
The Involvement of Ubiquitination Machinery in Cell Cycle Regulation and Cancer Progression


Abstract
1. Introduction
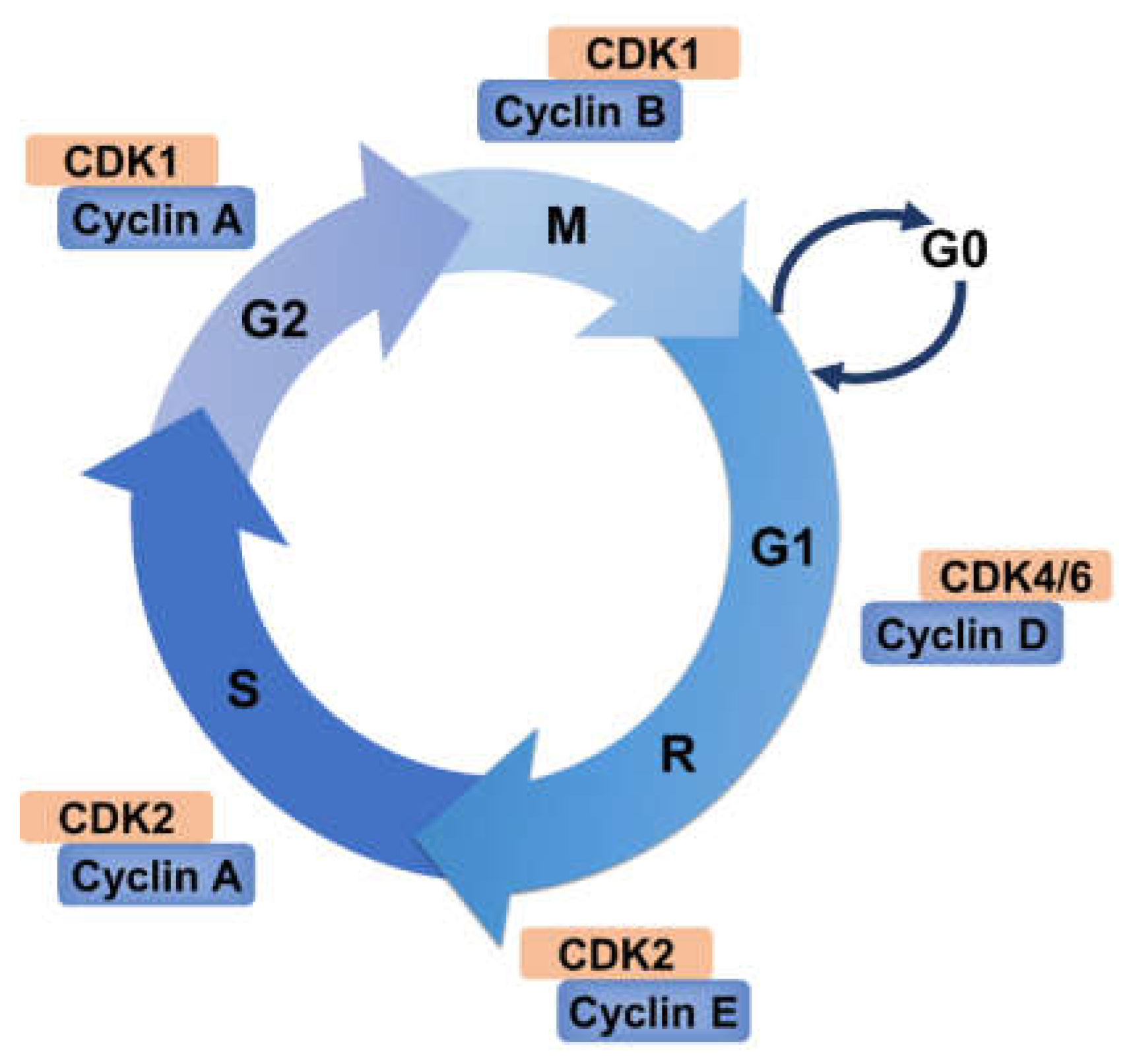
2. Cell Cycle and Its Regulation
2.1. Cyclin–CDK Complexes Regulation
2.2. CKIs Regulation
2.3. The Restriction Point and Checkpoints
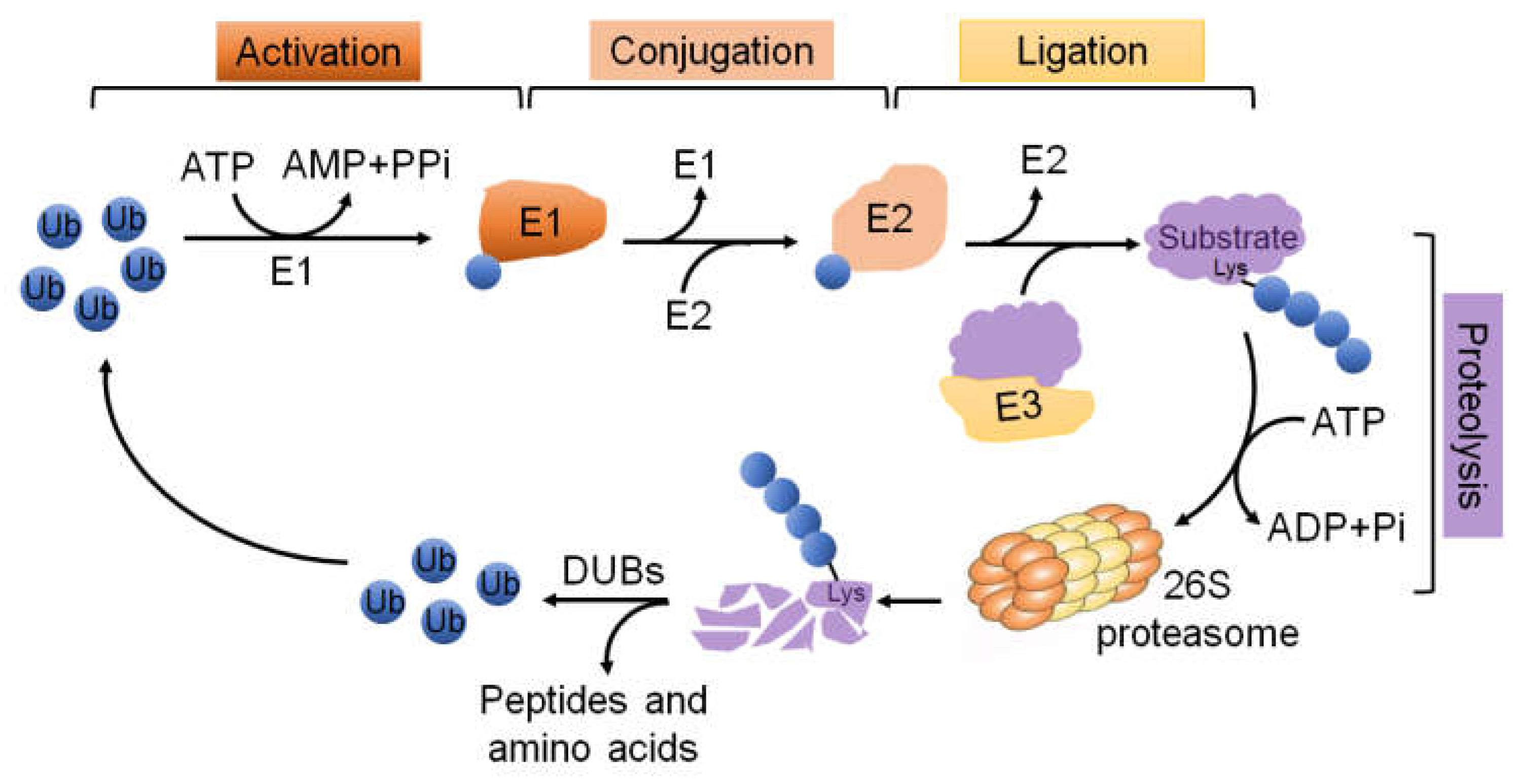
3. The UPS Molecular Machinery
4. The Functional Roles of UPS in Cyclins
4.1. Cyclin A
4.2. Cyclin B
4.3. Cyclin D
4.4. Cyclin E
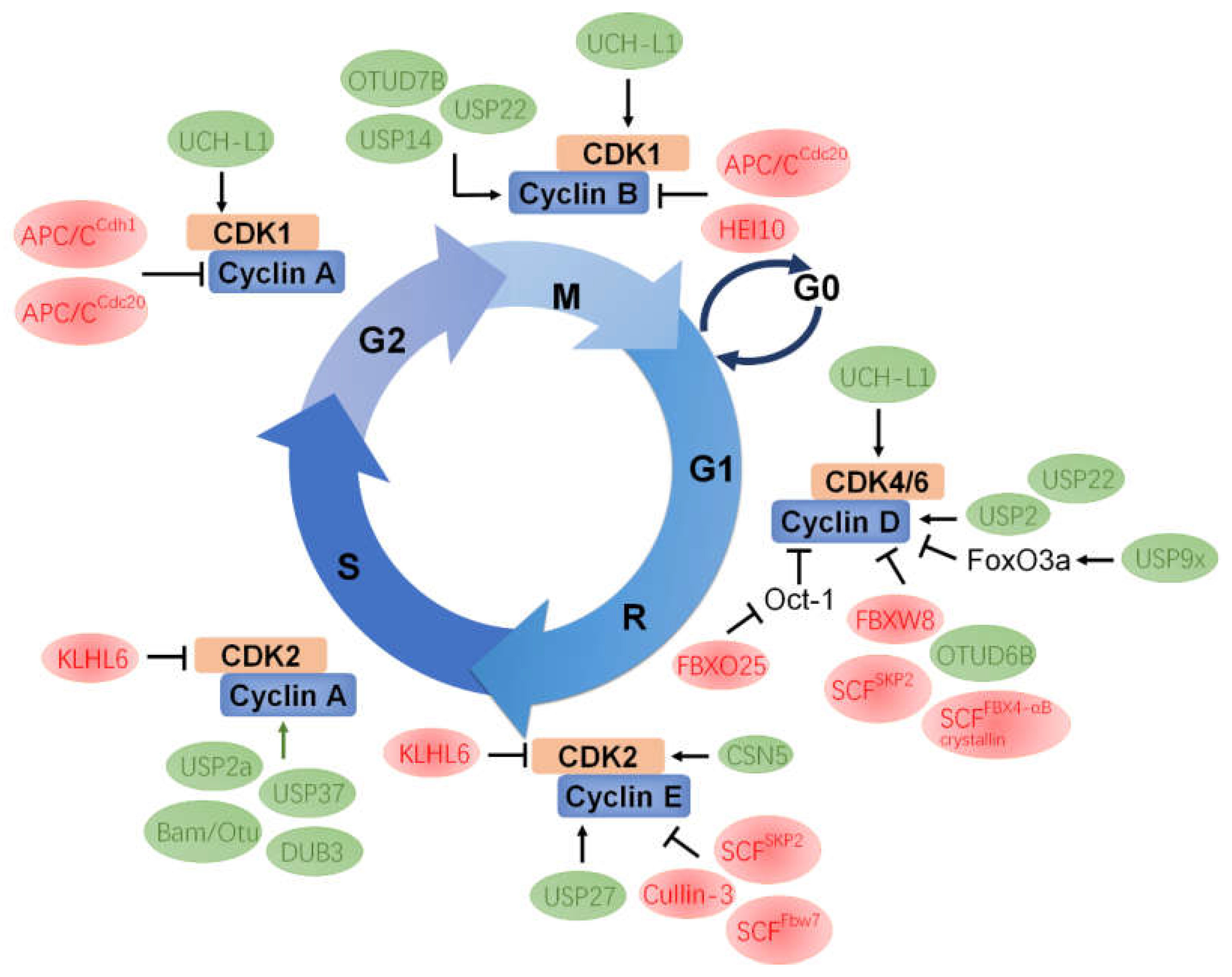
5. Mutual Regulation between CDK and UPS Components
5.1. CDK1
5.2. CDK2
5.3. CDK4/6
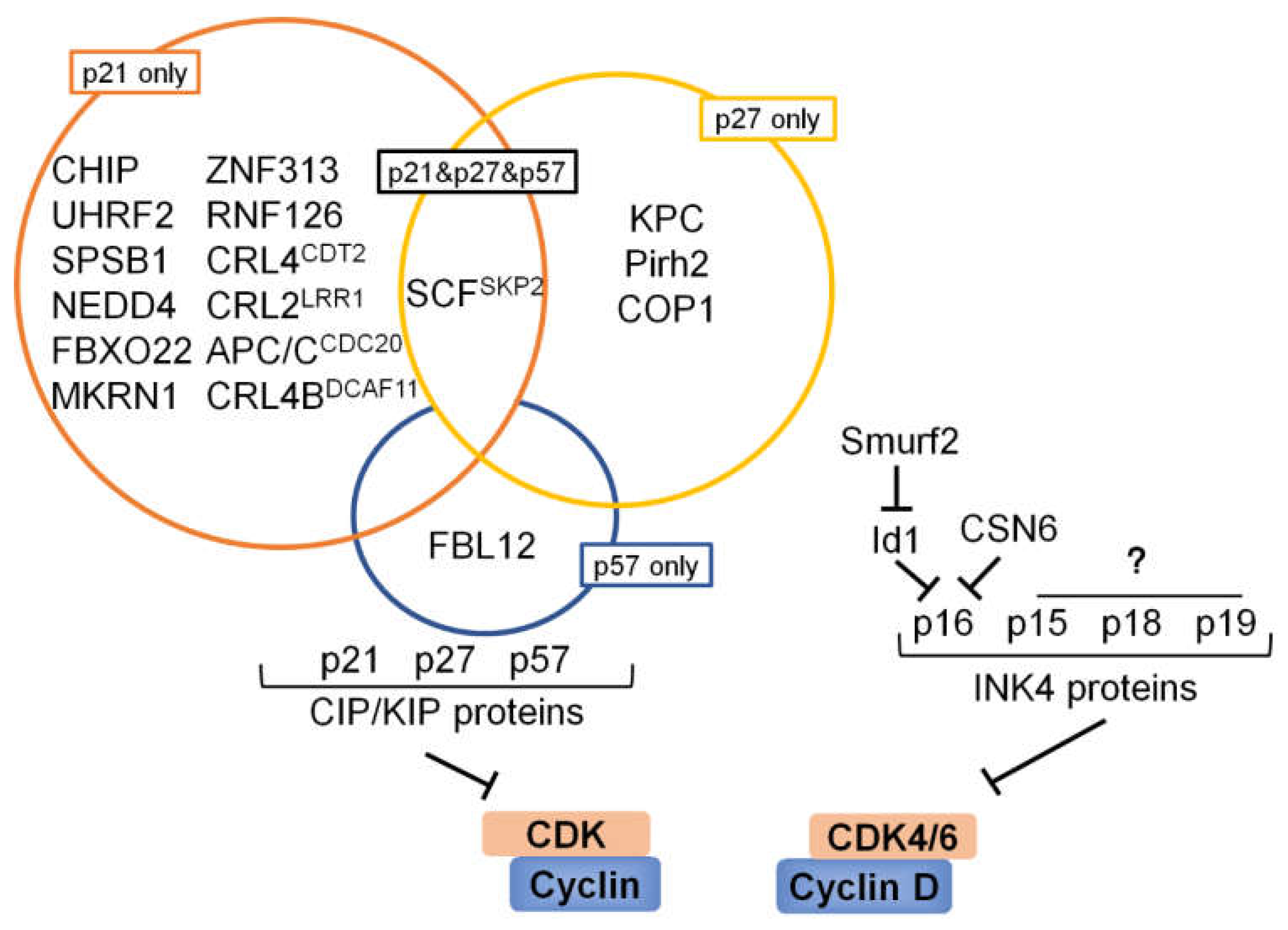
6. Ubiquitination and Deubiquitination Involved in CKIs
6.1. p53 and Its Downstream Target p21
6.2. p27
6.3. p57
6.4. INK4 Proteins
7. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Schafer, K.A. The cell cycle: A review. Vet. Pathol. 1998, 35, 461–478. [Google Scholar] [CrossRef]
- Poon, R.Y. Cell Cycle Control: A System of Interlinking Oscillators. Methods Mol. Biol. 2016, 1342, 3–19. [Google Scholar]
- Teixeira, L.K.; Reed, S.I. Ubiquitin ligases and cell cycle control. Annu. Rev. Biochem. 2013, 82, 387–414. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef]
- Schachter, M.M.; Fisher, R.P. The CDK-activating kinase Cdk7: Taking yes for an answer. Cell Cycle 2013, 12, 3239–3240. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, S.; Merrick, K.A.; Terret, M.E.; Wohlbold, L.; Barboza, N.M.; Zhang, C.; Shokat, K.M.; Jallepalli, P.V.; Fisher, R.P. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol. Cell 2007, 25, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M.M.; Merrick, K.A.; Larochelle, S.; Hirschi, A.; Zhang, C.; Shokat, K.M.; Rubin, S.M.; Fisher, R.P. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol. Cell 2013, 50, 250–260. [Google Scholar] [CrossRef]
- Bisteau, X.; Paternot, S.; Colleoni, B.; Ecker, K.; Coulonval, K.; De Groote, P.; Declercq, W.; Hengst, L.; Roger, P.P. CDK4 T172 phosphorylation is central in a CDK7-dependent bidirectional CDK4/CDK2 interplay mediated by p21 phosphorylation at the restriction point. PLoS Genet. 2013, 9, e1003546. [Google Scholar] [CrossRef] [PubMed]
- Van Vugt, M.A.; Bras, A.; Medema, R.H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 2004, 15, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Macurek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008, 455, 119–123. [Google Scholar] [CrossRef] [PubMed]
- McGowan, C.H.; Russell, P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993, 12, 75–85. [Google Scholar] [CrossRef]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.; Hardwick, K.; Javerzat, J.P. Fission yeast bub1 is a mitotic centromere protein essential for the spindle checkpoint and the preservation of correct ploidy through mitosis. J. Cell Biol. 1998, 143, 1775–1787. [Google Scholar] [CrossRef] [PubMed]
- Palframan, W.J.; Meehl, J.B.; Jaspersen, S.L.; Winey, M.; Murray, A.W. Anaphase inactivation of the spindle checkpoint. Science 2006, 313, 680–684. [Google Scholar] [CrossRef]
- Fry, A.M.; O’Regan, L.; Sabir, S.R.; Bayliss, R. Cell cycle regulation by the NEK family of protein kinases. J. Cell Sci. 2012, 125, 4423–4433. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Falck, J.; Lukas, J. CHK2 kinase--a busy messenger. Nat. Rev. Mol. Cell Biol. 2001, 2, 877–886. [Google Scholar] [CrossRef] [PubMed]
- McGowan, C.H. Checking in on Cds1 (Chk2): A checkpoint kinase and tumor suppressor. Bioessays 2002, 24, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar]
- Fasanaro, P.; Capogrossi, M.C.; Martelli, F. Regulation of the endothelial cell cycle by the ubiquitin-proteasome system. Cardiovasc Res. 2010, 85, 272–280. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Kouranti, I.; Peyroche, A. Protein degradation in DNA damage response. Semin. Cell Dev. Biol. 2012, 23, 538–545. [Google Scholar] [CrossRef]
- Coffman, J.A. Cell cycle development. Dev. Cell 2004, 6, 321–327. [Google Scholar] [CrossRef]
- Bai, J.; Li, Y.; Zhang, G. Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med. 2017, 14, 348–362. [Google Scholar]
- Norbury, C.; Nurse, P. Animal-Cell Cycles and Their Control. Annu. Rev. Biochem. 1992, 61, 441–470. [Google Scholar] [CrossRef] [PubMed]
- Figel, S.; Fenstermaker, R.A. Chapter 18 Cell-Cycle Regulation. In Handbook of Brain Tumor Chemotherapy, Molecular Therapeutics, and Immunotherapy, 2nd ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 257–269. [Google Scholar]
- Massague, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef]
- Chotiner, J.Y.; Wolgemuth, D.J.; Wang, P.J. Functions of cyclins and CDKs in mammalian gametogenesisdagger. Biol. Reprod. 2019, 101, 591–601. [Google Scholar] [CrossRef]
- Pines, J. Cyclins and Their Associated Cyclin-Dependent Kinases in the Human Cell-Cycle. Biochem. Soc. T 1993, 21, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.; Rosenthal, E.T.; Youngblom, J.; Distel, D.; Hunt, T. Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 1983, 33, 389–396. [Google Scholar] [CrossRef]
- Cao, L.; Chen, F.; Yang, X.; Xu, W.; Xie, J.; Yu, L. Phylogenetic analysis of CDK and cyclin proteins in premetazoan lineages. BMC Evol. Biol. 2014, 14, 10. [Google Scholar] [CrossRef]
- Nakamura, T.; Sanokawa, R.; Sasaki, Y.F.; Ayusawa, D.; Oishi, M.; Mori, N. Cyclin I: A new cyclin encoded by a gene isolated from human brain. Exp. Cell Res. 1995, 221, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, M.; Stacey, D.W. Cyclin D1 production in cycling cells depends on ras in a cell-cycle-specific manner. Curr. Biol. 1999, 9, 1075–1084. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Nabel, E.G. CDKs and CKIs: Molecular targets for tissue remodelling. Nat. Rev. Drug Discov. 2002, 1, 587–598. [Google Scholar] [CrossRef]
- Lundberg, A.S.; Weinberg, R.A. Control of the cell cycle and apoptosis. Eur. J. Cancer 1999, 35, 531–539. [Google Scholar] [CrossRef]
- Pardee, A.B. A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. USA 1974, 71, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, A.; Larsson, O.; Wiman, K.G. What is the restriction point? Curr. Opin. Cell Biol. 1995, 7, 835–842. [Google Scholar] [CrossRef]
- Elledge, S.J. Cell cycle checkpoints: Preventing an identity crisis. Science 1996, 274, 1664–1672. [Google Scholar] [CrossRef]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [PubMed]
