induced pluripotent stem cell lines. Nat. Biotechnol. 2009, 27, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Germain, N.D.; Hartman, N.W.; Cai, C.; Becker, S.; Naegele, J.R.; Grabel, L.B. Teratocarcinoma formation in embryonic stem cell-derived neural progenitor hippocampal transplants. Cell Transpl. 2012, 21, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 2011, 11, 268–277. [Google Scholar] [CrossRef]
- Ben-David, U.; Gan, Q.F.; Golan-Lev, T.; Arora, P.; Yanuka, O.; Oren, Y.S.; Leikin-Frenkel, A.; Graf, M.; Garippa, R.; Boehringer, M.; et al. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell 2013, 12, 167–179. [Google Scholar] [CrossRef]
- Zhao, M.-T.; Chen, H.; Liu, Q.; Shao, N.-Y.; Sayed, N.; Wo, H.-T.; Zhang, J.Z.; Ong, S.-G.; Liu, C.; Kim, Y.; et al. Molecular and functional resemblance of differentiated cells derived from isogenic human iPSCs and SCNT-derived ESCs. Proc. Natl. Acad. Sci. USA 2017, 114, E11111. [Google Scholar] [CrossRef]
- Guo, G.; von Meyenn, F.; Rostovskaya, M.; Clarke, J.; Dietmann, S.; Baker, D.; Sahakyan, A.; Myers, S.; Bertone, P.; Reik, W.; et al. Epigenetic resetting of human pluripotency. Development 2017, 144, 2748. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, K.; Yabuta, Y.; Ohinata, Y.; Saitou, M. Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis. Nat. Protoc. 2007, 2, 739–752. [Google Scholar] [CrossRef]
- Frumkin, D.; Wasserstrom, A.; Itzkovitz, S.; Harmelin, A.; Rechavi, G.; Shapiro, E. Amplification of multiple genomic loci from single cells isolated by laser micro-dissection of tissues. BMC Biotechnol. 2008, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef]
- Frieda, K.L.; Linton, J.M.; Hormoz, S.; Choi, J.; Chow, K.K.; Singer, Z.S.; Budde, M.W.; Elowitz, M.B.; Cai, L. Synthetic recording and in situ readout of lineage information in single cells. Nature 2017, 541, 107–111. [Google Scholar] [CrossRef]
- Svensson, V.; Vento-Tormo, R.; Teichmann, S.A. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 2018, 13, 599–604. [Google Scholar] [CrossRef]
- Yuan, J.; Sims, P.A. An Automated Microwell Platform for Large-Scale Single Cell RNA-Seq. Sci. Rep. 2016, 6, 33883. [Google Scholar] [CrossRef]
- Zhang, X.; Li, T.; Liu, F.; Chen, Y.; Yao, J.; Li, Z.; Huang, Y.; Wang, J. Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems. Mol. Cell 2019, 73, 130–142.e5. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Do, D.; Ramakrishnan, R. Single-Cell mRNA-Seq Using the Fluidigm C1 System and Integrated Fluidics Circuits. Methods Mol. Biol. 2018, 1783, 193–207. [Google Scholar]
- Der, E.; Suryawanshi, H.; Morozov, P.; Kustagi, M.; Goilav, B.; Ranabothu, S.; Izmirly, P.; Clancy, R.; Belmont, H.M.; Koenigsberg, M.; et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat. Immunol. 2019, 20, 915–927. [Google Scholar] [CrossRef]
- Nelson, L.; Tighe, A.; Golder, A.; Littler, S.; Bakker, B.; Moralli, D.; Murtuza Baker, S.; Donaldson, I.J.; Spierings, D.C.J.; Wardenaar, R.; et al. A living biobank of ovarian cancer ex vivo models reveals profound mitotic heterogeneity. Nat. Commun. 2020, 11, 822. [Google Scholar] [CrossRef]
- Mazutis, L.; Gilbert, J.; Ung, W.L.; Weitz, D.A.; Griffiths, A.D.; Heyman, J.A. Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 2013, 8, 870–891. [Google Scholar] [CrossRef]
- Matuła, K.; Rivello, F.; Huck, W.T.S. Single-Cell Analysis Using Droplet Microfluidics. Adv. Biosyst. 2020, 4, e1900188. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. TIG 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Tambe, A.; Pachter, L. Barcode identification for single cell genomics. BMC Bioinform. 2019, 20, 32. [Google Scholar] [CrossRef]
- Chen, W.; Li, Y.; Easton, J.; Finkelstein, D.; Wu, G.; Chen, X. UMI-count modeling and differential expression analysis for single-cell RNA sequencing. Genome Biol. 2018, 19, 70. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ilicic, T.; Kim, J.K.; Kolodziejczyk, A.A.; Bagger, F.O.; McCarthy, D.J.; Marioni, J.C.; Teichmann, S.A. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 2016, 17, 29. [Google Scholar] [CrossRef]
