
The Complexities of Organ Crosstalk in Phosphate Homeostasis: Time to Put Phosphate Sensing Back in the Limelight


Abstract
1. Introduction
2. Phosphate Homeostasis in Health and Disease
3. Phosphate Transporters: Knowledge from Animal Models and Human Physiology
3.1. SlC34 Sodium Phosphate Co-Transporters
3.2. Slc20 Sodium Phosphate Co-Transporters
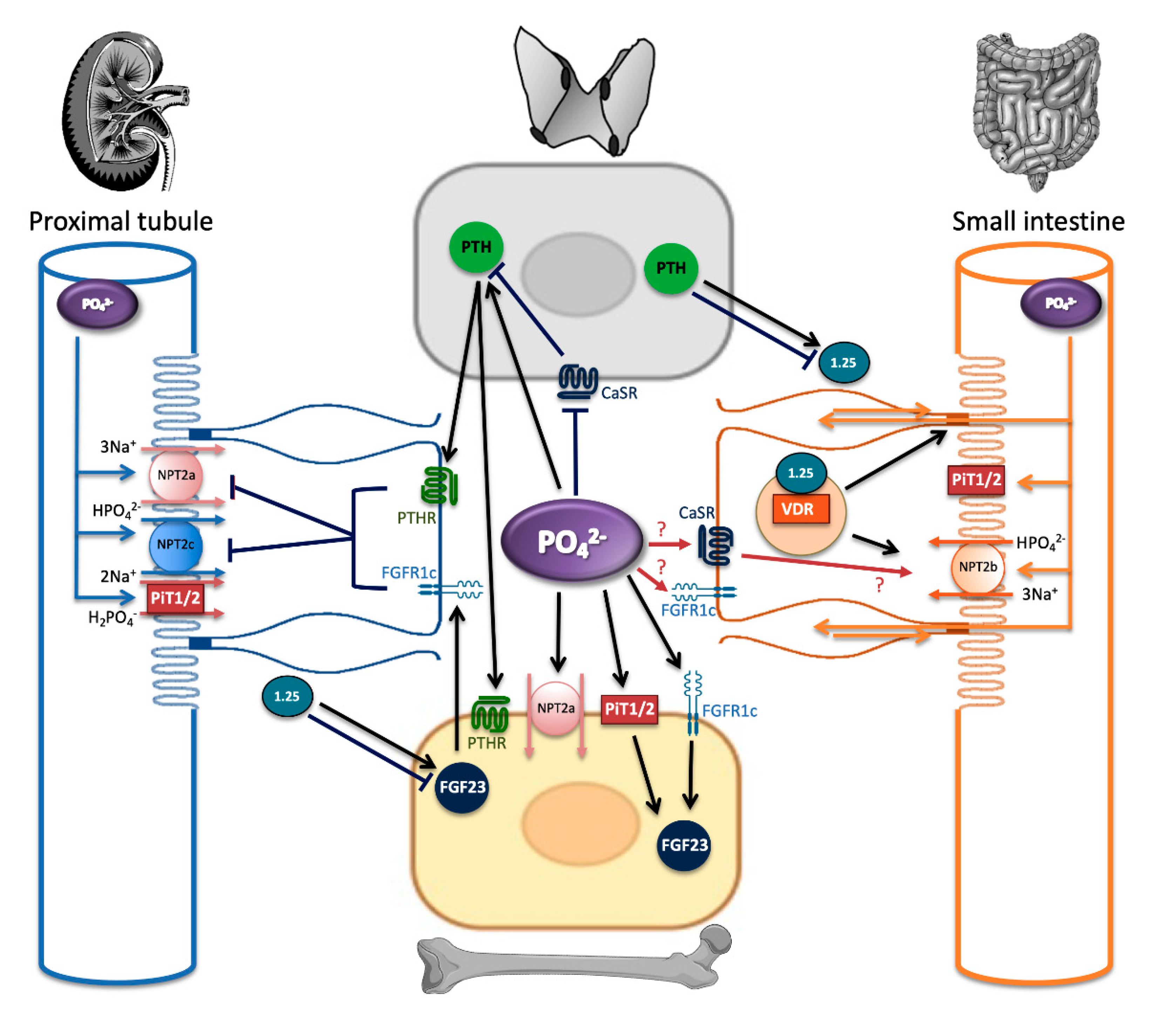
4. Phosphate Sensing: The Knowns and Unknowns
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Burtis, C.A.; Burtis, C.A.; Ashwood, E.R.; Bruns, D.E.; Sawyer, B.G. Tietz Fundamentals of Clinical Chemistry, 6th ed.; Saunders/Elsevier: St. Louis, MO, USA; Amsterdam, The Netherlands, 2008; p. 952. [Google Scholar]
- Mace, M.L.; Olgaard, K.; Lewin, E. New Aspects of the Kidney in the Regulation of Fibroblast Growth Factor 23 (FGF23) and Mineral Homeostasis. Int. J. Mol. Sci. 2020, 21, 8810. [Google Scholar] [CrossRef]
- Chande, S.; Bergwitz, C. Role of phosphate sensing in bone and mineral metabolism. Nat. Rev. Endocrinol. 2018, 11, 637–655. [Google Scholar] [CrossRef]
- Carney, E.F. The impact of chronic kidney disease on global health. Nat. Rev. Nephrol. 2020, 14, 251. [Google Scholar] [CrossRef]
- Copland, M.; Komenda, P.; Weinhandl, E.D.; McCullough, P.A.; Morfin, J.A. Intensive Hemodialysis, Mineral and Bone Disorder, and Phosphate Binder Use. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2016, 16 (Suppl. S1), S24–S32. [Google Scholar] [CrossRef]
- Moe, S.M.; Chen, N.X. Mechanisms of Vascular Calcification in Chronic Kidney Disease: Figure 1. J. Am. Soc. Nephrol. 2008, 19, 213–216. [Google Scholar] [CrossRef]
- Stevens, L.A. Calcium, Phosphate, and Parathyroid Hormone Levels in Combination and as a Function of Dialysis Duration Predict Mortality: Evidence for the Complexity of the Association between Mineral Metabolism and Outcomes. J. Am. Soc. Nephrol. 2004, 15, 770–779. [Google Scholar] [CrossRef]
- Ritter, C.S.; Slatopolsky, E. Phosphate Toxicity in CKD: The Killer among Us. Clin. J. Am. Soc. Nephrol. 2016, 11, 1088–1100. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO); CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2009, 113, S1–S130. [Google Scholar]
- Cooke, A. Dietary Food-Additive Phosphate and Human Health Outcomes: Dietary phosphate and human health. Compr. Rev. Food Sci. Food Saf. 2017, 16, 906–1021. [Google Scholar] [CrossRef]
- Calvo, M.S.; Moshfegh, A.J.; Tucker, K.L. Assessing the Health Impact of Phosphorus in the Food Supply: Issues and Considerations. Adv. Nutr. 2014, 5, 104–113. [Google Scholar] [CrossRef]