- Bharadwaj, R.; Yu, H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004, 23, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Le, W.D. Autophagy and Ubiquitin-Proteasome System. Adv. Exp. Med. Biol. 2019, 1206, 527–550. [Google Scholar]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 2004, 1695, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ma, L.; Wang, B.; Liu, J.; Wei, W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 2017, 36, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Bulatov, E.; Valiullina, A.; Sayarova, R.; Rizvanov, A. Promising new therapeutic targets for regulation of inflammation and immunity: RING-type E3 ubiquitin ligases. Immunol. Lett. 2018, 202, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Fournane, S.; Krupina, K.; Kleiss, C.; Sumara, I. Decoding ubiquitin for mitosis. Genes Cancer 2012, 3, 697–711. [Google Scholar] [CrossRef]
- Schrock, M.S.; Stromberg, B.R.; Scarberry, L.; Summers, M.K. APC/C ubiquitin ligase: Functions and mechanisms in tumorigenesis. Semin. Cancer Biol. 2020, 67, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Kramer, E.R.; Scheuringer, N.; Podtelejnikov, A.V.; Mann, M.; Peters, J.M. Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell 2000, 11, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Pesin, J.A.; Orr-Weaver, T.L. Regulation of APC/C activators in mitosis and meiosis. Annu. Rev. Cell Dev. Biol. 2008, 24, 475–499. [Google Scholar] [CrossRef] [PubMed]
- Vodermaier, H.C. APC/C and SCF: Controlling each other and the cell cycle. Curr. Biol. 2004, 14, R787–R796. [Google Scholar] [CrossRef]
- Kraft, C.; Herzog, F.; Gieffers, C.; Mechtler, K.; Hagting, A.; Pines, J.; Peters, J.M. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 2003, 22, 6598–6609. [Google Scholar] [CrossRef] [PubMed]
- Fujimitsu, K.; Grimaldi, M.; Yamano, H. Cyclin-dependent kinase 1-dependent activation of APC/C ubiquitin ligase. Science 2016, 352, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Tong, L. Structural biology of the separase-securin complex with crucial roles in chromosome segregation. Curr. Opin. Struct. Biol. 2018, 49, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Zachariae, W.; Nasmyth, K. Whose end is destruction: Cell division and the anaphase-promoting complex. Genes Dev. 1999, 13, 2039–2058. [Google Scholar] [CrossRef] [PubMed]
- Zachariae, W.; Schwab, M.; Nasmyth, K.; Seufert, W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science 1998, 282, 1721–1724. [Google Scholar] [CrossRef] [PubMed]
- Bashir, T.; Dorrello, N.V.; Amador, V.; Guardavaccaro, D.; Pagano, M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 2004, 428, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ayad, N.G.; Wan, Y.; Zhang, G.J.; Kirschner, M.W.; Kaelin, W.G., Jr. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 2004, 428, 194–198. [Google Scholar] [CrossRef]
- Deshaies, R.J. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu. Rev. Cell Dev. Biol. 1999, 15, 435–467. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Cardozo, T.; Lovering, R.C.; Elledge, S.J.; Pagano, M.; Harper, J.W. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004, 18, 2573–2580. [Google Scholar] [CrossRef] [PubMed]
- Okabe, H.; Lee, S.H.; Phuchareon, J.; Albertson, D.G.; McCormick, F.; Tetsu, O. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS ONE 2006, 1, e128. [Google Scholar] [CrossRef] [PubMed]
- Kuzmanov, A.; Johansen, P.; Hofbauer, G. FBXO25 Promotes Cutaneous Squamous Cell Carcinoma Growth and Metastasis through Cyclin D1. J. Investig. Dermatol. 2020, 140, 2496–2504. [Google Scholar] [CrossRef]
- Koepp, D.M.; Schaefer, L.K.; Ye, X.; Keyomarsi, K.; Chu, C.; Harper, J.W.; Elledge, S.J. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001, 294, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.K.; Gervais, J.L.; Zhang, H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 11324–11329. [Google Scholar] [CrossRef]
- Bornstein, G.; Bloom, J.; Sitry-Shevah, D.; Nakayama, K.; Pagano, M.; Hershko, A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J. Biol. Chem. 2003, 278, 25752–25757. [Google Scholar] [CrossRef]
- Wang, W.; Nacusi, L.; Sheaff, R.J.; Liu, X. Ubiquitination of p21Cip1/WAF1 by SCFSkp2: Substrate requirement and ubiquitination site selection. Biochemistry 2005, 44, 14553–14564. [Google Scholar] [CrossRef]
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999, 1, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Reimann, J.D.; Freed, E.; Hsu, J.Y.; Kramer, E.R.; Peters, J.M.; Jackson, P.K. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell 2001, 105, 645–655. [Google Scholar] [CrossRef]
- Margottin-Goguet, F.; Hsu, J.Y.; Loktev, A.; Hsieh, H.M.; Reimann, J.D.; Jackson, P.K. Prophase destruction of Emi1 by the SCF(betaTrCP/Slimb) ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev. Cell 2003, 4, 813–826. [Google Scholar] [CrossRef]
- Pagano, M.; Pepperkok, R.; Verde, F.; Ansorge, W.; Draetta, G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992, 11, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Geley, S.; Kramer, E.; Gieffers, C.; Gannon, J.; Peters, J.M.; Hunt, T. Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol. 2001, 153, 137–148. [Google Scholar] [CrossRef]
- Minshull, J.; Golsteyn, R.; Hill, C.S.; Hunt, T. The A- and B-type cyclin associated cdc2 kinases in Xenopus turn on and off at different times in the cell cycle. EMBO J. 1990, 9, 2865–2875. [Google Scholar] [CrossRef]
- Howe, J.A.; Howell, M.; Hunt, T.; Newport, J.W. Identification of a developmental timer regulating the stability of embryonic cyclin A and a new somatic A-type cyclin at gastrulation. Genes Dev. 1995, 9, 1164–1176. [Google Scholar] [CrossRef]
- Sweeney, C.; Murphy, M.; Kubelka, M.; Ravnik, S.E.; Hawkins, C.F.; Wolgemuth, D.J.; Carrington, M. A distinct cyclin A is expressed in germ cells in the mouse. Development 1996, 122, 53–64. [Google Scholar] [CrossRef]
- Yang, R.; Morosetti, R.; Koeffler, H.P. Characterization of a second human cyclin A that is highly expressed in testis and in several leukemic cell lines. Cancer Res. 1997, 57, 913–920. [Google Scholar] [PubMed]
- Huang, X.; Summers, M.K.; Pham, V.; Lill, J.R.; Liu, J.; Lee, G.; Kirkpatrick, D.S.; Jackson, P.K.; Fang, G.; Dixit, V.M. Deubiquitinase USP37 is activated by CDK2 to antagonize APC(CDH1) and promote S phase entry. Mol. Cell 2011, 42, 511–523. [Google Scholar] [CrossRef]
