
Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer


Abstract
1. Introduction
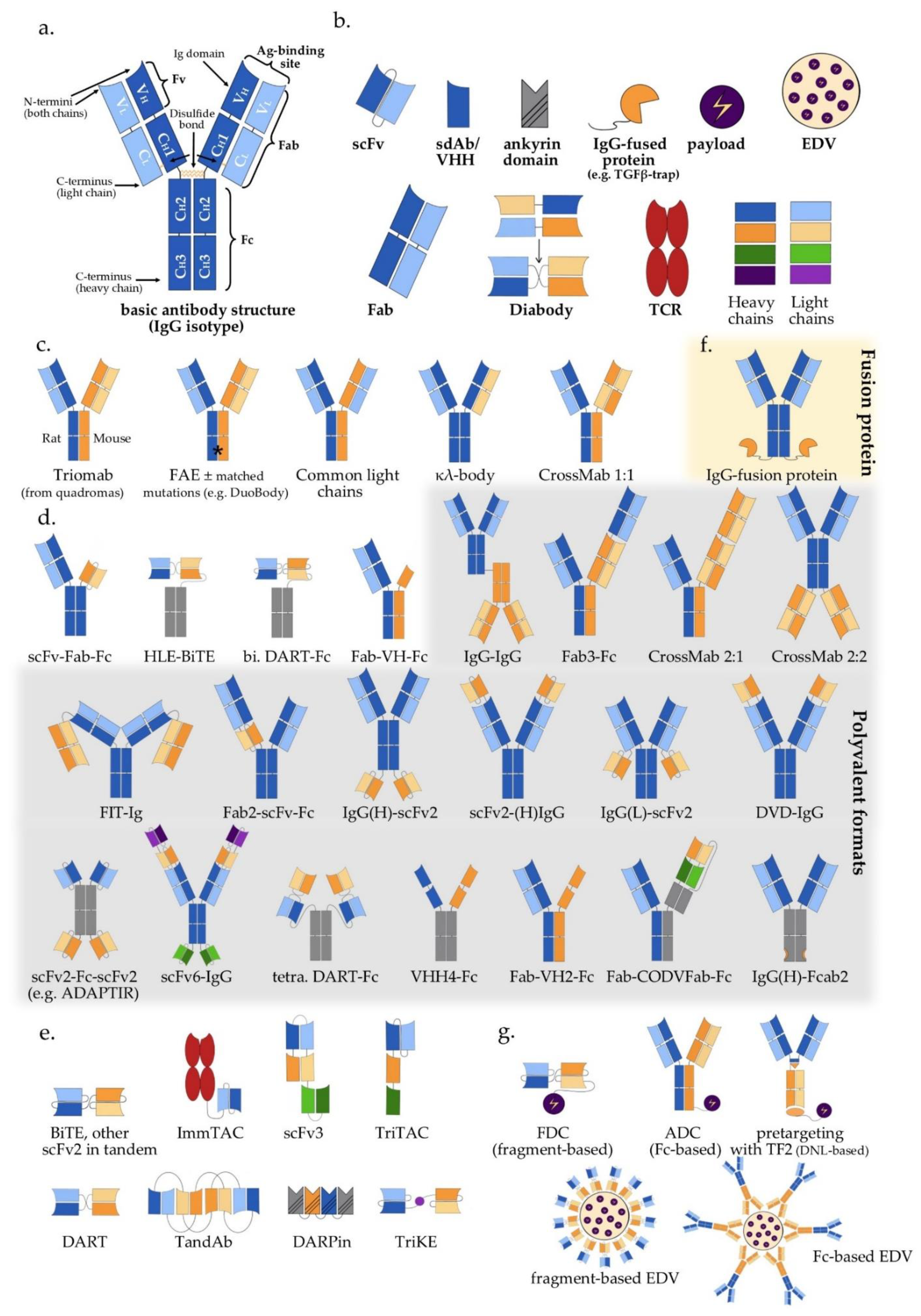
2. Antibody Structure and Approaches to Multispecificity
3. Bispecific Antibodies
3.1. IgG-Like Antibodies
3.2. IgG-Modified Bispecific Antibodies, Bivalent or Multivalent
3.3. Fragment-Based Bispecific Antibodies (Fc-Free), Bivalent or Multivalent
3.4. Antibody-Based Bispecific Fusion Proteins
4. Trispecific and Other Multispecific Antibodies
5. Payload Delivery Using Multispecific Constructs
6. Challenges and Perspectives
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Oreste, U.; Ametrano, A.; Coscia, M.R. On Origin and Evolution of the Antibody Molecule. Biology 2021, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Vos, M.R.; Kalisman, N.; Sherman, N.E.; Rachel, R.; Wirth, R.; Schröder, G.F.; Egelman, E.H. Archaeal flagellin combines a bacterial type IV pilin domain with an Ig-like domain. Proc. Natl. Acad. Sci. USA 2016, 113, 10352–10357. [Google Scholar] [CrossRef] [PubMed]
- Tilson, M.D.; Rzhetsky, A. A Novel Hypothesis Regarding the Evolutionary Origins of the Immunoglobulin Fold. Curr. Med. Res. Opin. 2000, 16, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Souriau, C.; Hudson, P.J. Recombinant antibodies for cancer diagnosis and therapy. Expert Opin. Biol. Ther. 2003, 3, 305–318. [Google Scholar] [CrossRef]
- Scott, A.M.; Allison, J.P.; Wolchok, J.D. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012, 12, 14. [Google Scholar]
- Rabia, L.A.; Desai, A.A.; Jhajj, H.S.; Tessier, P.M. Understanding and overcoming trade-offs between antibody affinity, specificity, stability and solubility. Biochem. Engin. J. 2018, 137, 365–374. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody humanization methods—A review and update. Biotechnol. Genet. Eng. Rev. 2013, 29, 175–186. [Google Scholar] [CrossRef]
- Parray, H.A.; Shukla, S.; Samal, S.; Shrivastava, T.; Ahmed, S.; Sharma, C.; Kumar, R. Hybridoma technology a versatile method for isolation of monoclonal antibodies, its applicability across species, limitations, advancement and future perspectives. Int. Immunopharmacol. 2020, 85, 106639. [Google Scholar] [CrossRef]
- Margulies, D.H.; Michael Kuehl, W.; Scharff, M.D. Somatic cell hybridization of mouse myeloma cells. Cell 1976, 8, 405–415. [Google Scholar] [CrossRef]
- Milstein, C.; Cuello, A.C. Hybrid hybridomas and their use in immunohistochemistry. Nature 1983, 305, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Casali, P.; Schettino, E.W. Structure and function of natural antibodies. Curr. Top Microbiol. Immunol. 1996, 210, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Norman, D.J. Mechanisms of action and overview of OKT3. Ther. Drug Monit. 1995, 17, 615–620. [Google Scholar] [CrossRef]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Gritti, G.; Rambaldi, A. Immunotherapy of Acute Lymphoblastic Leukemia and Lymphoma with T Cell–Redirected Bispecific Antibodies. JCO 2021, 39, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Spiess, C.; Merchant, M.; Huang, A.; Zheng, Z.; Yang, N.-Y.; Peng, J.; Ellerman, D.; Shatz, W.; Reilly, D.; Yansura, D.G.; et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat. Biotechnol. 2013, 31, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef]
- Chon, J.H.; Zarbis-Papastoitsis, G. Advances in the production and downstream processing of antibodies. Nat. Biotechnol. 2011, 28, 458–463. [Google Scholar] [CrossRef]
- Demarest, S.J.; Glaser, S.M. Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr. Opin. Drug Discov. Dev. 2008, 11, 675–687. [Google Scholar]
- Kelley, B. Industrialization of mAb production technology: The bioprocessing industry at a crossroads. mAbs 2009, 1, 443–452. [Google Scholar] [CrossRef]
- Schroeder, H.W.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed]
- Silverton, E.W.; Navia, M.A.; Davies, D.R. Three-dimensional structure of an intact human immunoglobulin. Proc. Natl. Acad. Sci. USA 1977, 74, 5140–5144. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, J.B.; Porter, R.R.; Press, E.M. The arrangement of the peptide chains in gamma-globulin. Biochem. J. 1963, 88, 220–228. [Google Scholar] [CrossRef]
- Bork, P.; Holm, L.; Sander, C. The immunoglobulin fold. Structural classification, sequence patterns and common core. J. Mol. Biol. 1994, 242, 309–320. [Google Scholar] [CrossRef]
- Porter, R.R. The hydrolysis of rabbit y-globulin and antibodies with crystalline papain. Biochem. J. 1959, 73, 119–126. [Google Scholar] [CrossRef]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.M.; Hamid, M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol. 2012, 2012, 980250. [Google Scholar] [CrossRef]
- Bever, C.S.; Dong, J.-X.; Vasylieva, N.; Barnych, B.; Cui, Y.; Xu, Z.-L.; Hammock, B.D.; Gee, S.J. VHH antibodies: Emerging reagents for the analysis of environmental chemicals. Anal. Bioanal. Chem. 2016, 408, 5985–6002. [Google Scholar] [CrossRef]
- Harmsen, M.M.; de Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef]
- Heidelberger, M. The Molecular Composition of Specific Immune Precipitates from Rabbit Sera. J. Am. Chem. Soc. 1938, 60, 242. [Google Scholar] [CrossRef]
- Van der Neut Kolfschoten, M.; Schuurman, J.; Losen, M.; Bleeker, W.K.; Martínez-Martínez, P.; Vermeulen, E.; den Bleker, T.H.; Wiegman, L.; Vink, T.; Aarden, L.A.; et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 2007, 317, 1554–1557. [Google Scholar] [CrossRef] [PubMed]
- Sedykh, S.E.; Lekchnov, E.A.; Prince, V.V.; Buneva, V.N.; Nevinsky, G.A. Half molecular exchange of IgGs in the blood of healthy humans: Chimeric lambda-kappa-immunoglobulins containing HL fragments of antibodies of different subclasses (IgG1-IgG4). Mol. Biosyst. 2016, 12, 3186–3195. [Google Scholar] [CrossRef] [PubMed]
- Nisonoff, A.; Rivers, M.M. Recombination of a mixture of univalent antibody fragments of different specificity. Arch. Biochem. Biophys. 1961, 93, 460–462. [Google Scholar] [CrossRef]
- Nisonoff, A.; Wissler, F.C.; Lipman, L.N. Properties of the major component of a peptic digest of rabbit antibody. Science 1960, 132, 1770–1771. [Google Scholar] [CrossRef]
- Suresh, M.R.; Cuello, A.C.; Milstein, C. Advantages of bispecific hybridomas in one-step immunocytochemistry and immunoassays. Proc. Natl. Acad. Sci. USA 1986, 83, 7989–7993. [Google Scholar] [CrossRef]
- Lindhofer, H.; Mocikat, R.; Steipe, B.; Thierfelder, S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J. Immunol. 1995, 155, 219–225. [Google Scholar]
- Ollier, R.; Wassmann, P.; Monney, T.; Ries Fecourt, C.; Gn, S.; Ca, V.; Ayoub, D.; Stutz, C.; Gudi, G.S.; Blein, S. Single-step Protein A and Protein G avidity purification methods to support bispecific antibody discovery and development. mAbs 2019, 11, 1464–1478. [Google Scholar] [CrossRef]
- Ridgway, J.B.; Presta, L.G.; Carter, P. ’Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef]
- Atwell, S.; Ridgway, J.B.; Wells, J.A.; Carter, P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J. Mol. Biol. 1997, 270, 26–35. [Google Scholar] [CrossRef]