- Patrick, R.; Humphreys, D.T.; Janbandhu, V.; Oshlack, A.; Ho, J.W.K.; Harvey, R.P.; Lo, K.K. Sierra: Discovery of differential transcript usage from polyA-captured single-cell RNA-seq data. Genome Biol. 2020, 21, 167. [Google Scholar] [CrossRef]
- Cole, C.; Byrne, A.; Beaudin, A.E.; Forsberg, E.C.; Vollmers, C. Tn5Prime, a Tn5 based 5′ capture method for single cell RNA-seq. Nucleic Acids Res. 2018, 46, e62. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zheng, L.; Zhu, Y.; Li, N.; Jia, H.; Ai, R.; Wildberg, A.; Wang, W. Normalization and noise reduction for single cell RNA-seq experiments. Bioinformatics 2015, 31, 2225–2227. [Google Scholar] [CrossRef] [PubMed]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef]
- Cakir, B.; Prete, M.; Huang, N.; van Dongen, S.; Pir, P.; Kiselev, V.Y. Comparison of visualization tools for single-cell RNAseq data. NAR Genom. Bioinform. 2020, 2, lqaa052. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.N.; Ang, K.S.; Chevrier, M.; Zhang, X.; Lee, N.Y.S.; Goh, M.; Chen, J. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020, 21, 12. [Google Scholar] [CrossRef]
- Tan, Y.; Cahan, P. SingleCellNet: A Computational Tool to Classify Single Cell RNA-Seq Data across Platforms and Across Species. Cell Syst. 2019, 9, 207–213.e2. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Levenberg, S.; Golub, J.S.; Amit, M.; Itskovitz-Eldor, J.; Langer, R. Endothelial cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 4391–4396. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.J.; Behfar, A.; Yamada, S.; Martinez-Fernandez, A.; Terzic, A. Stem cell platforms for regenerative medicine. Clin. Transl. Sci. 2009, 2, 222–227. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Ramsköld, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Huang, K.; Cai, C.; Cai, L.; Jiang, C.Y.; Feng, Y.; Liu, Z.; Zeng, Q.; Cheng, L.; Sun, Y.E.; et al. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature 2013, 500, 593–597. [Google Scholar] [CrossRef]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Lecault, V.; White, A.K.; Singhal, A.; Hansen, C.L. Microfluidic single cell analysis: From promise to practice. Curr. Opin. Chem. Biol. 2012, 16, 381–390. [Google Scholar] [CrossRef]
- Wu, A.R.; Neff, N.F.; Kalisky, T.; Dalerba, P.; Treutlein, B.; Rothenberg, M.E.; Mburu, F.M.; Mantalas, G.L.; Sim, S.; Clarke, M.F.; et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 2014, 11, 41–46. [Google Scholar] [CrossRef]
- Messmer, T.; von Meyenn, F.; Savino, A.; Santos, F.; Mohammed, H.; Lun, A.T.L.; Marioni, J.C.; Reik, W. Transcriptional Heterogeneity in Naive and Primed Human Pluripotent Stem Cells at Single-Cell Resolution. Cell Rep. 2019, 26, 815–824.e4. [Google Scholar] [CrossRef]
- Boroviak, T.; Nichols, J. Primate embryogenesis predicts the hallmarks of human naïve pluripotency. Development 2017, 144, 175–186. [Google Scholar] [CrossRef]
- Ehnes, D.D.; Hussein, A.M.; Ware, C.B.; Mathieu, J.; Ruohola-Baker, H. Combinatorial metabolism drives the naive to primed pluripotent chromatin landscape. Exp. Cell Res. 2020, 389, 111913. [Google Scholar] [CrossRef]
- Smith, A. Formative pluripotency: The executive phase in a developmental continuum. Development 2017, 144, 365–373. [Google Scholar] [CrossRef]
- Nguyen, Q.H.; Lukowski, S.W.; Chiu, H.S.; Senabouth, A.; Bruxner, T.J.C.; Christ, A.N.; Palpant, N.J.; Powell, J.E. Single-cell RNA-seq of human induced pluripotent stem cells reveals cellular heterogeneity and cell state transitions between subpopulations. Genome Res. 2018, 28, 1053–1066. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Buganim, Y.; Faddah, D.A.; Jaenisch, R. Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 2013, 14, 427–439. [Google Scholar] [CrossRef]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef]