- Kim, J.; Kim, W.J.; Liu, Z.; Loda, M.; Freeman, M.R. The ubiquitin-specific protease USP2a enhances tumor progression by targeting cyclin A1 in bladder cancer. Cell Cycle 2012, 11, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Deng, T.; Ma, H.; Liu, Y.; Feng, P.; Wei, D.; Ling, N.; Li, L.; Qiu, S.; Zhang, L.; et al. Deubiquitinase DUB3 Regulates Cell Cycle Progression via Stabilizing Cyclin A for Proliferation of Non-Small Cell Lung Cancer Cells. Cells 2019, 8, 297. [Google Scholar] [CrossRef]
- Wang, Z.; Lin, H. The division of Drosophila germline stem cells and their precursors requires a specific cyclin. Curr. Biol. 2005, 15, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Lilly, M.A.; de Cuevas, M.; Spradling, A.C. Cyclin A associates with the fusome during germline cyst formation in the Drosophila ovary. Dev. Biol. 2000, 218, 53–63. [Google Scholar] [CrossRef]
- Chen, D.; Wang, Q.; Huang, H.; Xia, L.; Jiang, X.; Kan, L.; Sun, Q.; Chen, D. Effete-mediated degradation of Cyclin A is essential for the maintenance of germline stem cells in Drosophila. Development 2009, 136, 4133–4142. [Google Scholar] [CrossRef] [PubMed]
- Ables, E.T.; Drummond-Barbosa, D. Cyclin E controls Drosophila female germline stem cell maintenance independently of its role in proliferation by modulating responsiveness to niche signals. Development 2013, 140, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Li, C.; Hu, L.; Liu, K.; Mei, J.; Luo, Y.; Tao, Y.; Xia, Z.; Sun, Q.; Chen, D. Bam-dependent deubiquitinase complex can disrupt germ-line stem cell maintenance by targeting cyclin A. Proc. Natl. Acad. Sci. USA 2017, 114, 6316–6321. [Google Scholar] [CrossRef] [PubMed]
- Irniger, S.; Nasmyth, K. The anaphase-promoting complex is required in G1 arrested yeast cells to inhibit B-type cyclin accumulation and to prevent uncontrolled entry into S-phase. J. Cell Sci. 1997, 110, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Toby, G.G.; Gherraby, W.; Coleman, T.R.; Golemis, E.A. A novel RING finger protein, human enhancer of invasion 10, alters mitotic progression through regulation of cyclin B levels. Mol. Cell Biol. 2003, 23, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Brockman, J.L.; Schuler, L.A. Prolactin signals via Stat5 and Oct-1 to the proximal cyclin D1 promoter. Mol. Cell Endocrinol. 2005, 239, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.I.; Barbash, O.; Kumar, K.G.; Weber, J.D.; Harper, J.W.; Klein-Szanto, A.J.; Rustgi, A.; Fuchs, S.Y.; Diehl, J.A. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol. Cell 2006, 24, 355–366. [Google Scholar] [CrossRef]
- Singer, J.D.; Gurian-West, M.; Clurman, B.; Roberts, J.M. Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev. 1999, 13, 2375–2387. [Google Scholar] [CrossRef]
- Yeh, K.H.; Kondo, T.; Zheng, J.; Tsvetkov, L.M.; Blair, J.; Zhang, H. The F-box protein SKP2 binds to the phosphorylated threonine 380 in cyclin E and regulates ubiquitin-dependent degradation of cyclin E. Biochem. Biophys. Res. Commun. 2001, 281, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, Y.; Wang, Y.; Xie, C.; Gan, M.; Han, T.; Cao, J.; Wang, J. CyclinB1 deubiquitination by USP14 regulates cell cycle progression in breast cancer. Pathol. Res. Pract. 2019, 215, 152592. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Tan, C.; Qiu, Q.; Kong, S.; Yang, H.; Zhao, F.; Liu, Z.; Li, J.; Kong, Q.; Gao, B.; et al. Ubiquitin-specific protease 22 is a deubiquitinase of CCNB1. Cell Discov. 2015, 1, 15028. [Google Scholar] [CrossRef] [PubMed]
- Bonacci, T.; Suzuki, A.; Grant, G.D.; Stanley, N.; Cook, J.G.; Brown, N.G.; Emanuele, M.J. Cezanne/OTUD7B is a cell cycle-regulated deubiquitinase that antagonizes the degradation of APC/C substrates. EMBO J. 2018, 37, e98701. [Google Scholar] [CrossRef]
- Shan, J.; Zhao, W.; Gu, W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol. Cell 2009, 36, 469–476. [Google Scholar] [CrossRef]
- Zheng, X.; Zhai, B.; Koivunen, P.; Shin, S.J.; Lu, G.; Liu, J.; Geisen, C.; Chakraborty, A.A.; Moslehi, J.J.; Smalley, D.M.; et al. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev. 2014, 28, 1429–1444. [Google Scholar] [CrossRef]
- Schmidt, M.; Fernandez de Mattos, S.; van der Horst, A.; Klompmaker, R.; Kops, G.J.; Lam, E.W.; Burgering, B.M.; Medema, R.H. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zheng, Y.; Zhu, Y.; Kong, X.; Hu, L. Evidence for OTUD-6B participation in B lymphocytes cell cycle after cytokine stimulation. PLoS ONE 2011, 6, e14514. [Google Scholar] [CrossRef]
- Gennaro, V.J.; Stanek, T.J.; Peck, A.R.; Sun, Y.; Wang, F.; Qie, S.; Knudsen, K.E.; Rui, H.; Butt, T.; Diehl, J.A.; et al. Control of CCND1 ubiquitylation by the catalytic SAGA subunit USP22 is essential for cell cycle progression through G1 in cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E9298–E9307. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Yu, L.; Bai, C.; Liu, L.; Long, H.; Shi, L.; Lin, Z. USP27-mediated Cyclin E stabilization drives cell cycle progression and hepatocellular tumorigenesis. Oncogene 2018, 37, 2702–2713. [Google Scholar] [CrossRef]
- Petri, E.T.; Errico, A.; Escobedo, L.; Hunt, T.; Basavappa, R. The crystal structure of human cyclin B. Cell Cycle 2007, 6, 1342–1349. [Google Scholar] [CrossRef] [PubMed]
- Lara-Gonzalez, P.; Moyle, M.W.; Budrewicz, J.; Mendoza-Lopez, J.; Oegema, K.; Desai, A. The G2-to-M Transition Is Ensured by a Dual Mechanism that Protects Cyclin B from Degradation by Cdc20-Activated APC/C. Dev. Cell 2019, 51, 313–325.e10. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Lee, M.J.; Park, S.; Oh, D.C.; Elsasser, S.; Chen, P.C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef]
- Chen, P.C.; Bhattacharyya, B.J.; Hanna, J.; Minkel, H.; Wilson, J.A.; Finley, D.; Miller, R.J.; Wilson, S.M. Ubiquitin homeostasis is critical for synaptic development and function. J. Neurosci. 2011, 31, 17505–17513. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Lu, Y.; Prado, M.A.; Shi, Y.; Tian, G.; Sun, S.; Elsasser, S.; Gygi, S.P.; King, R.W.; Finley, D. USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 2016, 532, 398–401. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Shan, J.; Gu, W. Targeting the degradation of cyclin D1 will help to eliminate oncogene addiction. Cell Cycle 2010, 9, 857–858. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, Q.; Geng, Y.; Sicinski, P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001, 411, 1017–1021. [Google Scholar] [CrossRef]