- Ugrica, M.; Bettoni, C.; Bourgeois, S.; Daryadel, A.; Pastor-Arroyo, E.-M.; Gehring, N.; Hernando, N.; Wagner, C.A.; Rubio-Aliaga, I. A chronic high phosphate intake in mice is detrimental for bone health without major renal alterations. Nephrol. Dial. Transplant. 2021. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Brandi, M.L. Congenital Conditions of Hypophosphatemia Expressed in Adults. Calcif. Tissue Int. 2021, 108, 91–103. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Sorribas, V. Compensatory regulation of the sodium/phosphate cotransporters NaPi-IIc (SCL34A3) and Pit-2 (SLC20A2) during Pi deprivation and acidosis. Pflügers Arch. Eur. J. Physiol. 2010, 459, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Lanzano, L.; Lei, T.; Okamura, K.; Giral, H.; Caldas, Y.; Masihzadeh, O.; Gratton, E.; Levi, M.; Blaine, J. Differential modulation of the molecular dynamics of the type IIa and IIc sodium phosphate cotransporters by parathyroid hormone. Am. J. Physiol. Cell Physiol. 2011, 301, C850–C861. [Google Scholar] [CrossRef]
- Sneddon, W.B.; Ruiz, G.W.; Gallo, L.I.; Xiao, K.; Zhang, Q.; Rbaibi, Y.; Weisz, O.A.; Apodaca, G.L.; Friedman, P. A Convergent Signaling Pathways Regulate Parathyroid Hormone and Fibroblast Growth Factor-23 Action on NPT2A-mediated Phosphate Transport. J. Biol. Chem. 2016, 291, 18632–18642. [Google Scholar] [CrossRef]
- Blaine, J.; Okamura, K.; Giral, H.; Breusegem, S.; Caldas, Y.; Millard, A.; Barry, N.; Levi, M. PTH-induced internalization of apical membrane NaPi2a: Role of actin and myosin VI. Am. J. Physiol. Cell Physiol. 2009, 297, C1339–C1346. [Google Scholar] [CrossRef]
- Gattineni, J.; Bates, C.; Twombley, K.; Dwarakanath, V.; Robinson, M.L.; Goetz, R.; Mohammadi, M.; Baum, M. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am. J. Physiol. Ren. Physiol. 2009, 297, F282–F291. [Google Scholar] [CrossRef]
- Beck, L.; Karaplis, A.C.; Amizuka, N.; Hewson, A.S.; Ozawa, H.; Tenenhouse, H.S. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc. Natl. Acad. Sci. USA 1998, 95, 5372–5377. [Google Scholar] [CrossRef]
- Prié, D.; Huart, V.; Bakouh, N.; Planelles, G.; Dellis, O.; Gérard, B.; Hulin, P.; Benque-Blanchet, F.; Silve, C.; Grandchamp, B.; et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N. Engl. J. Med. 2002, 347, 983–991. [Google Scholar] [CrossRef]
- Lorenz-Depiereux, B.; Benet-Pages, A.; Eckstein, G.; Tenenbaum-Rakover, Y.; Wagenstaller, J.; Tiosano, D.; Gershoni-Baruch, R.; Albers, N.; Lichter, P.; Schnabel, D.; et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am. J. Hum. Genet. 2006, 78, 193–201. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; Ruminska, J.; Kaufmann, M.; Dursun, I.; Patti, M.; Kranz, B.; Pronicka, E.; Ciara, E.; Akcay, T.; Bulus, D.; et al. Autosomal-Recessive Mutations in SLC34A1 Encoding Sodium-Phosphate Cotransporter 2A Cause Idiopathic Infantile Hypercalcemia. J. Am. Soc. Nephrol. 2016, 27, 604–614. [Google Scholar] [CrossRef]