- Kuglstatter, A.; Stihle, M.; Neumann, C.; Müller, C.; Schaefer, W.; Klein, C.; Benz, J. Structural differences between glycosylated, disulfide-linked heterodimeric Knob-into-Hole Fc fragment and its homodimeric Knob-Knob and Hole-Hole side products. Protein Eng. Des. Sel. 2017, 30, 649–656. [Google Scholar] [CrossRef]
- Moore, G.L.; Bautista, C.; Pong, E.; Nguyen, D.-H.T.; Jacinto, J.; Eivazi, A.; Muchhal, U.S.; Karki, S.; Chu, S.Y.; Lazar, G.A. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. mAbs 2011, 3, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Meesters, J.I.; de Goeij, B.E.C.G.; van den Bremer, E.T.J.; Neijssen, J.; van Kampen, M.D.; Strumane, K.; Verploegen, S.; Kundu, A.; Gramer, M.J.; et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 5145–5150. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Fischer, N.; Elson, G.; Magistrelli, G.; Dheilly, E.; Fouque, N.; Laurendon, A.; Gueneau, F.; Ravn, U.; Depoisier, J.-F.; Moine, V.; et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat. Commun. 2015, 6, 6113. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T.; Dumontet, C.; Brinkmann, U.; Bacac, M.; Umaña, P. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods 2019, 154, 21–31. [Google Scholar] [CrossRef]
- Fenn, S.; Schiller, C.B.; Griese, J.J.; Duerr, H.; Imhof-Jung, S.; Gassner, C.; Moelleken, J.; Regula, J.T.; Schaefer, W.; Thomas, M.; et al. Crystal structure of an anti-Ang2 CrossFab demonstrates complete structural and functional integrity of the variable domain. PLoS ONE 2013, 8, e61953. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef]
- Strop, P.; Ho, W.-H.; Boustany, L.M.; Abdiche, Y.N.; Lindquist, K.C.; Farias, S.E.; Rickert, M.; Appah, C.T.; Pascua, E.; Radcliffe, T.; et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J. Mol. Biol. 2012, 420, 204–219. [Google Scholar] [CrossRef]
- Davis, J.H.; Aperlo, C.; Li, Y.; Kurosawa, E.; Lan, Y.; Lo, K.-M.; Huston, J.S. SEEDbodies: Fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng. Des. Sel. 2010, 23, 195–202. [Google Scholar] [CrossRef]
- Von Kreudenstein, T.S.; Escobar-Carbrera, E.; Lario, P.I.; D’Angelo, I.; Brault, K.; Kelly, J.; Durocher, Y.; Baardsnes, J.; Woods, R.J.; Xie, M.H.; et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: Quality by molecular design. mAbs 2013, 5, 646–654. [Google Scholar] [CrossRef]
- Bostrom, J.; Yu, S.-F.; Kan, D.; Appleton, B.A.; Lee, C.V.; Billeci, K.; Man, W.; Peale, F.; Ross, S.; Wiesmann, C.; et al. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science 2009, 323, 1610–1614. [Google Scholar] [CrossRef]
- Schaefer, G.; Haber, L.; Crocker, L.M.; Shia, S.; Shao, L.; Dowbenko, D.; Totpal, K.; Wong, A.; Lee, C.V.; Stawicki, S.; et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 2011, 20, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, R. Dual Targeting. Patent Application No. 14/122,758, 24 July 2014. [Google Scholar]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.-R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Fu, W.; Xu, W.; Yang, Y.; Cruz, M.; Berezov, S.D.; Jorissen, D.; Takeda, H.; Zhu, W. Four-in-one antibodies have superior cancer inhibitory activity against EGFR, HER2, HER3, and VEGF through disruption of HER/MET crosstalk. Cancer Res. 2015, 75, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 2012, 18, 980–987. [Google Scholar] [CrossRef]
- Ravi, R.; Noonan, K.A.; Pham, V.; Bedi, R.; Zhavoronkov, A.; Ozerov, I.V.; Makarev, E.; Artemov, A.V.; Wysocki, P.T.; Mehra, R.; et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat. Commun. 2018, 9, 741. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, T.R.; Davis, Z.B.; Miller, J.S.; Vallera, D.A. Generation of BiKEs and TriKEs to Improve NK Cell-Mediated Targeting of Tumor Cells. Methods Mol. Biol. 2016, 1441, 333–346. [Google Scholar] [CrossRef]
- De Goeij, B.E.C.G.; Vink, T.; Ten Napel, H.; Breij, E.C.W.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P.W.H.I. Efficient Payload Delivery by a Bispecific Antibody-Drug Conjugate Targeting HER2 and CD63. Mol. Cancer Ther. 2016, 15, 2688–2697. [Google Scholar] [CrossRef]
- MacDiarmid, J.A.; Mugridge, N.B.; Weiss, J.C.; Phillips, L.; Burn, A.L.; Paulin, R.P.; Haasdyk, J.E.; Dickson, K.-A.; Brahmbhatt, V.N.; Pattison, S.T.; et al. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell 2007, 11, 431–445. [Google Scholar] [CrossRef] [PubMed]
- EDV Technology—EnGeneIC. Available online: https://engeneic.com/technology/edv-technology/ (accessed on 21 March 2021).
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies to extend plasma half-lives of recombinant antibodies. BioDrugs 2009, 23, 93–109. [Google Scholar] [CrossRef]
- Kellner, C.; Derer, S.; Valerius, T.; Peipp, M. Boosting ADCC and CDC activity by Fc engineering and evaluation of antibody effector functions. Methods 2014, 65, 105–113. [Google Scholar] [CrossRef]
- Ruf, P.; Lindhofer, H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood 2001, 98, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Saxena, V.; Ayyar, B.V. Affinity chromatography: A versatile technique for antibody purification. Methods 2017, 116, 84–94. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef]
- Van Witteloostuijn, S.B.; Pedersen, S.L.; Jensen, K.J. Half-Life Extension of Biopharmaceuticals using Chemical Methods: Alternatives to PEGylation. ChemMedChem 2016, 11, 2474–2495. [Google Scholar] [CrossRef]
- Lorenczewski, G.; Friedrich, M.; Kischel, R.; Dahlhoff, C.; Anlahr, J.; Balazs, M.; Rock, D.; Boyle, M.C.; Goldstein, R.; Coxon, A.; et al. Generation of a Half-Life Extended Anti-CD19 BiTE® Antibody Construct Compatible with Once-Weekly Dosing for Treatment of CD19-Positive Malignancies. Blood 2017, 130, 2815. [Google Scholar] [CrossRef]
- Li, Z.; Krippendorff, B.-F.; Sharma, S.; Walz, A.C.; Lavé, T.; Shah, D.K. Influence of molecular size on tissue distribution of antibody fragments. mAbs 2016, 8, 113–119. [Google Scholar] [CrossRef]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988, 242, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Gaissmaier, L.; Christopoulos, P. Immune Modulation in Lung Cancer: Current Concepts and Future Strategies. Respiration 2020, 1–27. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Laura Wood, Senior Press Manager. Global Bispecific Antibody Market to 2028—Opportunity, Drug Sales, Price & Clinical Trials Insights. Available online: https://www.businesswire.com/news/home/20210323005567/en/Global-Bispecific-Antibody-Market-to-2028---Opportunity-Drug-Sales-Price-Clinical-Trials-Insights---ResearchAndMarkets.com (accessed on 17 April 2021).
- Thakur, A.; Lum, L.G. “NextGen“ Biologics: Bispecific Antibodies and Emerging Clinical Results. Expert Opin. Biol. Ther. 2016, 16, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Chelius, D.; Ruf, P.; Gruber, P.; Plöscher, M.; Liedtke, R.; Gansberger, E.; Hess, J.; Wasiliu, M.; Lindhofer, H. Structural and functional characterization of the trifunctional antibody catumaxomab. mAbs 2010, 2, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Removab|European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/removab (accessed on 12 April 2021).