- Magiera, K.; Tomala, M.; Kubica, K.; De Cesare, V.; Trost, M.; Zieba, B.J.; Kachamakova-Trojanowska, N.; Les, M.; Dubin, G.; Holak, T.A.; et al. Lithocholic Acid Hydroxyamide Destabilizes Cyclin D1 and Induces G0/G1 Arrest by Inhibiting Deubiquitinase USP2a. Cell Chem. Biol. 2017, 24, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.; Leach, C.A.; Goldenberg, S.J.; Francis, D.M.; Kodrasov, M.P.; Tian, X.; Shanks, J.; Sterner, D.E.; Bernal, A.; Mattern, M.R.; et al. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein. Sci. 2008, 17, 1035–1043. [Google Scholar] [CrossRef]
- Issaenko, O.A.; Amerik, A.Y. Chalcone-based small-molecule inhibitors attenuate malignant phenotype via targeting deubiquitinating enzymes. Cell Cycle 2012, 11, 1804–1817. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Pragani, R.; Fox, J.T.; Shen, M.; Parmar, K.; Gaudiano, E.F.; Liu, L.; Tanega, C.; McGee, L.; Hall, M.D.; et al. Small Molecule Inhibition of the Ubiquitin-specific Protease USP2 Accelerates cyclin D1 Degradation and Leads to Cell Cycle Arrest in Colorectal Cancer and Mantle Cell Lymphoma Models. J. Biol. Chem. 2016, 291, 24628–24640. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, M.; Roberts, J.M. Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science 1993, 259, 1908–1912. [Google Scholar] [CrossRef] [PubMed]
- Spruck, C.H.; Won, K.A.; Reed, S.I. Deregulated cyclin E induces chromosome instability. Nature 1999, 401, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Keyomarsi, K.; Conte, D., Jr.; Toyofuku, W.; Fox, M.P. Deregulation of cyclin E in breast cancer. Oncogene 1995, 11, 941–950. [Google Scholar]
- Davidge, B.; Rebola, K.G.O.; Agbor, L.N.; Sigmund, C.D.; Singer, J.D. Cul3 regulates cyclin E1 protein abundance via a degron located within the N-terminal region of cyclin E. J. Cell Sci. 2019, 132, jcs233049. [Google Scholar] [CrossRef]
- Clurman, B.E.; Sheaff, R.J.; Thress, K.; Groudine, M.; Roberts, J.M. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 1996, 10, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Won, K.A.; Reed, S.I. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J. 1996, 15, 4182–4193. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, H.; Joo, H.Y.; Yu, J.H.; Smith, A.D.t.; Schneider, D.; Chow, L.T.; Renfrow, M.; Wang, H. Ubp-M serine 552 phosphorylation by cyclin-dependent kinase 1 regulates cell cycle progression. Cell Cycle 2013, 12, 3219–3227. [Google Scholar] [CrossRef]
- Larsen, C.N.; Krantz, B.A.; Wilkinson, K.D. Substrate specificity of deubiquitinating enzymes: Ubiquitin C-terminal hydrolases. Biochemistry 1998, 37, 3358–3368. [Google Scholar] [CrossRef] [PubMed]
- Kabuta, T.; Mitsui, T.; Takahashi, M.; Fujiwara, Y.; Kabuta, C.; Konya, C.; Tsuchiya, Y.; Hatanaka, Y.; Uchida, K.; Hohjoh, H.; et al. Ubiquitin C-terminal hydrolase L1 (UCH-L1) acts as a novel potentiator of cyclin-dependent kinases to enhance cell proliferation independently of its hydrolase activity. J. Biol. Chem. 2013, 288, 12615–12626. [Google Scholar] [CrossRef]
- Yoshida, A.; Yoneda-Kato, N.; Kato, J.Y. CSN5 specifically interacts with CDK2 and controls senescence in a cytoplasmic cyclin E-mediated manner. Sci. Rep. 2013, 3, 1054. [Google Scholar] [CrossRef] [PubMed]
- Dubiel, W.; Chaithongyot, S.; Dubiel, D.; Naumann, M. The COP9 Signalosome: A Multi-DUB Complex. Biomolecules 2020, 10, 1082. [Google Scholar] [CrossRef] [PubMed]
- Groisman, R.; Polanowska, J.; Kuraoka, I.; Sawada, J.; Saijo, M.; Drapkin, R.; Kisselev, A.F.; Tanaka, K.; Nakatani, Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 2003, 113, 357–367. [Google Scholar] [CrossRef]
- Ying, M.; Shao, X.; Jing, H.; Liu, Y.; Qi, X.; Cao, J.; Chen, Y.; Xiang, S.; Song, H.; Hu, R.; et al. Ubiquitin-dependent degradation of CDK2 drives the therapeutic differentiation of AML by targeting PRDX2. Blood 2018, 131, 2698–2711. [Google Scholar] [CrossRef]
- Liu, T.; Yu, J.; Deng, M.; Yin, Y.; Zhang, H.; Luo, K.; Qin, B.; Li, Y.; Wu, C.; Ren, T.; et al. CDK4/6-dependent activation of DUB3 regulates cancer metastasis through SNAIL1. Nat. Commun. 2017, 8, 13923. [Google Scholar] [CrossRef] [PubMed]
- Szynglarewicz, B.; Kasprzak, P.; Donizy, P.; Biecek, P.; Halon, A.; Matkowski, R. Biological Aggressiveness of Subclinical No-Mass Ductal Carcinoma In Situ (DCIS) Can Be Reflected by the Expression Profiles of Epithelial-Mesenchymal Transition Triggers. Int. J. Mol. Sci. 2018, 19, 3941. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Picksley, S.M.; Lane, D.P. The p53-mdm2 autoregulatory feedback loop: A paradigm for the regulation of growth control by p53? Bioessays 1993, 15, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lin, J.; Levine, A.J. Regulation of transcription functions of the p53 tumor suppressor by the mdm-2 oncogene. Mol. Med. 1995, 1, 142–152. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Linares, L.K.; Kiernan, R.; Triboulet, R.; Chable-Bessia, C.; Latreille, D.; Cuvier, O.; Lacroix, M.; Le Cam, L.; Coux, O.; Benkirane, M. Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nat. Cell Biol. 2007, 9, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, H.; Tseng, A.; Gao, D.; Zhai, B.; Zhang, Q.; Shaik, S.; Wan, L.; Ang, X.L.; Mock, C.; Yin, H.; et al. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(beta-TRCP) ubiquitin ligase. Cancer Cell 2010, 18, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wu, Z.; Mei, Y.; Wu, M. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J. 2013, 32, 2204–2216. [Google Scholar] [CrossRef]
- Joo, H.M.; Kim, J.Y.; Jeong, J.B.; Seong, K.M.; Nam, S.Y.; Yang, K.H.; Kim, C.S.; Kim, H.S.; Jeong, M.; An, S.; et al. Ret finger protein 2 enhances ionizing radiation-induced apoptosis via degradation of AKT and MDM2. Eur. J. Cell Biol. 2011, 90, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tan, Y.; Zhang, C.; Zhang, Y.; Zhang, L.; Ren, P.; Deng, H.; Luo, J.; Ke, Y.; Du, X. NAT10 regulates p53 activation through acetylating p53 at K120 and ubiquitinating Mdm2. EMBO Rep. 2016, 17, 349–366. [Google Scholar] [CrossRef]
- Zhao, K.; Yang, Y.; Zhang, G.; Wang, C.; Wang, D.; Wu, M.; Mei, Y. Regulation of the Mdm2-p53 pathway by the ubiquitin E3 ligase MARCH7. EMBO Rep. 2018, 19, 305–319. [Google Scholar] [CrossRef]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef]