- Magen, D.; Berger, L.; Coady, M.J.; Ilivitzki, A.; Militianu, D.; Tieder, M.; Selig, S.; Lapointe, J.Y.; Zelikovic, I.; Skorecki, K. A loss-of-function mutation in NaPi-IIa and renal Fanconi’s syndrome. N. Engl. J. Med. 2010, 362, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.J.; Li, D.; Doyle, D.; Zaritsky, J.; Levine, M.A. Digenic Heterozygous Mutations in SLC34A3 and SLC34A1 Cause Dominant Hypophosphatemic Rickets with Hypercalciuria. J. Clin. Endocrinol. Metab. 2020, 105, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Corut, A.; Senyigit, A.; Ugur, S.A.; Altin, S.; Ozcelik, U.; Calisir, H.; Yildirim, Z.; Gocmen, A.; Tolun, A. Mutations in SLC34A2 Cause Pulmonary Alveolar Microlithiasis and Are Possibly Associated with Testicular Microlithiasis. Am. J. Hum. Genet. 2006, 79, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, Y.; Etoh, N.; Hayasaka, M.; Takahashi, M.; Kakitani, M.; Yamashita, T.; Tomizuka, K.; Hanaoka, K. Targeted deletion of the tybe IIb Na+-dependent Pi-co-transporter, NaPi-IIb, results in early embryonic lethality. Biochem. Biophys. Res. Commun. 2009, 381, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, Y.; O’Brien, S.P.; Song, W.; Boulanger, J.H.; Stockmann, A.; Arbeeny, C.; Schiavi, S.C. Intestinal Npt2b Plays a Major Role in Phosphate Absorption and Homeostasis. J. Am. Soc. Nephrol. 2009, 20, 2348–2358. [Google Scholar] [CrossRef]
- Ikuta, K.; Segawa, H.; Sasaki, S.; Hanazaki, A.; Fujii, T.; Kushi, A.; Kawabata, Y.; Kirino, R.; Sasaki, S.; Noguchi, M.; et al. Effect of Npt2b deletion on intestinal and renal inorganic phosphate (Pi) handling. Clin. Exp. Nephrol. 2018, 22, 517–528. [Google Scholar] [CrossRef]
- Bergwitz, C.; Roslin, N.M.; Tieder, M.; Loredo-Osti, J.C.; Bastepe, M.; Abu-Zahra, H.; Frappier, D.; Burkett, D.; Carpenter, T.O.; Anderson, D.; et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am. J. Hum. Genet. 2006, 78, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Myakala, K.; Motta, S.; Murer, H.; Wagner, C.A.; Koesters, R.; Biber, J.; Hernando, N. Renal-specific and inducible depletion of NaPi-IIc/Slc34a3, the cotransporter mutated in HHRH, does not affect phosphate or calcium homeostasis in mice. Am. J. Physiol. Ren. Physiol. 2014, 306, F833–F843. [Google Scholar] [CrossRef] [PubMed]
- Segawa, H.; Onitsuka, A.; Kuwahata, M.; Hanabusa, E.; Furutani, J.; Kaneko, I.; Tomoe, Y.; Aranami, F.; Matsumoto, N.; Ito, M.; et al. Type IIc Sodium–Dependent Phosphate Transporter Regulates Calcium Metabolism. J. Am. Soc. Nephrol. 2009, 20, 104–113. [Google Scholar] [CrossRef]
- Beck, L.; Leroy, C.; Beck-Cormier, S.; Forand, A.; Salaün, C.; Paris, N.; Bernier, A.; Ureña-Torres, P.; Prié, D.; Ollero, M.; et al. The phosphate transporter PiT1 (Slc20a1) revealed as a new essential gene for mouse liver development. PLoS ONE 2010, 5, e9148. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-X.; Zou, X.-H.; Wang, C.; Yao, X.-P.; Su, H.-Z.; Lai, L.-L.; Chen, H.-T.; Lai, J.-H.; Liu, Y.-B.; Chen, D.-P.; et al. Spectrum of SLC20A2, PDGFRB, PDGFB, and XPR1 mutations in a large cohort of patients with primary familial brain calcification. Hum. Mutat. 