- Technische Universität München. Phase I/II Dose-Escalation Study of the Investigational Trifunctional Bispecific Anti-CD20 x Anti-CD3 Antibody FBTA05 in Combination with Donor Lymphocyte Infusion (DLI) in Patients with CD20 Positive Chronic Lymphocytic Leukemia (CLL), Low and High Grade Non-Hodgkin´s Lymphoma (NHL) After Allogeneic Stem Cell Transplantation: NCT01138579, STP-LYM-01-V01. Available online: https://clinicaltrials.gov/ct2/show/NCT01138579 (accessed on 12 April 2021).
- Neovii Biotech; Fresenius Biotech North America. Phase II Study of The Trifunctional Bispecific Anti-HER-2/Neu x Anti-CD3 Antibody Ertumaxomab in Patients with HER-2/Neu Overexpressing (3+ Or 2+/FISH+) Metastatic Breast Cancer Progressing After Trastuzumab Treatment: NCT00522457, IV-ERT-BC-04. Available online: https://clinicaltrials.gov/ct2/show/NCT00522457 (accessed on 12 April 2021).
- Moores, S.L.; Chiu, M.L.; Bushey, B.S.; Chevalier, K.; Luistro, L.; Dorn, K.; Brezski, R.J.; Haytko, P.; Kelly, T.; Wu, S.-J.; et al. A Novel Bispecific Antibody Targeting EGFR and cMet Is Effective against EGFR Inhibitor-Resistant Lung Tumors. Cancer Res. 2016, 76, 3942–3953. [Google Scholar] [CrossRef]
- Yun, J.; Lee, S.-H.; Kim, S.-Y.; Jeong, S.-Y.; Kim, J.-H.; Pyo, K.-H.; Park, C.-W.; Heo, S.G.; Yun, M.R.; Lim, S.; et al. Antitumor Activity of Amivantamab (JNJ-61186372), an EGFR-MET Bispecific Antibody, in Diverse Models of EGFR Exon 20 Insertion-Driven NSCLC. Cancer Discov. 2020, 10, 1194–1209. [Google Scholar] [CrossRef]
- Sabari, J.K.; Shu, C.A.; Park, K.; Leighl, N.; Mitchell, P.; Kim, S.; Lee, J.; Kim, D.; Viteri, S.; Spira, A.; et al. OA04.04 Amivantamab in Post-platinum EGFR Exon 20 Insertion Mutant Non-Small Cell Lung Cancer. J. Thor. Oncol. 2021, 16, S108–S109. [Google Scholar] [CrossRef]
- Cho, B.C.; Lee, K.H.; Cho, E.K.; Kim, D.-W.; Lee, J.-S.; Han, J.-Y.; Kim, S.-W.; Spira, A.; Haura, E.B.; Sabari, J.K.; et al. 1258O Amivantamab (JNJ-61186372), an EGFR-MET bispecific antibody, in combination with lazertinib, a 3rd-generation tyrosine kinase inhibitor (TKI), in advanced EGFR NSCLC. Ann. Oncol. 2020, 31, S813. [Google Scholar] [CrossRef]
- Witkowska, M.; Smolewski, P.; Robak, T. Investigational therapies targeting CD37 for the treatment of B-cell lymphoid malignancies. Expert Opin. Investig. Drugs 2018, 27, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Oostindie, S.C.; van der Horst, H.J.; Kil, L.P.; Strumane, K.; Overdijk, M.B.; van den Brink, E.N.; van den Brakel, J.H.N.; Rademaker, H.J.; van Kessel, B.; van den Noort, J.; et al. DuoHexaBody-CD37®, a novel biparatopic CD37 antibody with enhanced Fc-mediated hexamerization as a potential therapy for B-cell malignancies. Blood Cancer J. 2020, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Engelberts, P.J.; Hiemstra, I.H.; de Jong, B.; Schuurhuis, D.H.; Meesters, J.; Beltran Hernandez, I.; Oostindie, S.C.; Neijssen, J.; van den Brink, E.N.; Horbach, G.J.; et al. DuoBody-CD3xCD20 induces potent T-cell-mediated killing of malignant B cells in preclinical models and provides opportunities for subcutaneous dosing. EBioMedicine 2020, 52, 102625. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, M.; Mous, R.; Clausen, M.R.; Johnson, P.; Linton, K.M.; Chamuleau, M.E.; Lewis, D.J.; Sureda Balari, A.; Cunningham, D.; Oliveri, R.S.; et al. Subcutaneous Epcoritamab Induces Complete Responses with an Encouraging Safety Profile across Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma Subtypes, Including Patients with Prior CAR-T Therapy: Updated Dose Escalation Data. Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Mateos, M.-V.; Nahi, H.; Krishnan, A.Y.; van de Donk, N.W.; San Miguel, J.; Oriol, A.; Rosiñol, L.; Chari, A.; Adams, H.; et al. Phase I study of teclistamab, a humanized B-cell maturation antigen (BCMA) x CD3 bispecific antibody, in relapsed/refractory multiple myeloma (R/R MM). JCO 2020, 38, 100. [Google Scholar] [CrossRef]
- Garfall, A. Updated Phase 1 Results of Teclistamab, a B-Cell Maturation Antigen (BCMA) x CD3 Bispecific Antibody, in Relapsed and/or Refractory Multiple Myeloma (RRMM). Blood 2020, 136 (Suppl. 1), 27. [Google Scholar] [CrossRef]
- Nair-Gupta, P.; Diem, M.; Reeves, D.; Wang, W.; Schulingkamp, R.; Sproesser, K.; Mattson, B.; Heidrich, B.; Mendonça, M.; Joseph, J.; et al. A novel C2 domain binding CD33xCD3 bispecific antibody with potent T-cell redirection activity against acute myeloid leukemia. Blood Adv. 2020, 4, 906–919. [Google Scholar] [CrossRef]
- Nair-Gupta, P.; Diem, M.; Reeves, D.; Wang, W.; Schulingkamp, R.; Sproesser, K.; Mattson, B.; Heidrich, B.; Joseph, J.; Sendecki, J.; et al. Abstract 5662: JNJ-67571244: A novel anti-CD33 C2 domain binding bispecific antibody with potent T cell redirection activity. Cancer Res. 2020, 80, 5662. [Google Scholar] [CrossRef]
- Muik, A.; Altintas, I.; Kosoff, R.; Gieseke, F.; Schödel, K.; Salcedo, T.; Burm, S.; Toker, A.; Kranz, L.; Vormehr, M.; et al. 561 DuoBody®-PD-L1×4–1BB (GEN1046) induces superior immune-cell activation, cytokine production and cytotoxicity by combining PD-L1 blockade with conditional 4–1BB co-stimulation. J. Immunother. Cancer 2020, 8, A595. [Google Scholar] [CrossRef]
- Garralda, E.; Geva, R.; Ben-Ami, E.; Maurice-Dror, C.; Calvo, E.; LoRusso, P.; Türeci, Ö.; Niewood, M.; Şahin, U.; Jure-Kunkel, M.; et al. 412 First-in-human phase I/IIa trial to evaluate the safety and initial clinical activity of DuoBody®-PD-L1×4–1BB (GEN1046) in patients with advanced solid tumors. J. Immunother. Cancer 2020, 8, A437. [Google Scholar] [CrossRef]
- Kotanides, H.; Li, Y.; Malabunga, M.; Carpenito, C.; Eastman, S.W.; Shen, Y.; Wang, G.; Inigo, I.; Surguladze, D.; Pennello, A.L.; et al. Bispecific Targeting of PD-1 and PD-L1 Enhances T-cell Activation and Antitumor Immunity. Cancer Immunol. Res. 2020, 8, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, B.; Pudi, S.; Wallace Moyer, I.; Zhong, L.; Prinz, B.; Baruah, H.; Lynaugh, H.; Kumar, S.; Wittrup, K.D.; Nett, J.H. Purification of common light chain IgG-like bispecific antibodies using highly linear pH gradients. mAbs 2017, 9, 257–268. [Google Scholar] [CrossRef]
- Yen, W.-C.; Fischer, M.M.; Argast, G.; Wallace, B.; Wang, M.; Meisner, R.; Lewicki, J.; Kapoun, A.M.; Gurney, A.; Hoey, T. Abstract C164: Dual targeting of the DLL4 and VEGF pathways with a bispecific monoclonal antibody inhibits tumor growth and reduces cancer stem cell frequency. Mol. Cancer Ther. 2015, 14, C164. [Google Scholar] [CrossRef]
- Jimeno, A.; Moore, K.N.; Gordon, M.; Chugh, R.; Diamond, J.R.; Aljumaily, R.; Mendelson, D.; Kapoun, A.M.; Xu, L.; Stagg, R.; et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Invest. New Drugs 2019, 37, 461–472. [Google Scholar] [CrossRef]
- Bannerji, R.; Allan, J.N.; Arnason, J.E.; Brown, J.R.; Advani, R.H.; Barnes, J.A.; Ansell, S.M.; O’Brien, S.M.; Chavez, J.; Duell, J.; et al. Clinical Activity of REGN1979, a Bispecific Human, Anti-CD20 x Anti-CD3 Antibody, in Patients with Relapsed/Refractory (R/R) B-Cell Non-Hodgkin Lymphoma (B-NHL). Blood 2019, 134, 762. [Google Scholar] [CrossRef]
- Moek, K.L.; Fehrmann, R.S.N.; van der Vegt, B.; de Vries, E.G.E.; de Groot, D.J.A. Glypican 3 Overexpression across a Broad Spectrum of Tumor Types Discovered with Functional Genomic mRNA Profiling of a Large Cancer Database. Am. J. Pathol. 2018, 188, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- Masternak, K.; Chauchet, X.; Buatois, V.; Salgado-Pires, S.; Shang, L.; Johnson, Z.; Dheilly, E.; Moine, V.; Ferlin, W.G.; Kosco-Vilbois, M.H.; et al. Abstract B37: NI-1701, a bispecific antibody for selective neutralization of CD47 in B cell malignancies. Cancer Immunol. Res. 2017, 5, B37. [Google Scholar]
- Olszewski, A. Single-Agent Mosunetuzumab Is a Promising Safe and Efficacious Chemotherapy-Free Regimen for Elderly/Unfit Patients with Previously Untreated Diffuse Large B-Cell Lymphoma. In Proceedings of the 62nd ASH Annual Meeting and Exposition, San Diego, CA, USA, 5–8 December 2020. [Google Scholar]
- Bendell, J.C.; Sauri, T.; Gracián, A.C.; Alvarez, R.; López-López, C.; García-Alfonso, P.; Hussein, M.; Miron, M.-L.L.; Cervantes, A.; Montagut, C.; et al. The McCAVE Trial: Vanucizumab plus mFOLFOX-6 Versus Bevacizumab plus mFOLFOX-6 in Patients with Previously Untreated Metastatic Colorectal Carcinoma (mCRC). Oncologist 2020, 25, e451–e459. [Google Scholar] [CrossRef]