- Stevenson, L.F.; Sparks, A.; Allende-Vega, N.; Xirodimas, D.P.; Lane, D.P.; Saville, M.K. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 2007, 26, 976–986. [Google Scholar] [CrossRef]
- Zou, Q.; Jin, J.; Hu, H.; Li, H.S.; Romano, S.; Xiao, Y.; Nakaya, M.; Zhou, X.; Cheng, X.; Yang, P.; et al. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat. Immunol. 2014, 15, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648–653. [Google Scholar] [CrossRef]
- Hu, M.; Gu, L.; Li, M.; Jeffrey, P.D.; Gu, W.; Shi, Y. Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: Implications for the regulation of the p53-MDM2 pathway. PLoS Biol. 2006, 4, e27. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Saridakis, V.; Sarkari, F.; Duan, S.; Wu, T.; Arrowsmith, C.H.; Frappier, L. Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 2006, 13, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef]
- Boutell, C.; Everett, R.D. The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and Ubiquitinates p53. J. Biol. Chem. 2003, 278, 36596–36602. [Google Scholar] [CrossRef]
- Dornan, D.; Wertz, I.; Shimizu, H.; Arnott, D.; Frantz, G.D.; Dowd, P.; O’Rourke, K.; Koeppen, H.; Dixit, V.M. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004, 429, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Rajendra, R.; Malegaonkar, D.; Pungaliya, P.; Marshall, H.; Rasheed, Z.; Brownell, J.; Liu, L.F.; Lutzker, S.; Saleem, A.; Rubin, E.H. Topors functions as an E3 ubiquitin ligase with specific E2 enzymes and ubiquitinates p53. J. Biol. Chem. 2004, 279, 36440–36444. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Scheffner, M.; Hohfeld, J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem. 2005, 280, 27443–27448. [Google Scholar] [CrossRef]
- Chen, D.; Kon, N.; Li, M.; Zhang, W.; Qin, J.; Gu, W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 2005, 121, 1071–1083. [Google Scholar] [CrossRef]
- Allton, K.; Jain, A.K.; Herz, H.M.; Tsai, W.W.; Jung, S.Y.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Yagishita, N.; Sasaki, T.; Nakazawa, M.; Kato, Y.; Yamadera, T.; Bae, E.; Toriyama, S.; Ikeda, R.; Zhang, L.; et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ’Synoviolin’. EMBO J. 2007, 26, 113–122. [Google Scholar] [CrossRef]
- Lee, E.W.; Lee, M.S.; Camus, S.; Ghim, J.; Yang, M.R.; Oh, W.; Ha, N.C.; Lane, D.P.; Song, J. Differential regulation of p53 and p21 by MKRN1 E3 ligase controls cell cycle arrest and apoptosis. EMBO J. 2009, 28, 2100–2113. [Google Scholar] [CrossRef] [PubMed]
- Laine, A.; Ronai, Z. Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 2007, 26, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, N.J.; Chen, C.; Tang, W.; Kornbluth, S. Ubiquitylation of p53 by the APC/C inhibitor Trim39. Proc. Natl. Acad. Sci. USA 2012, 109, 20931–20936. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Zhang, S.; Qu, G.; Zhang, D.; Li, S.; Liu, S. Downregulation of ubiquitin E3 ligase TNF receptor-associated factor 7 leads to stabilization of p53 in breast cancer. Oncol. Rep. 2013, 29, 283–287. [Google Scholar] [CrossRef]
- Thirunavukarasou, A.; Singh, P.; Govindarajalu, G.; Bandi, V.; Baluchamy, S. E3 ubiquitin ligase Cullin4B mediated polyubiquitination of p53 for its degradation. Mol. Cell Biochem. 2014, 390, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.T.; Richter, D.; Michel, G.; Mitschka, S.; Kolanus, W.; Cuevas, E.; Wulczyn, F.G. The ubiquitin ligase LIN41/TRIM71 targets p53 to antagonize cell death and differentiation pathways during stem cell differentiation. Cell Death Differ. 2017, 24, 1063–1078. [Google Scholar] [CrossRef]
- Shen, J.; Li, P.; Shao, X.; Yang, Y.; Liu, X.; Feng, M.; Yu, Q.; Hu, R.; Wang, Z. The E3 Ligase RING1 Targets p53 for Degradation and Promotes Cancer Cell Proliferation and Survival. Cancer Res. 2018, 78, 359–371. [Google Scholar] [CrossRef]
- Tripathi, V.; Kaur, E.; Kharat, S.S.; Hussain, M.; Damodaran, A.P.; Kulshrestha, S.; Sengupta, S. Abrogation of FBW7alpha-dependent p53 degradation enhances p53’s function as a tumor suppressor. J. Biol. Chem. 2019, 294, 13224–13232. [Google Scholar] [CrossRef]
- Rong, X.; Rao, J.; Li, D.; Jing, Q.; Lu, Y.; Ji, Y. TRIM69 inhibits cataractogenesis by negatively regulating p53. Redox. Biol. 2019, 22, 101157. [Google Scholar] [CrossRef]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.X.; Challagundla, K.B.; Dai, M.S. Positive regulation of p53 stability and activity by the deubiquitinating enzyme Otubain 1. EMBO J. 2012, 31, 576–592. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chung, H.J.; Vogt, M.; Jin, Y.; Malide, D.; He, L.; Dundr, M.; Levens, D. JTV1 co-activates FBP to induce USP29 transcription and stabilize p53 in response to oxidative stress. EMBO J. 2011, 30, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.K.; Vigneron, A.M.; Carter, S.; Ludwig, R.L.; Vousden, K.H. Regulation of p53 stability and function by the deubiquitinating enzyme USP42. EMBO J. 2011, 30, 4921–4930. [Google Scholar] [CrossRef]
- Ke, J.Y.; Dai, C.J.; Wu, W.L.; Gao, J.H.; Xia, A.J.; Liu, G.P.; Lv, K.S.; Wu, C.L. USP11 regulates p53 stability by deubiquitinating p53. J. Zhejiang Univ. Sci. B 2014, 15, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, W.; Liang, C.; Chen, B.W.; Zhi, X.; Zhang, S.; Zheng, X.; Bai, X.; Liang, T. WP1130 increases doxorubicin sensitivity in hepatocellular carcinoma cells through usp9x-dependent p53 degradation. Cancer Lett. 2015, 361, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gong, F. Involvement of USP24 in the DNA damage response. Mol. Cell Oncol. 2016, 3, e1011888. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, L.; Nemzow, L.; Chen, H.; Lubin, A.; Rong, X.; Sun, Z.; Harris, T.K.; Gong, F. The deubiquitinating enzyme USP24 is a regulator of the UV damage response. Cell Rep. 2015, 10, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, X.; Ning, G.; Zhu, S.; Ma, X.; Liu, X.; Liu, C.; Huang, M.; Schmitt, I.; Wullner, U.; et al. The Machado-Joseph Disease Deubiquitinase Ataxin-3 Regulates the Stability and Apoptotic Function of p53. PLoS Biol. 2016, 14, e2000733. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Brugarolas, J.; Chandrasekaran, C.; Gordon, J.I.; Beach, D.; Jacks, T.; Hannon, G.J. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 1995, 377, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Zhang, P.; Harper, J.W.; Elledge, S.J.; Leder, P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995, 82, 675–684. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Amador, V.; Ge, S.; Santamaria, P.G.; Guardavaccaro, D.; Pagano, M. APC/C(Cdc20) controls the ubiquitin-mediated degradation of p21 in prometaphase. Mol. Cell 2007, 27, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Sivaprasad, U.; Terai, K.; Amador, V.; Pagano, M.; Dutta, A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008, 22, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Starostina, N.G.; Kipreos, E.T. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008, 22, 2507–2519. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, H.; Shiomi, Y.; Iida, H.; Michishita, M.; Takami, T.; Tsurimoto, T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J. Biol. Chem. 2008, 283, 29045–29052. [Google Scholar] [CrossRef] [PubMed]