2019, 40, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Beck-Cormier, S.; Lelliott, C.J.; Logan, J.G.; Lafont, D.T.; Merametdjian, L.; Leitch, V.D.; Butterfield, N.C.; Protheroe, H.J.; Croucher, P.I.; Baldock, P.A.; et al. Slc20a2, Encoding the Phosphate Transporter PiT2, Is an Important Genetic Determinant of Bone Quality and Strength. J. Bone Miner. Res. 2019, 34, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M.R.; Brown, E.M.; Chou, Y.-H.W.; Hebert, S.C.; Marx, S.J.; Stelnmann, B.; Levi, T.; Seidman, C.E.; Seidman, J. Mutations in the human Ca2+-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 1993, 75, 1297–1303. [Google Scholar] [CrossRef]
- Ho, C.; Conner, D.A.; Pollak, M.R.; Ladd, D.J.; Kifor, O.; Warren, H.B.; Brown, E.M.; Seidman, J.G.; Seidman, C.E. A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat. Genet. 1995, 11, 389–394. [Google Scholar] [CrossRef]
- Garner, S.C.; Pi, M.; Tu, Q.; Quarles, L.D. Rickets in cation-sensing receptor-deficient mice: An unexpected skeletal phenotype. Endocrinology 2001, 142, 3996–4005. [Google Scholar] [CrossRef]
- Dershem, R.; Gorvin, C.M.; Metpally, R.P.R.; Krishnamurthy, S.; Smelser, D.T.; Hannan, F.M.; Carey, D.C.; Thakker, R.V.; Breitweisser, G.E. Familial Hypocalciuric Hypercalcemia Type 1 and Autosomal-Dominant Hypocalcemia Type 1: Prevalence in a Large Healthcare Population. Am. J. Hum. Genet. 2020, 106, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Beck, L. Expression and function of Slc34 sodium–phosphate co-transporters in skeleton and teeth. Pflügers Arch. Eur. J. Physiol. 2019, 471, 175–184. [Google Scholar] [CrossRef]
- Lundquist, P.; Murer, H.; Biber, J. Type II Na+-Pi Cotransporters in Osteoblast Mineral Formation: Regulation by Inorganic Phosphate. Cell Physiol. Biochem. 2007, 19, 43–56. [Google Scholar] [CrossRef]
- Segawa, H.; Kaneko, I.; Yamanaka, S.; Ito, M.; Kuwahata, M.; Inoue, Y.; Kato, S.; Miyamoto, K.-I. Intestinal Na-Pi cotransporter adaptation to dietary Pi content in vitamin D receptor null mice. Am. J. Physiol. Ren. Physiol. 2004, 287, F39–F47. [Google Scholar] [CrossRef]
- Nishimura, M.; Naito, S. Tissue-specific mRNA Expression Profiles of Human Solute Carrier Transporter Superfamilies. Drug Metab. Pharmacokinet. 2008, 23, 22–44. [Google Scholar] [CrossRef]
- Motta, S.E.; Imenez Silva, P.H.; Daryadel, A.; Haykir, B.; Pastor-Arroyo, E.M.; Bettoni, C.; Hernando, N.; Wagner, C.A. Expression of NaPi-IIb in rodent and human kidney and upregulation in a model of chronic kidney disease. Pflügers Arch. Eur. J. Physiol. 