- Kang, T.H.; Jung, S.T. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hamilton, E.P.; Beeram, M.; Hanna, D.L.; El-Khoueiry, A.B.; Kang, Y.-K.; Lee, K.W.; Lee, J.; Rha, S.Y.; Chaves, J.M.; et al. Zanidatamab (ZW25) in HER2-expressing gastroesophageal adenocarcinoma (GEA): Results from a phase I study. JCO 2021, 39, 164. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hanna, D.L.; El-Khoueiry, A.B.; Kang, Y.-K.; Oh, D.-Y.; Chaves, J.M.; Rha, S.Y.; Hamilton, E.P.; Pant, S.; Javle, M.M.; et al. Zanidatamab (ZW25) in HER2-positive biliary tract cancers (BTCs): Results from a phase I study. JCO 2021, 39, 299. [Google Scholar] [CrossRef]
- Moore, G.L.; Bernett, M.J.; Rashid, R.; Pong, E.W.; Nguyen, D.-H.T.; Jacinto, J.; Eivazi, A.; Nisthal, A.; Diaz, J.E.; Chu, S.Y.; et al. A robust heterodimeric Fc platform engineered for efficient development of bispecific antibodies of multiple formats. Methods 2019, 154, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yi, J.; Zhou, P. Development of bispecific antibodies in China: Overview and prospects. Antib. Ther. 2020, 3, 126–145. [Google Scholar] [CrossRef]
- Moretti, P.; Skegro, D.; Ollier, R.; Wassmann, P.; Aebischer, C.; Laurent, T.; Schmid-Printz, M.; Giovannini, R.; Blein, S.; Bertschinger, M. BEAT® the bispecific challenge: A novel and efficient platform for the expression of bispecific IgGs. BMC Proc. 2013, 7. [Google Scholar] [CrossRef]
- Moore, P.A.; Shah, K.; Yang, Y.; Alderson, R.; Roberts, P.; Long, V.; Liu, D.; Li, J.C.; Burke, S.; Ciccarone, V.; et al. Development of MGD007, a gpA33 x CD3-Bispecific DART Protein for T-Cell Immunotherapy of Metastatic Colorectal Cancer. Mol. Cancer Ther. 2018, 17, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Arvedson, T.L.; Balazs, M.; Bogner, P.; Black, K.; Graham, K.; Henn, A.; Friedrich, M.; Hoffmann, P.; Kischel, R.; Kufer, P.; et al. Abstract 55: Generation of half-life extended anti-CD33 BiTE® antibody constructs compatible with once-weekly dosing. Cancer Res. 2017, 77, 55. [Google Scholar] [CrossRef]
- Subklewe, M.; Stein, A.; Walter, R.B.; Bhatia, R.; Wei, A.H.; Ritchie, D.; Bücklein, V.; Vachhani, P.; Dai, T.; Hindoyan, A.; et al. Preliminary Results from a Phase 1 First-in-Human Study of AMG 673, a Novel Half-Life Extended (HLE) Anti-CD33/CD3 BiTE® (Bispecific T-Cell Engager) in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML). Blood 2019, 134, 833. [Google Scholar] [CrossRef]
- Liu, L.; Lam, C.-Y.K.; Long, V.; Widjaja, L.; Yang, Y.; Li, H.; Jin, L.; Burke, S.; Gorlatov, S.; Brown, J.; et al. MGD011, A CD19 x CD3 Dual-Affinity Retargeting Bi-specific Molecule Incorporating Extended Circulating Half-life for the Treatment of B-Cell Malignancies. Clin. Cancer Res. 2017, 23, 1506–1518. [Google Scholar] [CrossRef]
- Van der Linden, R.; Frenken, L.; de Geus, B.; Harmsen, M.M.; Ruuls, R.C.; Stok, W.; de Ron, L.; Wilson, S.; Davis, P.; Verrips, C.T. Comparison of physical chemical properties of llama VHH antibody fragments and mouse monoclonal antibodies. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1999, 1431, 37–46. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, T.R.; Kodal, B.; Lenvik, A.J.; Hinderlie, P.; Bendzick, L.E.; Schirm, D.K.; Kaminski, M.F.; McElmurry, R.T.; Geller, M.A.; et al. Potent Cytolytic Activity and Specific IL15 Delivery in a Second-Generation Trispecific Killer Engager. Cancer Immunol. Res. 2020, 8, 1139–1149. [Google Scholar] [CrossRef]
- Clarke, S.; Dang, K.; Li, Y.; Sankaran, P.; Pham, D.; Balasubramani, A.; Davison, L.; Harris, K.; Jorgensen, B.; Schellenberger, U.; et al. A novel CD3xPSMA bispecific antibody for efficient T cell mediated killing of prostate tumor cells with minimal cytokine release. JCO 2019, 37, 324. [Google Scholar] [CrossRef]
- Malik, H.; Buelow, B.; Avanzino, B.; Balasubramani, A.; Boudreau, A.; Clarke, S.; Dang, K.; Davison, L.; Force Aldred, S.; Harris, K.; et al. A Novel Fully Human Bispecific CD19 x CD3 Antibody That Kills Lymphoma Cells with Minimal Cytokine Secretion. Blood 2018, 132, 1671. [Google Scholar] [CrossRef]
- Costa, L.J.; Wong, S.W.; Bermúdez, A.; de La Rubia, J.; Mateos, M.-V.; Ocio, E.M.; Rodríguez-Otero, P.; San-Miguel, J.; Li, S.; Sarmiento, R.; et al. First Clinical Study of the B-Cell Maturation Antigen (BCMA) 2 + 1 T Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Interim Results of a Phase 1 Multicenter Trial. Blood 2019, 134, 143. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. mAbs 2016, 8, 1010–1020. [Google Scholar] [CrossRef]
- Bacac, M.; Colombetti, S.; Herter, S.; Sam, J.; Perro, M.; Chen, S.; Bianchi, R.; Richard, M.; Schoenle, A.; Nicolini, V.; et al. CD20-TCB with Obinutuzumab Pretreatment as Next-Generation Treatment of Hematologic Malignancies. Clin. Cancer Res. 2018, 24, 4785–4797. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, M.; Morschhauser, F.; Iacoboni, G.; Carlo-Stella, C.; Offner, F.C.; Sureda, A.; Salles, G.; Martínez-Lopez, J.; Crump, M.; Thomas, D.N.; et al. Glofitamab, a Novel, Bivalent CD20-Targeting T-Cell-Engaging Bispecific Antibody, Induces Durable Complete Remissions in Relapsed or Refractory B-Cell Lymphoma: A Phase I Trial. JCO 2021. [Google Scholar] [CrossRef]
- Budde, L.E.; Sehn, L.H.; Assouline, S.; Flinn, I.W.; Isufi, I.; Yoon, S.-S.; Kim, W.-S.; Matasar, M.J.; Nastoupil, L.J.; Santiago, R.; et al. Mosunetuzumab, a Full-Length Bispecific CD20/CD3 Antibody, Displays Clinical Activity in Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma (NHL): Interim Safety and Efficacy Results from a Phase 1 Study. Blood 2018, 132, 399. [Google Scholar] [CrossRef]
- Brünker, P.; Wartha, K.; Friess, T.; Grau-Richards, S.; Waldhauer, I.; Koller, C.F.; Weiser, B.; Majety, M.; Runza, V.; Niu, H.; et al. RG7386, a Novel Tetravalent FAP-DR5 Antibody, Effectively Triggers FAP-Dependent, Avidity-Driven DR5 Hyperclustering and Tumor Cell Apoptosis. Mol. Cancer Ther. 2016, 15, 946–957. [Google Scholar] [CrossRef]
- Gong, S.; Ren, F.; Wu, D.; Wu, X.; Wu, C. Fabs-in-tandem immunoglobulin is a novel and versatile bispecific design for engaging multiple therapeutic targets. mAbs 2017, 9, 1118–1128. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O. Abstract DDT02-03: AMG 509: A novel, humanized, half-Life extended, bispecific STEAP1 × CD3 T cell recruiting XmAb ® 2+1 antibody. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Ji, J. Abstract CT120: AK104, a PD-1/CTLA-4 bispecific antibody in combination with chemotherapy as first-line therapy in a phase Ib trial in patients (pts) with advanced gastric (G) or gastroesophageal junction (GEJ) cancer. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Liu, L.; Zeng, W.; Chedid, M.; Zeng, Y.; Tschang, S.; Tian, Y.; Tang, Y.; Lu, J. Abstract 873: A novel MET-EGFR bispecific antibody LY3164530 shows advantage over combining MET and EGFR antibodies in tumor inhibition and overcome resistance. Cancer Res. 2016, 76, 873. [Google Scholar] [CrossRef]
- Patnaik, A.; Gordon, M.; Tsai, F.; Papadopoulos, K.P.; Rasco, D.; Beeram, M.; Fu, S.; Janku, F.; Hynes, S.M.; Gundala, S.R.; et al. A phase I study of LY3164530, a bispecific antibody targeting MET and EGFR, in patients with advanced or metastatic cancer. Cancer Chemother. Pharmacol. 2018, 82, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, M.; Guo, H.; Chen, Y.; Huse, M.; Cheung, N.-K.V. Retargeting T cells to GD2 pentasaccharide on human tumors using Bispecific humanized antibody. Cancer Immunol. Res. 2015, 3, 266–277. [Google Scholar] [CrossRef]
- Li, Y.; Hickson, J.A.; Ambrosi, D.J.; Haasch, D.L.; Foster-Duke, K.D.; Eaton, L.J.; DiGiammarino, E.L.; Panchal, S.C.; Jiang, F.; Mudd, S.R.; et al. ABT-165, a Dual Variable Domain Immunoglobulin (DVD-Ig) Targeting DLL4 and VEGF, Demonstrates Superior Efficacy and Favorable Safety Profiles in Preclinical Models. Mol. Cancer Ther. 2018, 17, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Hoyos, G.; Sewell, T.; Bader, R.; Bannink, J.; Chenault, R.A.; Daugherty, M.; Dasovich, M.; Fang, H.; Gottschalk, R.; Kumer, J.; et al. MOR209/ES414, a Novel Bispecific Antibody Targeting PSMA for the Treatment of Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2016, 15, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Aptevo Therapeutics Inc. Aptevo Therapeutics and MorphoSys End Joint Development and Commercialization Agreement for MOR209/ES414. Aptevo Therapeutics Inc. Available online: https://www.globenewswire.com/news-release/2017/08/31/1106420/0/en/Aptevo-Therapeutics-and-MorphoSys-End-Joint-Development-and-Commercialization-Agreement-for-MOR209-ES414.html (accessed on 25 April 2021).