- Starostina, N.G.; Simpliciano, J.M.; McGuirk, M.A.; Kipreos, E.T. CRL2(LRR-1) targets a CDK inhibitor for cell cycle control in C. elegans and actin-based motility regulation in human cells. Dev. Cell 2010, 19, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kim, Y.L.; Lee, K.W.; Her, N.G.; Ha, T.K.; Yoon, S.; Jeong, S.I.; Lee, J.H.; Kang, M.J.; Lee, M.G.; et al. ZNF313 is a novel cell cycle activator with an E3 ligase activity inhibiting cellular senescence by destabilizing p21(WAF1.). Cell Death Differ. 2013, 20, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhi, X.; Zhao, D.; Wang, Z.; Zhou, Z.; Wang, C.; Chen, W.; Liu, R.; Chen, C. E3 ubiquitin ligase RNF126 promotes cancer cell proliferation by targeting the tumor suppressor p21 for ubiquitin-mediated degradation. Cancer Res. 2013, 73, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.; Sarkar, S.; Du, K.; Brautigan, D.L.; Abbas, T.; Larner, J.M. The E3 Ligase CHIP Mediates p21 Degradation to Maintain Radioresistance. Mol. Cancer Res. 2017, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, K.; Hou, C.; Jiang, K.; Chen, B.; Chen, J.; Lao, L.; Qian, L.; Zhong, G.; Liu, Z.; et al. CRL4B(DCAF11) E3 ligase targets p21 for degradation to control cell cycle progression in human osteosarcoma cells. Sci. Rep. 2017, 7, 1175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yan, X.; Zeng, S.; Zhang, T.; Cheng, F.; Chen, R.; Duan, C. UHRF2 promotes DNA damage response by decreasing p21 via RING finger domain. Biotechnol. Lett. 2018, 40, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, H.J.; Kim, M.K.; Bae, M.K.; Sung, H.Y.; Ahn, J.H.; Kim, Y.H.; Kim, S.C.; Ju, W. SPSB1 enhances ovarian cancer cell survival by destabilizing p21. Biochem. Biophys. Res. Commun. 2019, 510, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yu, C.; Yang, X.; Hong, H.; Lu, J.; Hu, W.; Hao, X.; Li, S.; Aikemu, B.; Yang, G.; et al. N-myc downstream-regulated gene 1 inhibits the proliferation of colorectal cancer through emulative antagonizing NEDD4-mediated ubiquitylation of p21. J. Exp. Clin. Cancer Res. 2019, 38, 490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, J.; Ning, D.; Liu, Q.; Wang, C.; Zhang, Z.; Chu, L.; Yu, C.; Liang, H.F.; Zhang, B.; et al. FBXO22 promotes the development of hepatocellular carcinoma by regulating the ubiquitination and degradation of p21. J. Exp. Clin. Cancer Res. 2019, 38, 101. [Google Scholar] [CrossRef]
- Deng, T.; Yan, G.; Song, X.; Xie, L.; Zhou, Y.; Li, J.; Hu, X.; Li, Z.; Hu, J.; Zhang, Y.; et al. Deubiquitylation and stabilization of p21 by USP11 is critical for cell-cycle progression and DNA damage responses. Proc. Natl. Acad. Sci. USA 2018, 115, 4678–4683. [Google Scholar] [CrossRef]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef]
- Li, X.; Amazit, L.; Long, W.; Lonard, D.M.; Monaco, J.J.; O’Malley, B.W. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Mol. Cell 2007, 26, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Lotem, J.; Cohen, B.; Sachs, L.; Shaul, Y. Regulation of p53 stability and p53-dependent apoptosis by NADH quinone oxidoreductase 1. Proc. Natl. Acad. Sci. USA 2001, 98, 1188–1193. [Google Scholar] [CrossRef]
- Asher, G.; Lotem, J.; Sachs, L.; Kahana, C.; Shaul, Y. Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQO1. Proc. Natl. Acad. Sci. USA 2002, 99, 13125–13130. [Google Scholar] [CrossRef] [PubMed]
- Dubiel, W.; Pratt, G.; Ferrell, K.; Rechsteiner, M. Purification of an 11 S regulator of the multicatalytic protease. J. Biol. Chem. 1992, 267, 22369–22377. [Google Scholar] [CrossRef]
- Sgambato, A.; Cittadini, A.; Faraglia, B.; Weinstein, I.B. Multiple functions of p27(Kip1) and its alterations in tumor cells: A review. J. Cell Physiol. 2000, 183, 18–27. [Google Scholar] [CrossRef]
- Tsvetkov, L.M.; Yeh, K.H.; Lee, S.J.; Sun, H.; Zhang, H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 1999, 9, 661–664. [Google Scholar] [CrossRef]
- Sutterluty, H.; Chatelain, E.; Marti, A.; Wirbelauer, C.; Senften, M.; Muller, U.; Krek, W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat. Cell Biol. 1999, 1, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Auld, C.A.; Fernandes, K.M.; Morrison, R.F. Skp2-mediated p27(Kip1) degradation during S/G2 phase progression of adipocyte hyperplasia. J. Cell Physiol. 2007, 211, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Hara, T.; Matsumoto, M.; Ishida, N.; Okumura, F.; Hatakeyama, S.; Yoshida, M.; Nakayama, K.; Nakayama, K.I. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat. Cell Biol. 2004, 6, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Isobe, T.; Abe, K.; Kikuchi, H.; Kitagawa, K.; Oda, T.; Uchida, C.; Kitagawa, M. Pirh2 promotes ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor p27Kip1. Cancer Res. 2007, 67, 10789–10795. [Google Scholar] [CrossRef]
- Choi, H.H.; Phan, L.; Chou, P.C.; Su, C.H.; Yeung, S.C.; Chen, J.S.; Lee, M.H. COP1 enhances ubiquitin-mediated degradation of p27Kip1 to promote cancer cell growth. Oncotarget 2015, 6, 19721–19734. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Yeung, S.C.; Chen, J.; Iwakuma, T.; Su, C.H.; Chen, B.; Qu, C.; Zhang, F.; Chen, Y.T.; Lin, Y.L.; et al. Subunit 6 of the COP9 signalosome promotes tumorigenesis in mice through stabilization of MDM2 and is upregulated in human cancers. J. Clin. Investig. 2011, 121, 851–865. [Google Scholar] [CrossRef]
- Choi, H.H.; Guma, S.; Fang, L.; Phan, L.; Ivan, C.; Baggerly, K.; Sood, A.; Lee, M.H. Regulating the stability and localization of CDK inhibitor p27(Kip1) via CSN6-COP1 axis. Cell Cycle 2015, 14, 2265–2273. [Google Scholar] [CrossRef]