2020, 472, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Schlingmann, K.P.; Kaufmann, M.; Weber, S.; Irwin, A.; Goos, C.; John, U.; Misselwitz, J.; Klaus, G.; Kuwertz-Bröking, E.; Fehrenbach, H.; et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N. Engl. J. Med. 2011, 365, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Ix, J.H.; Isakova, T.; Larive, B.; Raphael, K.L.; Raj, D.S.; Cheung, A.K.; Sprague, S.M.; Fried, L.F.; Gassman, J.J.; Middleton, J.P.; et al. Effects of Nicotinamide and Lanthanum Carbonate on Serum Phosphate and Fibroblast Growth Factor-23 in CKD: The COMBINE Trial. J. Am. Soc. Nephrol. 2019, 30, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Marks, J. The role of SLC34A2 in intestinal phosphate absorption and phosphate homeostasis. Pflügers Arch. Eur. J. Physiol. 2019, 471, 165–173. [Google Scholar] [CrossRef]
- Saurette, M.; Alexander, R.T. Intestinal phosphate absorption: The paracellular pathway predominates? Exp. Biol. Med. 2019, 244, 646–654. [Google Scholar] [CrossRef]
- Hashimoto, N.; Matsui, I.; Ishizuka, S.; Inoue, K.; Matsumoto, A.; Shimada, K.; Hori, S.; Lee, D.G.; Yasuda, S.; Katsuma, Y.; et al. Lithocholic acid increases intestinal phosphate and calcium absorption in a vitamin D receptor dependent but transcellular pathway independent manner. Kidney Int. 2020, 97, 1164–1180. [Google Scholar] [CrossRef]
- Asowata, E.O.; Olusanya, O.; Abaakil, K.; Chichger, H.; Srai, S.K.; Unwin, R.J.; Marks, J. Diet-induced iron deficiency in rats impacts small intestinal calcium and phosphate absorption. Acta Physiol. 2021, e13650. [Google Scholar] [CrossRef]
- Walton, J.; Gray, T.K. Absorption of Inorganic Phosphate in the Human Small Intestine. Clin. Sci. 1979, 56, 407–412. [Google Scholar] [CrossRef]
- Marks, J.; Srai, S.K.; Biber, J.; Murer, H.; Unwin, R.J.; Debnam, E.S. Intestinal phosphate absorption and the effect of vitamin D: A comparison of rats with mice: Intestinal phosphate transport in rats and mice. Exp. Physiol. 2006, 91, 531–537. [Google Scholar] [CrossRef]
- Kavanaugh, M.P.; Miller, D.G.; Zhang, W.; Law, W.; Kozak, S.L.; Kabat, D.; Miller, A.D. Cell-surface receptors for gibbon ape leukemia virus and amphotropic murine retrovirus are inducible sodium-dependent phosphate symporters. Proc. Natl. Acad. Sci. USA 1994, 91, 7071–7075. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.G.; Miller, A.D. A family of retroviruses that utilize related phosphate transporters for cell entry. J. Virol. 1994, 68, 8270–8276. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.G.; Edwards, R.H.; Miller, A.D. Cloning of the cellular receptor for amphotropic murine retroviruses reveals homology to that for gibbon ape leukemia virus. Proc. Natl. Acad. Sci. USA 1994, 91, 78–82. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, B.; Johann, S.V.; Klinger, H.P.; Blair, D.G.; Rubinson, H.; Dunn, K.J.; Sass, P.; Vitek, S.M.; Robins, T. Characterization of a human gene conferring sensitivity to infection by gibbon ape leukemia virus. Cell Growth Differ. 1990, 1. Available online: https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.327.7077&rep=rep1&type=pdf (accessed on 21 May 2021).