- La Motte-Mohs, R.; Shah, K.; Brown, J.G.; Smith, D.; Gorlatov, S.; Ciccarone, V.; Tamura, J.K.; Li, H.; Rillema, J.R.; Licea, M.; et al. Preclinical characterization of MGD013, a PD-1 x LAG-3 bispecific DART® molecule; 32nd Annual Meeting and Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2017): Part Two. J. Immunother. Cancer 2017, 5, 87. [Google Scholar] [CrossRef]
- Berezhnoy, A.; Sumrow, B.J.; Stahl, K.; Shah, K.; Liu, D.; Li, J.; Hao, S.-S.; de Costa, A.; Kaul, S.; Bendell, J.; et al. Development and Preliminary Clinical Activity of PD-1-Guided CTLA-4 Blocking Bispecific DART Molecule. Cell Rep. Med. 2020, 1, 100163. [Google Scholar] [CrossRef]
- Luke, J.J.; Patel, M.R.; Hamilton, E.P.; Chmielowski, B.; Ulahannan, S.V.; Kindler, H.L.; Bahadur, S.W.; Clingan, P.R.; Mallesara, G.; Weickhardt, A.J.; et al. A phase I, first-in-human, open-label, dose-escalation study of MGD013, a bispecific DART molecule binding PD-1 and LAG-3, in patients with unresectable or metastatic neoplasms. JCO 2020, 38, 3004. [Google Scholar] [CrossRef]
- Sharma, M.; Sanborn, R.E.; Cote, G.M.; Bendell, J.C.; Kaul, S.; Chen, F.; Berezhnoy, A.; Moore, P.; Bonvini, E.; Sumrow, B.J.; et al. 1020O A phase I, first-in-human, open-label, dose escalation study of MGD019, an investigational bispecific PD-1 x CTLA-4 DART® molecule in patients with advanced solid tumours. Ann. Oncol. 2020, 31, S704–S705. [Google Scholar] [CrossRef]
- Inhibrx. INBRX-105|Inhibrx|Improving Checkpoint Inhibitor Therapy. Available online: https://inhibrx.com/inbrx-105/ (accessed on 25 April 2021).
- Gong, J.; Dong, Z.; Liu, D.; Xu, J.; Yang, J.; Yang, Y.; Qi, Y.; Men, J.; Kong, P.; Xu, T.; et al. 339 Preliminary safety, tolerability and efficacy results of KN026 (a HER2-targeted Bispecific Antibody) in combination with KN046 (an anti-PD-L1/CTLA-4 Bispecific Antibody) in patients (pts) with HER2 aberrated solid tumors. J. Immunother. Cancer 2020, 8, A364. [Google Scholar] [CrossRef]
- Wang, Y.; Ni, H.; Zhou, S.; He, K.; Gao, Y.; Wu, W.; Wu, M.; Wu, Z.; Qiu, X.; Zhou, Y.; et al. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol. Immunother. 2021, 70, 365–376. [Google Scholar] [CrossRef]
- Buelow, B.; D’Souza, A.; Rodriguez, C.; Vij, R.; Nath, R.; Snyder, M.; Pham, D.; Patel, A.; Iyer, S. TNB383B.0001: A Multicenter, Phase 1, Open-Label, Dose-Escalation Andexpansion Study of TNB-383B, a Bispecific Antibodytargeting BCMA in Subjects with Relapsed or Refractorymultiple Myeloma. Blood 2019, 134, 1874. [Google Scholar] [CrossRef]
- Rodriguez, C.; D’Souza, A.; Shah, N.; Voorhees, P.M.; Buelow, B.; Vij, R.; Kumar, S.K. Initial Results of a Phase I Study of TNB-383B, a BCMA x CD3 Bispecific T-Cell Redirecting Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 43–44. [Google Scholar] [CrossRef]
- Everett, K.L.; Kraman, M.; Wollerton, F.P.G.; Zimarino, C.; Kmiecik, K.; Gaspar, M.; Pechouckova, S.; Allen, N.L.; Doody, J.F.; Tuna, M. Generation of Fcabs targeting human and murine LAG-3 as building blocks for novel bispecific antibody therapeutics. Methods 2019, 154, 60–69. [Google Scholar] [CrossRef]
- Yap, T.; Wong, D.; Hu-Lieskovan, S.; Papadopoulos, K.; Morrow, M.; Grabowska, U.; Gliddon, D.; Holz, J.-B.; LoRusso, P. 395 A first-in-human study of FS118, a tetravalent bispecific antibody targeting LAG-3 and PD-L1, in patients with advanced cancer and resistance to PD-(L)1 therapy. J. Immunother. Cancer 2020, 8, A420. [Google Scholar] [CrossRef]
- Kraman, M.; Faroudi, M.; Allen, N.L.; Kmiecik, K.; Gliddon, D.; Seal, C.; Koers, A.; Wydro, M.M.; Batey, S.; Winnewisser, J.; et al. FS118, a Bispecific Antibody Targeting LAG-3 and PD-L1, Enhances T-Cell Activation Resulting in Potent Antitumor Activity. Clin. Cancer Res. 2020, 26, 3333–3344. [Google Scholar] [CrossRef]
- Jen, E.Y.; Xu, Q.; Schetter, A.; Przepiorka, D.; Shen, Y.L.; Roscoe, D.; Sridhara, R.; Deisseroth, A.; Philip, R.; Farrell, A.T.; et al. FDA Approval: Blinatumomab for Patients with B-cell Precursor Acute Lymphoblastic Leukemia in Morphologic Remission with Minimal Residual Disease. Clin. Cancer Res. 2019, 25, 473–477. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. JCO 2020, 38, 775–783. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). JCO 2020, 38, 7508. [Google Scholar] [CrossRef]
- Feldmann, A.; Arndt, C.; Töpfer, K.; Stamova, S.; Krone, F.; Cartellieri, M.; Koristka, S.; Michalk, I.; Lindemann, D.; Schmitz, M.; et al. Novel humanized and highly efficient bispecific antibodies mediate killing of prostate stem cell antigen-expressing tumor cells by CD8+ and CD4+ T cells. J. Immunol. 2012, 189, 3249–3259. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, U.; Huhalov, A.; Harms, B.; Paragas, V.; Adams, S.; Gu, J.; Nguyen, S.; Luus, L.; Oyama, S.; Razlog, M.; et al. MM-111: A novel bispecific antibody targeting ErbB3 with potent anti-tumor activity in ErbB2 over-expressing malignancies. SABCS. Cancer Res. 2009. [Google Scholar] [CrossRef]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Fu, J.; Zhang, M.; Liu, D. AFM13: A first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J. Hematol. Oncol. 2015, 8, 96. [Google Scholar] [CrossRef]
- Kipriyanov, S.M.; Moldenhauer, G.; Schuhmacher, J.; Cochlovius, B.; von der Lieth, C.W.; Matys, E.R.; Little, M. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J. Mol. Biol. 1999, 293, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Rothe, A.; Sasse, S.; Topp, M.S.; Eichenauer, D.A.; Hummel, H.; Reiners, K.S.; Dietlein, M.; Kuhnert, G.; Kessler, J.; Buerkle, C.; et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 2015, 125, 4024–4031. [Google Scholar] [CrossRef]
- Sasse, S. AFM13 in Patients with Relapsed or Refractory Hodgkin Lymphoma: Final Results of an Open-Label, Randomized, Multicenter Phase II Trial. Blood 2020, 136 (Suppl. 1), 31–32. [Google Scholar] [CrossRef]
- Bartlett, N.L.; Herrera, A.F.; Domingo-Domenech, E.; Mehta, A.; Forero-Torres, A.; Garcia-Sanz, R.; Armand, P.; Devata, S.; Izquierdo, A.R.; Lossos, I.S.; et al. A phase 1b study of AFM13 in combination with pembrolizumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2020, 136, 2401–2409. [Google Scholar] [CrossRef]
- Fury, M.G.; Lipton, A.; Smith, K.M.; Winston, C.B.; Pfister, D.G. A phase-I trial of the epidermal growth factor receptor directed bispecific antibody MDX-447 without and with recombinant human granulocyte-colony stimulating factor in patients with advanced solid tumors. Cancer Immunol. Immunother. 2008, 57, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Hovest, L.; Hartlapp, I.; Marwan, W.; Brem, G.; Rammensee, H.-G.; Jung, G. A recombinant bispecific single-chain antibody induces targeted, supra-agonistic CD28-stimulation and tumor cell killing. Eur. J. Immunol. 2003, 33, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Kim, T.M.; Vicente, D.; Felip, E.; Lee, D.H.; Lee, K.H.; Lin, C.-C.; Flor, M.J.; Di Nicola, M.; Alvarez, R.M.; et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Second-Line Treatment of Patients With NSCLC: Results From an Expansion Cohort of a Phase 1 Trial. J. Thor. Oncol. 2020, 15, 1210–1222. [Google Scholar] [CrossRef]
- Merck KGaA. Merck KGaA, Darmstadt, Germany, Reports Topline Data for Bintrafusp Alfa as Second-Line Monotherapy Treatment in Biliary Tract Cancer. Available online: https://www.emdgroup.com/en/news/bintrafusp-topline-data-biliary-tract-cancer-16-03-2021.html (accessed on 29 April 2021).