- Choi, H.H.; Gully, C.; Su, C.H.; Velazquez-Torres, G.; Chou, P.C.; Tseng, C.; Zhao, R.; Phan, L.; Shaiken, T.; Chen, J.; et al. COP9 signalosome subunit 6 stabilizes COP1, which functions as an E3 ubiquitin ligase for 14-3-3sigma. Oncogene 2011, 30, 4791–4801. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Das, C.M.; Taylor, P.; Gireud, M.; Singh, A.; Lee, D.; Fuller, G.; Ji, L.; Fangusaro, J.; Rajaram, V.; Goldman, S.; et al. The deubiquitylase USP37 links REST to the control of p27 stability and cell proliferation. Oncogene 2013, 32, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Creff, J.; Besson, A. Functional Versatility of the CDK Inhibitor p57(Kip2). Front. Cell Dev. Biol. 2020, 8, 584590. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Hara, T.; Kotoshiba, S.; Yada, M.; Ishida, N.; Imaki, H.; Hatakeyama, S.; Nakayama, K.; Nakayama, K.I. Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proc. Natl. Acad. Sci. USA 2003, 100, 10231–10236. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Nakamoto, T.; Nishimori, S.; Tanaka, K.; Chiba, T. A new ubiquitin ligase involved in p57KIP2 proteolysis regulates osteoblast cell differentiation. EMBO Rep. 2008, 9, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhao, R.; Su, C.H.; Linan, M.; Tseng, C.; Phan, L.; Fang, L.; Yang, H.Y.; Yang, H.; Wang, W.; et al. CDK inhibitor p57 (Kip2) is negatively regulated by COP9 signalosome subunit 6. Cell Cycle 2012, 11, 4633–4641. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Nita, A.; Nishiyama, M.; Muto, Y.; Shimizu, H.; Nakatsumi, H.; Nakayama, K.I. Skp2 contributes to cell cycle progression in trophoblast stem cells and to placental development. Genes Cells 2020, 25, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Cui, H.; Zhang, H. Smurf2-mediated ubiquitination and degradation of Id1 regulates p16 expression during senescence. Aging Cell 2011, 10, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Canepa, E.T.; Scassa, M.E.; Ceruti, J.M.; Marazita, M.C.; Carcagno, A.L.; Sirkin, P.F.; Ogara, M.F. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life 2007, 59, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Latres, E.; Malumbres, M.; Sotillo, R.; Martin, J.; Ortega, S.; Martin-Caballero, J.; Flores, J.M.; Cordon-Cardo, C.; Barbacid, M. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000, 19, 3496–3506. [Google Scholar] [CrossRef] [PubMed]
- Franklin, D.S.; Godfrey, V.L.; Lee, H.; Kovalev, G.I.; Schoonhoven, R.; Chen-Kiang, S.; Su, L.; Xiong, Y. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998, 12, 2899–2911. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Cunningham, J.J.; Sherr, C.J.; Jogal, S.; Smeyne, R.J.; Roussel, M.F. Postnatal neuronal proliferation in mice lacking Ink4d and Kip1 inhibitors of cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA 1999, 96, 13462–13467. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; den Besten, W.; Chen, B.; Rehg, J.E.; Latres, E.; Barbacid, M.; Pollard, J.W.; Sherr, C.J.; Cohen, P.E.; Roussel, M.F. Control of spermatogenesis in mice by the cyclin D-dependent kinase inhibitors p18(Ink4c) and p19(Ink4d). Mol. Cell Biol. 2001, 21, 3244–3255. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zindy, F.; Abdala, C.; Liu, F.; Li, X.; Roussel, M.F.; Segil, N. Progressive hearing loss in mice lacking the cyclin-dependent kinase inhibitor Ink4d. Nat. Cell Biol. 2003, 5, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Roussel, M.F.; Kato, J.Y.; Ashmun, R.A.; Sherr, C.J. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol. Cell Biol. 1995, 15, 2672–2681. [Google Scholar] [CrossRef]
- Thullberg, M.; Bartek, J.; Lukas, J. Ubiquitin/proteasome-mediated degradation of p19INK4d determines its periodic expression during the cell cycle. Oncogene 2000, 19, 2870–2876. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat. Res. 2005, 576, 22–38. [Google Scholar] [CrossRef]
- Du, W.; Liu, Z.; Zhu, W.; Li, T.; Zhu, Z.; Wei, L.; Song, J.; Pei, D. CSN6 promotes tumorigenesis of gastric cancer by ubiquitin-independent proteasomal degradation of p16(INK4a). Cancer Biol. Med. 2019, 16, 514–529. [Google Scholar]
- Alani, R.M.; Young, A.Z.; Shifflett, C.B. Id1 regulation of cellular senescence through transcriptional repression of p16/Ink4a. Proc. Natl. Acad. Sci. USA 2001, 98, 7812–7816. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Zebedee, Z.; Huot, T.J.; Stinson, J.A.; Sugimoto, M.; Ohashi, Y.; Sharrocks, A.D.; Peters, G.; Hara, E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 2001, 409, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- De Luca, M.; Lavia, P.; Guarguaglini, G. A functional interplay between Aurora-A, Plk1 and TPX2 at spindle poles: Plk1 controls centrosomal localization of Aurora-A and TPX2 spindle association. Cell Cycle 2006, 5, 296–303. [Google Scholar] [CrossRef]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Ozaki, T.; Takada, Y.; Kageyama, H.; Nakamura, Y.; Hata, A.; Zhang, J.H.; Simonds, W.F.; Nakagawara, A.; Koseki, H. topors, a p53 and topoisomerase I-binding RING finger protein, is a coactivator of p53 in growth suppression induced by DNA damage. Oncogene 2005, 24, 3385–3396. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E3 Ligase/DUB | Target | Function | Reference |
|---|---|---|---|
| E3 ligase | |||
| APC/CCdc20 APC/CCdh1 | cyclin A | Promotes the onset of anaphase and exit from Mitosis | [78] |
| APC/CCdc20 | cyclin B | Inhibits B-type cyclin accumulation and to prevent uncontrolled entry into S phase | [91] |
| HEI10 | cyclin B1 | Functions in progression through G2/M | [92] |
| SCFSKP2 | cyclin D | Regulates the mammalian G1/S transition | [71] |
| FBXW8 | cyclin D1 | Promotes cancer cell proliferation | [68] |
| FBXO25 | Oct-1 | Promotes tumor growth and metastasis | [69,93] |
| SCFFBX4-αB crystallin | cyclin D1 | Knockdown of the SCFFBX4-αB crystallin ligase accelerates G1 phase progression | [94] |
| Cullin-3 | cyclin E1 | Controls S phase in mammalian cells | [95] |
| SCFFbw7 | cyclin E | Inhibits cancer cell proliferation | [70] |
| SCFSKP2 | cyclin E | Plays a role in the S phase progression | [96] |
| DUB | |||
| USP37 | cyclin A | Accelerates S phase entry | [83] |
| USP2a | cyclin A1 | Increases cell proliferation | [84] |
| DUB3 (USP17) | cyclin A | Be critical for G1/S transition | [85] |