- Van Zeijl, M.; Johann, S.V.; Closs, E.; Cunningham, J.; Eddy, R.; Shows, T.B.; O’Hara, B. A human amphotropic retrovirus receptor is a second member of the gibbon ape leukemia virus receptor family. Proc. Natl. Acad. Sci. USA 1994, 91, 1168–1172. [Google Scholar] [CrossRef]
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transporters of the SLC20 and SLC34 families. Mol. Aspects Med. 2013, 34, 386–395. [Google Scholar] [CrossRef]
- Couasnay, G.; Bon, N.; Devignes, C.-S.; Sourice, S.; Bianchi, A.; Véziers, J.; Weiss, P.; Elefteriou, F.; Provot, S.; Guicheux, J.; et al. PiT1/Slc20a1 Is Required for Endoplasmic Reticulum Homeostasis, Chondrocyte Survival, and Skeletal Development. J. Bone Miner. Res. 2019, 34, 387–398. [Google Scholar] [CrossRef]
- Beck-Cormier, S.; Beck, L. The Need of a Paradigm Shift to Better Understand PiT1 and PiT2 Biology: Response to “Why Is There No PiT1/SLC20A1 Pathogenic Variants Yet Linked to Primary Familial Brain Calcification?”. J. Bone Miner. Res. 2020, 35, 825–826. [Google Scholar] [CrossRef]
- Ramos, E.M.; The French PFBC Study Group; Carecchio, M.; Lemos, R.; Ferreira, J.; Legati, A.; Sears, R.L.; Hsu, S.C.; Panteghini, C.; Magistrelli, L.; et al. Primary brain calcification: An international study reporting novel variants and associated phenotypes. Eur. J. Hum. Genet. 2018, 26, 1462–1477. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker, H.; Hattenhauer, O.; Traebert, M.; Forster, I.; Murer, H.; Biber, J. Characterization of a murine type II sodium-phosphate cotransporter expressed in mammalian small intestine. Proc. Natl. Acad. Sci. USA 1998, 95, 14564–14569. [Google Scholar] [CrossRef] [PubMed]
- Giral, H.; Caldas, Y.; Sutherland, E.; Wilson, P.; Breusegem, S.; Barry, N.; Blaine, J.; Jiang, T.; Wang, X.X.; Levi, M. Regulation of rat intestinal Na-dependent phosphate transporters by dietary phosphate. Am. J. Physiol. Ren Physiol. 2009, 297, F1466–F1475. [Google Scholar] [CrossRef]
- Reining, S.C.; Liesegang, A.; Betz, H.; Biber, J.; Murer, H.; Hernando, N. Expression of renal and intestinal Na/Pi cotransporters in the absence of GABARAP. Pflügers Arch. Eur. J. Physiol. 2010, 460, 207–217. [Google Scholar] [CrossRef]
- Ichida, Y.; Ohtomo, S.; Yamamoto, T.; Murao, N.; Tsuboi, Y.; Kawabe, Y.; Segawa, H.; Horiba, N.; Miyamoto, K.-I.; Floege, J. Evidence of an intestinal phosphate transporter alternative to type IIb sodium-dependent phosphate transporter in rats with chronic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 68–75. [Google Scholar] [CrossRef]
- Pastor-Arroyo, E.M.; Knöpfel, T.; Silva, P.H.I.; Schnitzbauer, U.; Poncet, N.; Biber, J.; Wagner, C.A.; Hernando, N. Intestinal epithelial ablation of Pit-2/Slc20a2 in mice leads to sustained elevation of vitamin D3 upon dietary restriction of phosphate. Acta Physiol. 2020, 230, e13526. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Ohtomo, S.; Ichida, Y.; Hagita, H.; Ozawa, K.; Iida, M.; Nagao, S.; Ikegami, H.; Takahashi, T.; Horiba, N. EOS789, a novel pan-phosphate transporter inhibitor, is effective for the treatment of chronic kidney disease–mineral bone disorder. Kidney Int. 2020, 98, 343–354. [Google Scholar] [CrossRef]
- Berndt, T.; Thomas, L.F.; Craig, T.A.; Sommer, S.; Li, X.; Bergstralh, E.J.; Kumar, R. Evidence for a signaling axis by which intestinal phosphate rapidly modulates renal phosphate reabsorption. Proc. Natl. Acad. Sci. USA 2007, 104, 11085–11090. [Google Scholar] [CrossRef]