- Li, H.; Wang, C.; Guo, H.; Gu, L. The preclinical characterization of TST005, a bi-functional anti-PD-L1 and TGF-β trap fusion protein. AACR Ann. Meet. 2021. Abstract 972. [Google Scholar]
- Durm, G.; Frentzas, S.; Rasmussen, E.; Najmi, S.; Sadraei, N. 417 Design and rationale of a phase 1 study evaluating AMG 256, a novel, targeted, IL-21 receptor agonist and anti-PD-1 antibody, in patients with advanced solid tumors. J. Immunother. Cancer 2020, 8, A443. [Google Scholar] [CrossRef]
- Yu, J.; Song, Y.; Tian, W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol. 2020, 13, 45. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Hassel, J.C.; Rutkowski, P.; Baurain, J.-F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Phase 3 randomized trial comparing tebentafusp with investigator’s choice in first line metastatic uveal melanoma. AACR Ann. Meet. 2021. [Google Scholar]
- Ishihara, M.; Kageyama, S.; Miyahara, Y.; Ishikawa, T.; Ueda, S.; Soga, N.; Naota, H.; Mukai, K.; Harada, N.; Ikeda, H.; et al. MAGE-A4, NY-ESO-1 and SAGE mRNA expression rates and co-expression relationships in solid tumours. BMC Cancer 2020, 20, 606. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yin, J.; Zhong, J.; Yang, Z.; Tang, A.; Li, S. Clinicopathological and Prognostic Significance of PRAME Overexpression in Human Cancer: A Meta-Analysis. BioMed Res. Int. 2020, 2020, 8828579. [Google Scholar] [CrossRef]
- Ellis, J.M.; Henson, V.; Slack, R.; Ng, J.; Hartzman, R.J.; Katovich Hurley, C. Frequencies of HLA-A2 alleles in five U.S. population groups. Hum. Immunol. 2000, 61, 334–340. [Google Scholar] [CrossRef]
- Mullard, A. Trispecific antibodies take to the clinic. Nat. Rev. Drug Discov. 2020, 19, 657–658. [Google Scholar] [CrossRef]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Overview: GT Biopharma, Inc. (GTBP). Available online: https://www.gtbiopharma.com/product-pipeline/overview (accessed on 9 March 2021).
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef]
- Vallera, D.A.; Ferrone, S.; Kodal, B.; Hinderlie, P.; Bendzick, L.; Ettestad, B.; Hallstrom, C.; Zorko, N.A.; Rao, A.; Fujioka, N.; et al. NK-Cell-Mediated Targeting of Various Solid Tumors Using a B7-H3 Tri-Specific Killer Engager In Vitro and In Vivo. Cancers 2020, 12, 2659. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Austin, R.; Aaron, W.; Barath, M.; Callihan, E.; Gatto, S.; Hemmati, G.; Che-Leung, L.; Yu, T. FLT3-targeting TriTACs are T cell engagers for treatment of acute myeloid leukemia. AACR Ann. Meet. 2021. Abstract 2643. [Google Scholar]
- Molloy, M.; Valenzuela, L.; Basak, P.; Law, C. Combinatorial antitumor effects of CD3-based trispecific T cell activating constructs (TriTACs) and checkpoint inhibitors in preclinical models. AACR Ann. Meet 2021. Abstract 1573. [Google Scholar]
- Stumpp, M.T.; Dawson, K.M.; Binz, H.K. Beyond Antibodies: The DARPin® Drug Platform. BioDrugs 2020, 34, 423–433. [Google Scholar] [CrossRef]
- Liu, R.; Li, H.; Liu, L.; Yu, J.; Ren, X. Fibroblast activation protein: A potential therapeutic target in cancer. Cancer Biol. Ther. 2012, 13, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.A.; Goldenberg, D.M.; Cardillo, T.M.; McBride, W.J.; Sharkey, R.M.; Chang, C.-H. Stably tethered multifunctional structures of defined composition made by the dock and lock method for use in cancer targeting. Proc. Natl. Acad. Sci. USA 2006, 103, 6841–6846. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.A.; Goldenberg, D.M.; Cardillo, T.M.; Stein, R.; Chang, C.-H. Hexavalent bispecific antibodies represent a new class of anticancer therapeutics: 1. Properties of anti-CD20/CD22 antibodies in lymphoma. Blood 2009, 113, 6161–6171. [Google Scholar] [CrossRef] [PubMed]
- Pekar, L.; Busch, M.; Valldorf, B.; Hinz, S.C.; Toleikis, L.; Krah, S.; Zielonka, S. Biophysical and biochemical characterization of a VHH-based IgG-like bi- and trispecific antibody platform. mAbs 2020, 12, 1812210. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, L.; Wang-Rodriguez, J.; Zhang, L.; Cui, B.; Frankel, W.; Wu, R.; Kipps, T.J. The onco-embryonic antigen ROR1 is expressed by a variety of human cancers. Am. J. Pathol. 2012, 181, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Peddi, P.F.; Hurvitz, S.A. Trastuzumab emtansine: The first targeted chemotherapy for treatment of breast cancer. Future Oncol. 2013, 9, 319–326. [Google Scholar] [CrossRef]
- Linehan, A.S.; Fitzpatrick, O.M.; Morris, P.G. Profile of Trastuzumab Deruxtecan in the Management of Patients with HER2-Positive Unresectable or Metastatic Breast Cancer: An Evidence-Based Review. Breast Cancer 2021, 13, 151–159. [Google Scholar] [CrossRef]
- Scott, L.J. Brentuximab Vedotin: A Review in CD30-Positive Hodgkin Lymphoma. Drugs 2017, 77, 435–445. [Google Scholar] [CrossRef]
- Deonarain, M.P.; Yahioglu, G.; Stamati, I.; Pomowski, A.; Clarke, J.; Edwards, B.M.; Diez-Posada, S.; Stewart, A.C. Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours? Antibodies 2018, 7, 16. [Google Scholar] [CrossRef]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Sagnella, S.M.; Trieu, J.; Brahmbhatt, H.; MacDiarmid, J.A.; MacMillan, A.; Whan, R.M.; Fife, C.M.; McCarroll, J.A.; Gifford, A.J.; Ziegler, D.S.; et al. Targeted Doxorubicin-Loaded Bacterially Derived Nano-Cells for the Treatment of Neuroblastoma. Mol. Cancer Ther. 2018, 17, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Whittle, J.R.; Lickliter, J.D.; Gan, H.K.; Scott, A.M.; Simes, J.; Solomon, B.J.; MacDiarmid, J.A.; Brahmbhatt, H.; Rosenthal, M.A. First in human nanotechnology doxorubicin delivery system to target epidermal growth factor receptors in recurrent glioblastoma. J. Clin. Neurosci. 2015, 22, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Merck KGaA. M1231 M1231: Potential First in class bispecific antibody drug conjugate Targeting EGFR and MUC1. Available online: https://www.merckgroup.com/investors/events-and-presentations/webcasts-and-presentations/2020/en/200819_RD_UpdateCall_Final_EN%20FINAL.pdf (accessed on 21 March 2021).
- Hamblett, K.J.; Hammond, P.W.; Barnscher, S.D.; Fung, V.K.; Davies, R.H.; Wickman, G.R.; Hernandez, A.; Ding, T.; Galey, A.S.; Winters, G.C.; et al. Abstract 3914: ZW49, a HER2-targeted biparatopic antibody-drug conjugate for the treatment of HER2-expressing cancers. Cancer Res. 2018, 78, 3914. [Google Scholar] [CrossRef]
- Vallera, D.A.; Chen, H.; Sicheneder, A.R.; Panoskaltsis-Mortari, A.; Taras, E.P. Genetic alteration of a bispecific ligand-directed toxin targeting human CD19 and CD22 receptors resulting in improved efficacy against systemic B cell malignancy. Leuk. Res. 2009, 33, 1233–1242. [Google Scholar] [CrossRef]
- Masonic Cancer Center, University of Minnesota. HM2014-26 DT2219 Immunotoxin for the Treatment of Relapsed or Refractory CD19 (+) and/or CD 22 (+) B-lineage Leukemia or Lymphoma: NCT02370160, 2014LS093. Available online: https://clinicaltrials.gov/ct2/show/NCT02370160 (accessed on 19 April 2021).