- O’Malley, J.; Skylaki, S.; Iwabuchi, K.A.; Chantzoura, E.; Ruetz, T.; Johnsson, A.; Tomlinson, S.R.; Linnarsson, S.; Kaji, K. High-resolution analysis with novel cell-surface markers identifies routes to iPS cells. Nature 2013, 499, 88–91. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Trapnell, C.; Ziller, M.J.; Soumillon, M.; Cesana, M.; Karnik, R.; Donaghey, J.; Smith, Z.D.; Ratanasirintrawoot, S.; Zhang, X.; et al. Integrative Analyses of Human Reprogramming Reveal Dynamic Nature of Induced Pluripotency. Cell 2015, 162, 412–424. [Google Scholar] [CrossRef]
- Tanaka, Y.; Hysolli, E.; Su, J.; Xiang, Y.; Kim, K.-Y.; Zhong, M.; Li, Y.; Heydari, K.; Euskirchen, G.; Snyder, M.P.; et al. Transcriptome Signature and Regulation in Human Somatic Cell Reprogramming. Stem Cell Rep. 2015, 4, 1125–1139. [Google Scholar] [CrossRef] [PubMed]
- Parenti, A.; Halbisen, M.A.; Wang, K.; Latham, K.; Ralston, A. OSKM Induce Extraembryonic Endoderm Stem Cells in Parallel to Induced Pluripotent Stem Cells. Stem Cell Rep. 2016, 6, 447–455. [Google Scholar] [CrossRef]
- Chronis, C.; Fiziev, P.; Papp, B.; Butz, S.; Bonora, G.; Sabri, S.; Ernst, J.; Plath, K. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell 2017, 168, 442–459.e20. [Google Scholar] [CrossRef]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Plath, K.; Lowry, W.E. Progress in understanding reprogramming to the induced pluripotent state. Nat. Rev. Genet. 2011, 12, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Buganim, Y.; Faddah, D.A.; Cheng, A.W.; Itskovich, E.; Markoulaki, S.; Ganz, K.; Klemm, S.L.; van Oudenaarden, A.; Jaenisch, R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 2012, 150, 1209–1222. [Google Scholar] [CrossRef]
- Guo, L.; Lin, L.; Wang, X.; Gao, M.; Cao, S.; Mai, Y.; Wu, F.; Kuang, J.; Liu, H.; Yang, J.; et al. Resolving Cell Fate Decisions during Somatic Cell Reprogramming by Single-Cell RNA-Seq. Mol. Cell 2019, 73, 815–829.e7. [Google Scholar] [CrossRef]
- Lee, J.; Sayed, N.; Hunter, A.; Au, K.F.; Wong, W.H.; Mocarski, E.S.; Pera, R.R.; Yakubov, E.; Cooke, J.P. Activation of innate immunity is required for efficient nuclear reprogramming. Cell 2012, 151, 547–558. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef]
- Liang, Y. Revealing cell fate decisions during reprogramming by scRNA-seq. In Proceedings of the E3S Web of Conferences, Wuhan, China, 25–27 November 2020; Volume 145, p. 1033. [Google Scholar]
- Hanna, J.; Saha, K.; Pando, B.; van Zon, J.; Lengner, C.J.; Creyghton, M.P.; van Oudenaarden, A.; Jaenisch, R. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 2009, 462, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.; Bickmore, W. Nuclear organization of the genome and the potential for gene regulation. Nature 2007, 447, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.A.; Groves, A.K. Transcription Factor Reprogramming in the Inner Ear: Turning on Cell Fate Switches to Regenerate Sensory Hair Cells. Front. Cell. Neurosci. 2021, 15, 660748. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.K.; Ha, T.W.; Lee, M.R. Single-Cell Transcriptome Analysis as a Promising Tool to Study Pluripotent Stem Cell Reprogramming. Int. J. Mol. Sci. 2021, 22, 5988. https://doi.org/10.3390/ijms22115988
Kim HK, Ha TW, Lee MR. Single-Cell Transcriptome Analysis as a Promising Tool to Study Pluripotent Stem Cell Reprogramming. International Journal of Molecular Sciences. 2021; 22(11):5988. https://doi.org/10.3390/ijms22115988
Chicago/Turabian StyleKim, Hyun Kyu, Tae Won Ha, and Man Ryul Lee. 2021. "Single-Cell Transcriptome Analysis as a Promising Tool to Study Pluripotent Stem Cell Reprogramming" International Journal of Molecular Sciences 22, no. 11: 5988. https://doi.org/10.3390/ijms22115988
APA StyleKim, H. K., Ha, T. W., & Lee, M. R. (2021). Single-Cell Transcriptome Analysis as a Promising Tool to Study Pluripotent Stem Cell Reprogramming. International Journal of Molecular Sciences, 22(11), 5988. https://doi.org/10.3390/ijms22115988