- Lee, G.J.; Mossa-Al Hashimi, L.; Debnam, E.S.; Unwin, R.J.; Marks, J. Postprandial adjustments in renal phosphate excretion do not involve a gut-derived phosphaturic factor: No evidence for an intestinal phosphatonin. Exp. Physiol. 2017, 102, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Bettoni, C.; Knöpfel, T.; Hernando, N.; Biber, J.; Wagner, C.A. Acute Adaption to Oral or Intravenous Phosphate Requires Parathyroid Hormone. J. Am. Soc. Nephrol. 2017, 28, 903–914. [Google Scholar] [CrossRef]
- Bon, N.; Frangi, G.; Sourice, S.; Guicheux, J.; Beck-Cormier, S.; Beck, L. Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol. Metab. 2018, 11, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.A.U.; de Brauwere, D.P. Three feedback loops precisely regulating serum phosphate concentration. Kidney Int. 2011, 80, 443–445. [Google Scholar] [CrossRef]
- Kimata, M.; Michigami, T.; Tachikawa, K.; Okada, T.; Koshimizu, T.; Yamazaki, M.; Kogo, M.; Ozono, K. Signaling of extracellular inorganic phosphate up-regulates cyclin D1 expression in proliferating chondrocytes via the Na+/Pi cotransporter Pit-1 and Raf/MEK/ERK pathway. Bone 2010, 47, 938–947. [Google Scholar] [CrossRef]
- Chavkin, N.W.; Chia, J.J.; Crouthamel, M.H.; Giachelli, C.M. Phosphate uptake-independent signaling functions of the type III sodium-dependent phosphate transporter, PiT-1, in vascular smooth muscle cells. Exp. Cell Res. 2015, 333, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Bon, N.; Couasnay, G.; Bourgine, A.; Sourice, S.; Beck-Cormier, S.; Guicheux, J.; Beck, L. Phosphate (Pi)-regulated heterodimerization of the high-affinity sodium-dependent Pi transporters PiT1/Slc20a1 and PiT2/Slc20a2 underlies extracellular Pi sensing independently of Pi uptake. J. Biol. Chem. 2017, 12. [Google Scholar] [CrossRef]
- Beck, L.; Beck-Cormier, S. Extracellular phosphate sensing in mammals: What do we know? J. Mol. Endocrinol. 2020, 65, R53–R63. [Google Scholar] [CrossRef] [PubMed]
- Centeno, P.P.; Herberger, A.; Mun, H.-C.; Tu, C.; Nemeth, E.F.; Chang, W.; Conigrave, A.D.; Ward, D.T. Phosphate acts directly on the calcium-sensing receptor to stimulate parathyroid hormone secretion. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Geng, Y.; Mosyak, L.; Kurinov, I.; Zuo, H.; Sturchler, E.; Cheng, T.C.; Subramanyam, P.; Brown, A.P.; Brennan, S.C.; Mun, H.-C.; et al. Structural mechanism of ligand activation in human calcium-sensing receptor. eLife 2016, 5, e13662. [Google Scholar] [CrossRef]
- Loupy, A.; Ramakrishnan, S.K.; Wootla, B.; Chambrey, R.; De La Faille, R.; Bourgeois, S.; Bruneval, P.; Mandet, C.; Christensen, E.I.; Faure, H.; et al. PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor. J. Clin. Investig. 2012, 122, 3355–3367. [Google Scholar] [CrossRef]
- Good, D.W.; George, T.; Watts, B.A. Aldosterone potentiates 1,25-dihydroxyvitamin D3 action in renal thick ascending limb via a nongenomic, ERK-dependent pathway. Am. J. Physiol. Cell Physiol. 2003, 285, C1122–C1130. [Google Scholar] [CrossRef]
- Takashi, Y.; Kosako, H.; Sawatsubashi, S.; Kinoshita, Y.; Ito, N.; Tsoumpra, M.K.; Nangaku, M.; Abe, M.; Matsuhisa, M.; Kato, S.; et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc. Natl. Acad. Sci. USA 2019, 116, 11418–11427. [Google Scholar] [CrossRef]
- Takashi, Y.; Fukumoto, S. Phosphate-sensing and regulatory mechanism of FGF23 production. J. Endocrinol. Investig. 2020, 43, 877–883. [Google Scholar] [CrossRef]
- Block, G.A.; Rosenbaum, D.P.; Yan, A.; Chertow, G.M. Efficacy and Safety of Tenapanor in Patients with Hyperphosphatemia Receiving Maintenance Hemodialysis: A Randomized Phase 3 Trial. J. Am. Soc. Nephrol. 2019, 30, 641–652. [Google Scholar] [CrossRef]
- Thomas, L.; Xue, J.; Murali, S.K.; Fenton, R.A.; Dominguez Rieg, J.A.; Rieg, T. Pharmacological Npt2a Inhibition Causes Phosphaturia and Reduces Plasma Phosphate in Mice with Normal and Reduced Kidney Function. J. Am. Soc. Nephrol. 2019, 30, 2128–2139. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef]