| Bam/Otu complex | cyclin A | Contributes to germ-line stem cell fate determination | [90] |
| USP14 | cyclin B1 | Facilitates G2/M phase transition | [97] |
| USP22 | cyclin B1 | USP22 knockdown leads to slower cell growth | [98] |
| OTUD7B | cyclin B | Contributes to mitotic exit | [99] |
| USP2 | cyclin D1 | Accelerates the G1 to S phase progression | [100] |
| USP9x | FoxO3a | Suppresses cell proliferation by downregulating expression of cyclin D | [101,102] |
| OTUD6B | ? | Blocks cell proliferation by arresting cells in G1 phase | [103] |
| USP22 | cyclin D1 | Promotes G1/S cell cycle transition | [104] |
| USP27 | cyclin E | Increases the percentage of cells in G2/M phase and decreases the percentage of cells in G0/G1 phase | [105] |
| E3 Ligase/DUB | Target | Function | Reference |
|---|---|---|---|
| E3 ligase | |||
| KLHL6 | CDK2 | Drives granulocytic differentiation in human AML cells | [130] |
| DUB | |||
| Ubp-M | uH2A | Facilitates chromosome condensation and cell cycle progression | [124] |
| UCH-L1 | CDK1/4/5 | Enhances CDKs activity and promotes cell proliferation | [126] |
| CSN5 | CDK2 | Promotes cell cycle progression | [127] |
| DUB3 | SNAIL1 | Regulates cell migration and cancer metastasis | [131] |
| Substrate | E3 Ligase/DUB | Function | Reference | |
|---|---|---|---|---|
| E3 ligase | ||||
| p21CIP1/WAF1 | SCFSKP2 | Regulates the mammalian G1/S transition | [71,72,73] | |
| APC/CCDC20 | Contributes to the full activation of Cdk1 necessary for mitotic events and prevents mitotic slippage during spindle checkpoint activation | [186] | ||
| CRL4CDT2 | Promotes DNA repair by promoting the degradation of p21 | [187,188,189] | ||
| MKRN1 | Depletion of MKRN1 induces cell cycle arrest by activating p53 and p21 | [164] | ||
| CRL2LRR1 | Nematode CRL2LRR1 ensures G1-phase cell cycle progression in germ cells, whereas human CRL2LRR−1 has no significant effect on cell cycle | [190] | ||
| ZNF313 | Activates cell cycle progression and inhibits cellular senescence | [191] | ||
| RNF126 | Facilitates cell cycle G1/S progression and cell proliferation | [192] | ||
| CHIP | Knockdown of CHIP results in enhanced cellular senescence and increased sensitivity of lung cancer cells to ionizing radiation | [193] | ||
| CRL4BDCAF11 | Promotes S phase entry and osteosarcoma cell proliferation | [194] | ||
| UHRF2 | Promotes DNA damage response | [195] | ||
| SPSB1 | SPSB1 knockdown induced cell cycle arrest and apoptosis | [196] | ||
| NEDD4 | Promotes cell growth | [197] | ||
| FBXO22 | Promotes cell growth, and affects cell cycle and apoptosis | [198] | ||
| p27KIP1 | SCFSKP2 | Induces S phase in quiescent cells | [74,206,207,208] | |
| KPC | Controls cell cycle progression from G0 to S phase | [209] | ||
| Pirh2 | Depletion of Pirh2 induces an inhibition of cell cycle progression at G1/S transition | [210] | ||
| COP1 | Cell cycle progression is delayed with COP1 deficiency | [211] | ||
| p57KIP2 | SCFSKP2 | Contributes to cell cycle progression | [217,220] | |
| FBL12 | Regulates osteoblast cell differentiation | [218] | ||
| Id1 | Smurf2 | Provides a mechanistic link between Smurf2 and p16 during senescence | [221] | |
| DUB | ||||
| p21CIP1/WAF1 | USP11 | Regulates G1/S transition and the DNA damage response | [199] | |
| p27KIP1 | USP37 | Controls cell proliferation | [215] | |
| E3 Ligase/DUB | Function | Reference |
|---|---|---|
| E3 ligase | ||
| MDM2 | Blocks p53-dependent transcription, and promotes the rapid degradation of p53 | [140,141,142] |
| Pirh2 | Represses p53-dependent transactivation and cell cycle arrest | [156] |
| ICP0 | Inhibits the apoptotic response to DNA damage in irradiated U2OS cells | [157] |
| COP1 | Inhibits p53-dependent transcription, apoptosis, and cell cycle arrest | [158] |
| Topors | Acts as a coactivator of p53 in response to DNA damage | [159,236] |
| CHIP | Influences p53-mediated transcription | [160] |
| ARE-BP1 | Inhibits p53-dependent apoptosis | [161] |
| TRIM24 | Inhibits p53-dependent apoptosis | [162] |
| HRD1 | Directly regulates p53-dependent apoptotic pathway in Drosophila fly | [163] |
| MKRN1 | Leads cells to p53-dependent apoptosis by suppressing p21 | [164] |
| WWP1 | Increases p53 stability, and decreases p53 transcriptional activities | [165] |
| TRIM39 | Promotes G1/S transition and cell proliferation | [166] |
| TRAF7 | Promotes cell proliferation | [167] |
| Cullin4B | Promotes cell proliferation | [168] |
| TRIM71 | Antagonizes p53-dependent pro-apoptotic and pro-differentiation responses. | [169] |
| RING1 | Promotes cancer cell proliferation and survival | [170] |
| FBW7α | Responds to DNA damage | [171] |
| TRIM69 | Decreases cell apoptosis and ROS production after UVB irradiation | [172] |
| DUB | ||
| HAUSP (USP7) | Plays a dual role in the regulation of p53 function | [153,154,155] |
| USP10 | Potentiates p53-dependent transcription activity and apoptosis | [173] |
| Otub1 | Results in apoptosis and inhibition of cell proliferation in a p53-dependent manner | [174] |
| USP29 | Stabilizes p53 in response to oxidative stress | [175] |
| USP42 | Contributes to the repair and recovery of cells from mild or transient damage | [176] |
| USP11 | Promotes p53 activation in response to DNA damage | [177] |
| USP9x | Stabilizes p53 and increases p53-dependent apoptosis | [178] |
| USP24 | Regulates the DNA damage response | [179,180] |
| Ataxin-3 | Regulates the functions of p53 in transactivation and apoptosis | [181] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, T.; Lin, Z. The Involvement of Ubiquitination Machinery in Cell Cycle Regulation and Cancer Progression. Int. J. Mol. Sci. 2021, 22, 5754. https://doi.org/10.3390/ijms22115754
Zou T, Lin Z. The Involvement of Ubiquitination Machinery in Cell Cycle Regulation and Cancer Progression. International Journal of Molecular Sciences. 2021; 22(11):5754. https://doi.org/10.3390/ijms22115754
Chicago/Turabian StyleZou, Tingting, and Zhenghong Lin. 2021. "The Involvement of Ubiquitination Machinery in Cell Cycle Regulation and Cancer Progression" International Journal of Molecular Sciences 22, no. 11: 5754. https://doi.org/10.3390/ijms22115754
APA StyleZou, T., & Lin, Z. (2021). The Involvement of Ubiquitination Machinery in Cell Cycle Regulation and Cancer Progression. International Journal of Molecular Sciences, 22(11), 5754. https://doi.org/10.3390/ijms22115754