- Sharkey, R.M.; McBride, W.J.; Karacay, H.; Chang, K.; Griffiths, G.L.; Hansen, H.J.; Goldenberg, D.M. A Universal Pretargeting System for Cancer Detection and Therapy Using Bispecific Antibody. Cancer Res. 2003, 63, 354–363. [Google Scholar]
- Frampas, E.; Rousseau, C.; Bodet-Milin, C.; Barbet, J.; Chatal, J.-F.; Kraeber-Bodéré, F. Improvement of radioimmunotherapy using pretargeting. Front. Oncol. 2013, 3, 159. [Google Scholar] [CrossRef]
- Bodet-Milin, C.; Ferrer, L.; Rauscher, A.; Masson, D.; Rbah-Vidal, L.; Faivre-Chauvet, A.; Cerato, E.; Rousseau, C.; Hureaux, J.; Couturier, O.; et al. Pharmacokinetics and Dosimetry Studies for Optimization of Pretargeted Radioimmunotherapy in CEA-Expressing Advanced Lung Cancer Patients. Front. Med. 2015, 2, 84. [Google Scholar] [CrossRef]
- Karacay, H.; McBride, W.J.; Griffiths, G.L.; Sharkey, R.M.; Barbet, J.; Hansen, H.J.; Goldenberg, D.M. Experimental pretargeting studies of cancer with a humanized anti-CEA x murine anti-In-DTPA bispecific antibody construct and a (99m)Tc-/(188)Re-labeled peptide. Bioconjug. Chem. 2000, 11, 842–854. [Google Scholar] [CrossRef]
- Ding, L.; Tian, C.; Feng, S.; Fida, G.; Zhang, C.; Ma, Y.; Ai, G.; Achilefu, S.; Gu, Y. Small sized EGFR1 and HER2 specific bifunctional antibody for targeted cancer therapy. Theranostics 2015, 5, 378–398. [Google Scholar] [CrossRef]
- Frejd, F.Y.; Kim, K.-T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 49, e306. [Google Scholar] [CrossRef]
- Löfblom, J.; Feldwisch, J.; Tolmachev, V.; Carlsson, J.; Ståhl, S.; Frejd, F.Y. Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010, 584, 2670–2680. [Google Scholar] [CrossRef] [PubMed]
- Gaissmaier, L.; Elshiaty, M.; Christopoulos, P. Breaking Bottlenecks for the TCR Therapy of Cancer. Cells 2020, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- De Souza, J.E.S.; Galante, P.A.F.; de Almeida, R.V.B.; da Cunha, J.P.C.; Ohara, D.T.; Ohno-Machado, L.; Old, L.J.; de Souza, S.J. SurfaceomeDB: A cancer-orientated database for genes encoding cell surface proteins. Cancer Immun. 2012, 12, 15. [Google Scholar]
- Douglass, J.; Hsiue, E.H.-C.; Mog, B.J.; Hwang, M.S.; DiNapoli, S.R.; Pearlman, A.H.; Miller, M.S.; Wright, K.M.; Azurmendi, P.A.; Wang, Q.; et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol. 2021, 6. [Google Scholar] [CrossRef]
- Dao, T.; Pankov, D.; Scott, A.; Korontsvit, T.; Zakhaleva, V.; Xu, Y.; Xiang, J.; Yan, S.; de Morais Guerreiro, M.D.; Veomett, N.; et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat. Biotechnol. 2015, 33, 1079–1086. [Google Scholar] [CrossRef]
- Banaszek, A.; Bumm, T.G.P.; Nowotny, B.; Geis, M.; Jacob, K.; Wölfl, M.; Trebing, J.; Kucka, K.; Kouhestani, D.; Gogishvili, T.; et al. On-target restoration of a split T cell-engaging antibody for precision immunotherapy. Nat. Commun. 2019, 10, 5387. [Google Scholar] [CrossRef]
- Cheadle, E.J.; Gornall, H.; Baldan, V.; Hanson, V.; Hawkins, R.E.; Gilham, D.E. CAR T cells: Driving the road from the laboratory to the clinic. Immunol. Rev. 2014, 257, 91–106. [Google Scholar] [CrossRef]
- Subklewe, M. BiTEs better than CAR T cells. Blood Adv. 2021, 5, 607–612. [Google Scholar] [CrossRef]
- Molina, J.C.; Shah, N.N. CAR T cells better than BiTEs. Blood Adv. 2021, 5, 602–606. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Levy, M.Y.; Dalovisio, A.P.; Bahlis, N.J.; Solh, M.; Sebag, M.; Jakubowiak, A.; Jethava, Y.S.; Costello, C.L.; Chu, M.P.; et al. Preliminary Safety, Efficacy, Pharmacokinetics, and Pharmacodynamics of Subcutaneously (SC) Administered PF-06863135, a B-Cell Maturation Antigen (BCMA)-CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 8–9. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Singh, I.; Zudaire, E.; Yeh, T.-M.; Allred, A.J.; Olyslager, Y.; Banerjee, A.; Goldberg, J.D.; et al. Update of CARTITUDE-1: A phase Ib/II study of JNJ-4528, a B-cell maturation antigen (BCMA)-directed CAR-T-cell therapy, in relapsed/refractory multiple myeloma. JCO 2020, 38, 8505. [Google Scholar] [CrossRef]
- Jacobson, C.; Chavez, J.C.; Sehgal, A.R.; William, B.M.; Munoz, J.; Salles, G.; Munshi, P.N.; Casulo, C.; Maloney, D.; de Vos, S.; et al. Primary Analysis of Zuma-5: A Phase 2 Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients with Relapsed/Refractory (R/R) Indolent Non-Hodgkin Lymphoma (iNHL). Blood 2020, 136, 40–41. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients With Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results From a Phase I Study. JCO 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Bannerji, R.; Allan, J.N.; Arnason, J.E.; Brown, J.R.; Advani, R.; Ansell, S.M.; O’Brien, S.M.; Duell, J.; Martin, P.; Joyce, R.M.; et al. Odronextamab (REGN1979), a Human CD20 x CD3 Bispecific Antibody, Induces Durable, Complete Responses in Patients with Highly Refractory B-Cell Non-Hodgkin Lymphoma, Including Patients Refractory to CAR T Therapy. Blood 2020, 136, 42–43. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Einsele, H.; Rasche, L.; Topp, M.S.; Martin Kortüm, K.; Duell, J. The use of bispecific antibodies to optimize the outcome of patients with acute leukemia, lymphoma and multiple myeloma after SCT. Bone Marrow Transpl. 2019, 54, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.; Schmoor, C.; Waterhouse, M.; Marks, R.; Wäsch, R.; Bertz, H.; Finke, J. Reduced-intensity conditioning with fludarabine and thiotepa for second allogeneic transplantation of relapsed patients with AML. Bone Marrow Transpl. 2013, 48, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.; Bertz, H.; Ihorst, G.; Marks, R.; Wäsch, R.; Finke, J. Radiation-free allogeneic conditioning with fludarabine, carmustine, and thiotepa for acute lymphoblastic leukemia and other hematologic malignancies necessitating enhanced central nervous system activity. Biol. Blood Marrow Transpl. 2012, 18, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Kobold, S.; Pantelyushin, S.; Rataj, F.; vom Berg, J. Rationale for Combining Bispecific T Cell Activating Antibodies With Checkpoint Blockade for Cancer Therapy. Front. Oncol. 2018, 8, 285. [Google Scholar] [CrossRef] [PubMed]