- DeLacey, S.; Liu, Z.; Broyles, A.; El-Azab, S.A.; Guandique, C.F.; James, B.C.; Imel, E.A. Hyperparathyroidism and parathyroidectomy in X-linked hypophosphatemia patients. Bone 2019, 127, 386–392. [Google Scholar] [CrossRef]
- Lecoq, A.-L.; Chaumet-Riffaud, P.; Blanchard, A.; Dupeux, M.; Rothenbuhler, A.; Lambert, B.; Durand, E.; Boros, E.; Briot, K.; Silve, C.; et al. Hyperparathyroidism in Patients With X-Linked Hypophosphatemia. J. Bone Miner. Res. 2020, 35, 1263–1273. [Google Scholar] [CrossRef]
- Alon, U.S.; Levy-Olomucki, R.; Moore, W.V.; Stubbs, J.; Liu, S.; Quarles, L.D. Calcimimetics as an Adjuvant Treatment for Familial Hypophosphatemic Rickets. Clin. J. Am. Soc. Nephrol. 2008, 3, 658–664. [Google Scholar] [CrossRef]
- Roberts, M.S.; Gafni, R.I.; Brillante, B.; Guthrie, L.C.; Streit, J.; Gash, D.; Gelb, J.; Krusinska, E.; Brennan, S.C.; Schepelmann, M.; et al. Treatment of Autosomal Dominant Hypocalcemia Type 1 With the Calcilytic NPSP795 (SHP635). J. Bone Miner. Res. 2019, 34, 1609–1618. [Google Scholar] [CrossRef]
- Hannan, F.M.; Gorvin, C.M.; Babinsky, V.N.; Olesen, M.K.; Stewart, M.; Wells, S.; Cox, R.D.; Nemeth, E.F.; Thakker, R.V. Calcilytic NPSP795 Increases Plasma Calcium and PTH in an Autosomal Dominant Hypocalcemia Type 1 Mouse Model. JBMR Plus 2020, 4. Available online: https://onlinelibrary.wiley.com/doi/10.1002/jbm4.10402 (accessed on 19 January 2021). [CrossRef]


{kind=link}
| Gene | Human Inactivation | Same Phenotype Human/Murine Y/N | Murine Inactivation |
|---|---|---|---|
| SLC34A1 | Idiopathic hypercalcemia [22] | N | Hypophosphatemia High level of calcitriol Retarded secondary ossification [19] |
| Hypophosphatemic Nephrolithiasis/osteoporosis [20] | Y | ||
| Fanconi syndrome [23] | N | ||
| SLC34A2 | Normophosphatemia [25] | N | Embryonic lethal E10.5 [26] |
| N/A | Conditional knock out: Normophosphatemia Decreased intestinal absorption [27,28] | ||
| SLC34A3 | Hypophosphatemia Nephrolithiasis Impaired bone function [21,29] | N | Normophosphatemia No bone phenotype [30,31] |
| SLC20A1 | Unknown | N/A | Embryonic lethal E12.5 [32] |
| SLC20A2 | PFBC Normal phosphatemia No data about bone phenotype [33] | Y Y N/A | Brain calcifications Normal phosphatemia Decreased bone quality [34] |
| CaSR | Severe hyperparathyroidism Osteopenia Failure to thrive [35] | Y | Severe hyperparathyroidism and premature death Rickets [36,37] |
| Familial hypocalciuric hypercalcemia [38] | Y | Hypercalcemia Hypocalciuria [36] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueres, L.; Beck-Cormier, S.; Beck, L.; Marks, J. The Complexities of Organ Crosstalk in Phosphate Homeostasis: Time to Put Phosphate Sensing Back in the Limelight. Int. J. Mol. Sci. 2021, 22, 5701. https://doi.org/10.3390/ijms22115701
Figueres L, Beck-Cormier S, Beck L, Marks J. The Complexities of Organ Crosstalk in Phosphate Homeostasis: Time to Put Phosphate Sensing Back in the Limelight. International Journal of Molecular Sciences. 2021; 22(11):5701. https://doi.org/10.3390/ijms22115701
Chicago/Turabian StyleFigueres, Lucile, Sarah Beck-Cormier, Laurent Beck, and Joanne Marks. 2021. "The Complexities of Organ Crosstalk in Phosphate Homeostasis: Time to Put Phosphate Sensing Back in the Limelight" International Journal of Molecular Sciences 22, no. 11: 5701. https://doi.org/10.3390/ijms22115701
APA StyleFigueres, L., Beck-Cormier, S., Beck, L., & Marks, J. (2021). The Complexities of Organ Crosstalk in Phosphate Homeostasis: Time to Put Phosphate Sensing Back in the Limelight. International Journal of Molecular Sciences, 22(11), 5701. https://doi.org/10.3390/ijms22115701