- Barlabé, P.; de Sostoa, J.; Fajardo, C.A.; Alemany, R.; Moreno, R. Enhanced antitumor efficacy of an oncolytic adenovirus armed with an EGFR-targeted BiTE using menstrual blood-derived mesenchymal stem cells as carriers. Cancer Gene Ther. 2020, 27, 383–388. [Google Scholar] [CrossRef]
- Christopoulos, P.; Pfeifer, D.; Bartholomé, K.; Follo, M.; Timmer, J.; Fisch, P.; Veelken, H. Definition and characterization of the systemic T-cell dysregulation in untreated indolent B-cell lymphoma and very early CLL. Blood 2011, 117, 3836–3846. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.; Follo, M.; Fisch, P.; Veelken, H. The peripheral helper T-cell repertoire in untreated indolent B-cell lymphomas: Evidence for antigen-driven lymphomagenesis. Leukemia 2008, 22, 1952–1954. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.; Schneider, M.A.; Bozorgmehr, F.; Kuon, J.; Engel-Riedel, W.; Kollmeier, J.; Baum, V.; Muley, T.; Schnabel, P.A.; Bischoff, H.; et al. Large cell neuroendocrine lung carcinoma induces peripheral T-cell repertoire alterations with predictive and prognostic significance. Lung Cancer 2018, 119, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.; Fisch, P. Acquired T-Cell Immunodeficiency in Thymoma Patients. Crit. Rev. Immunol. 2016, 36, 315–327. [Google Scholar] [CrossRef]
- Christopoulos, P.; Dopfer, E.P.; Malkovsky, M.; Esser, P.R.; Schaefer, H.-E.; Marx, A.; Kock, S.; Rupp, N.; Lorenz, M.R.; Schwarz, K.; et al. A novel thymoma-associated immunodeficiency with increased naive T cells and reduced CD247 expression. J. Immunol. 2015, 194, 3045–3053. [Google Scholar] [CrossRef]
- Herrmann, M.; Krupka, C.; Deiser, K.; Brauchle, B.; Marcinek, A.; Ogrinc Wagner, A.; Rataj, F.; Mocikat, R.; Metzeler, K.H.; Spiekermann, K.; et al. Bifunctional PD-1 × αCD3 × αCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood 2018, 132, 2484–2494. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Bargou, R.C. T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef]
- Correnti, C.E.; Laszlo, G.S.; van der Schueren, W.J.; Godwin, C.D.; Bandaranayake, A.; Busch, M.A.; Gudgeon, C.J.; Bates, O.M.; Olson, J.M.; Mehlin, C.; et al. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia 2018, 32, 1239–1243. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. LBA51 KEYNOTE-024 5-year OS update: First-line (1L) pembrolizumab (pembro) vs. platinum-based chemotherapy (chemo) in patients (pts) with metastatic NSCLC and PD-L1 tumour proportion score (TPS) ≥50%. Ann. Oncol. 2020, 31, S1181–S1182. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
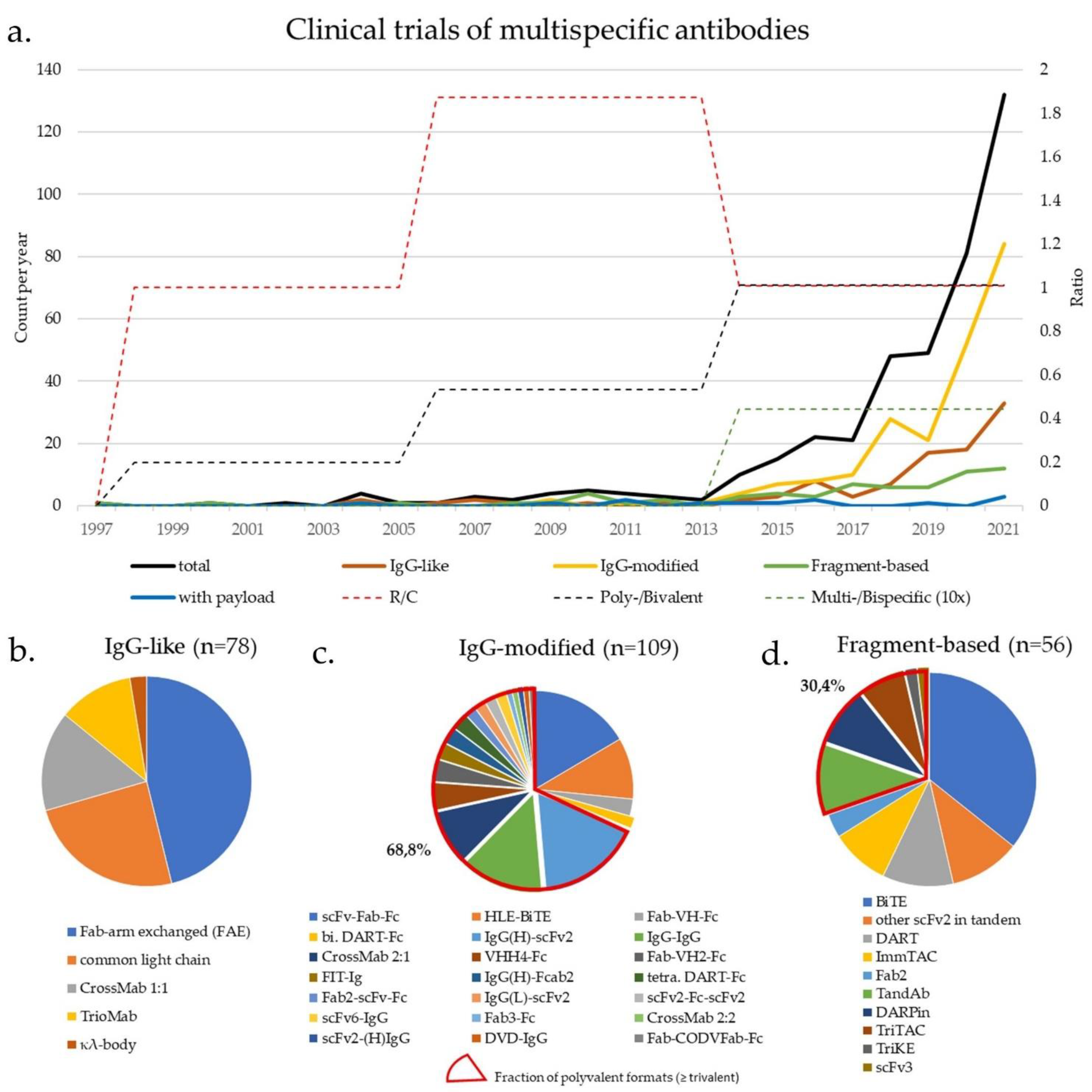
| Class | Specificity/ Valence | Action (C/R) | Format | No. of Clinical Trials | Comment | First Clinical Trial | |
|---|---|---|---|---|---|---|---|
| Fc-based | IgG-like (Section 3.1) (Figure 1c) | 2/2 | R | TrioMab | 9 | “two-halves” formats | 2004 |
| 2/2 | C/R | FAE | 36 | 2010 | |||
| 2/2 | C/R | common light chain | 19 | 2014 | |||
| 2/2 | C | κλ-body | 2 | 2019 | |||
| 2/2 | C/R | CrossMab 1:1 | 12 | 2012 | |||
| IgG-modified (Section 3.2 & Section 4) (Figure 1d) | 2/2 | C/R | scFv-Fab-Fc | 18 | scFv-monosubstituted | 2016 | |
| 2/2 | R | HLE-BiTE | 11 | scFv-bisubstituted | 2015 | ||
| 2/2 | R | DART-Fc | 2 | Db-bisubstituted | 2014 | ||
| 2/2 | R | Fab-VH-Fc ** | 3 | VH-monosubstituted | 2021 | ||
| 2/4 | R | IgG-IgG | 15 | IgG-IgG | 2004 | ||
| 2/3 | R | Fab3-Fc | 1 | Fab-appended | 2018 | ||
| 2/3 | R | CrossMab 2:1 | 10 | 2014 | |||
| 2/4 | C | CrossMab 2:2 | 1 | 2015 | |||
| 2/4 | C/R | FIT-Ig (Fabs-in-Tandem) | 3 | 2018 | |||
| 2/3 | R | Fab2-scFv-Fc | 2 | scFv-appended | 2020 | ||
| 2/4 | C/R | IgG(H)-scFv2 | 19 | 2017 | |||
| 2/4 | C | scFv2-(H)IgG | 1 | 2014 | |||
| 2/4 | R | IgG(L)-scFv2 | 2 | 2019 | |||
| 2/4 | C | DVD-IgG | 1 | V-appended | 2013 | ||
| 2/4 | R | scFv2-Fc-scFv2 | 2 | scFv-multisubstituted | 2015 | ||
| 4/8 | R | scFv6-IgG | 2 | 2020 | |||
| 2/4 | C | DART-Fc | 3 | Db-multisubstituted | 2017 | ||
| 2/4 | C | VHH4-Fc | 5 | V-multi substituted | 2019 | ||
| 2/3 | C/R | Fab-VH2-Fc ** | 4 | 2019 | |||
| 3/3 | R | Fab-CODVFab-Fc | 1 | 2020 | |||
| 2/3-4 * | C | IgG-fusion proteins | 55 | Fusion moiety | 2015 | ||
| 2/4 | C | IgG(H)-Fcab2 | 3 | Fc-modified | 2018 | ||
| Fc-free | Fv-based $ ($Section 3.3 & Section 4) ($ Figure 1e) | 2/2 | R | BiTE | 20 | scFv-based | 2008 |
| 2/2 | C/R | other scFv2 in tandem | 6 | 2005 | |||
| 2/2 * | R | ImmTAC | 5 | 2015 | |||
| 3/3 * | R | TriKE | 1 | 2020 | |||
| 3/3 | C | scFv3 | 1 | 2020 | |||
| 2/2 | R | DART | 6 | Db-based | 2014 | ||
| 2/4 | R | TandAb | 6 | 2010 | |||
| 3/3 | R | TriTAC | 4 | 2018 | |||
| ¾ * | C | DARPin | 5 | Ankyrin-based | 2014 | ||
| Fab-based $ | 2/2 | C | Fab2 | 2 | Fab-based | 1997 | |
| with payload (Section 5) (Figure 1f) | 2/2 # | C | Fc-based ADC/EDV | 4 | IgG-based | 2014 | |
| 2/2 # | C | Fc-free FDC/EDV | 3 | Fragment-based | 2013 | ||
| 2/2-3 (#) | C | ±pretargeting (±imaging) | 4 | 2004 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elshiaty, M.; Schindler, H.; Christopoulos, P. Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer. Int. J. Mol. Sci. 2021, 22, 5632. https://doi.org/10.3390/ijms22115632
Elshiaty M, Schindler H, Christopoulos P. Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer. International Journal of Molecular Sciences. 2021; 22(11):5632. https://doi.org/10.3390/ijms22115632
Chicago/Turabian StyleElshiaty, Mariam, Hannah Schindler, and Petros Christopoulos. 2021. "Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer" International Journal of Molecular Sciences 22, no. 11: 5632. https://doi.org/10.3390/ijms22115632
APA StyleElshiaty, M., Schindler, H., & Christopoulos, P. (2021). Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer. International Journal of Molecular Sciences, 22(11), 5632. https://doi.org/10.3390/ijms22115632