
Identification of Novel Genomic-Variant Patterns of OR56A5, OR52L1, and CTSD in Retinitis Pigmentosa Patients by Whole-Exome Sequencing

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Results
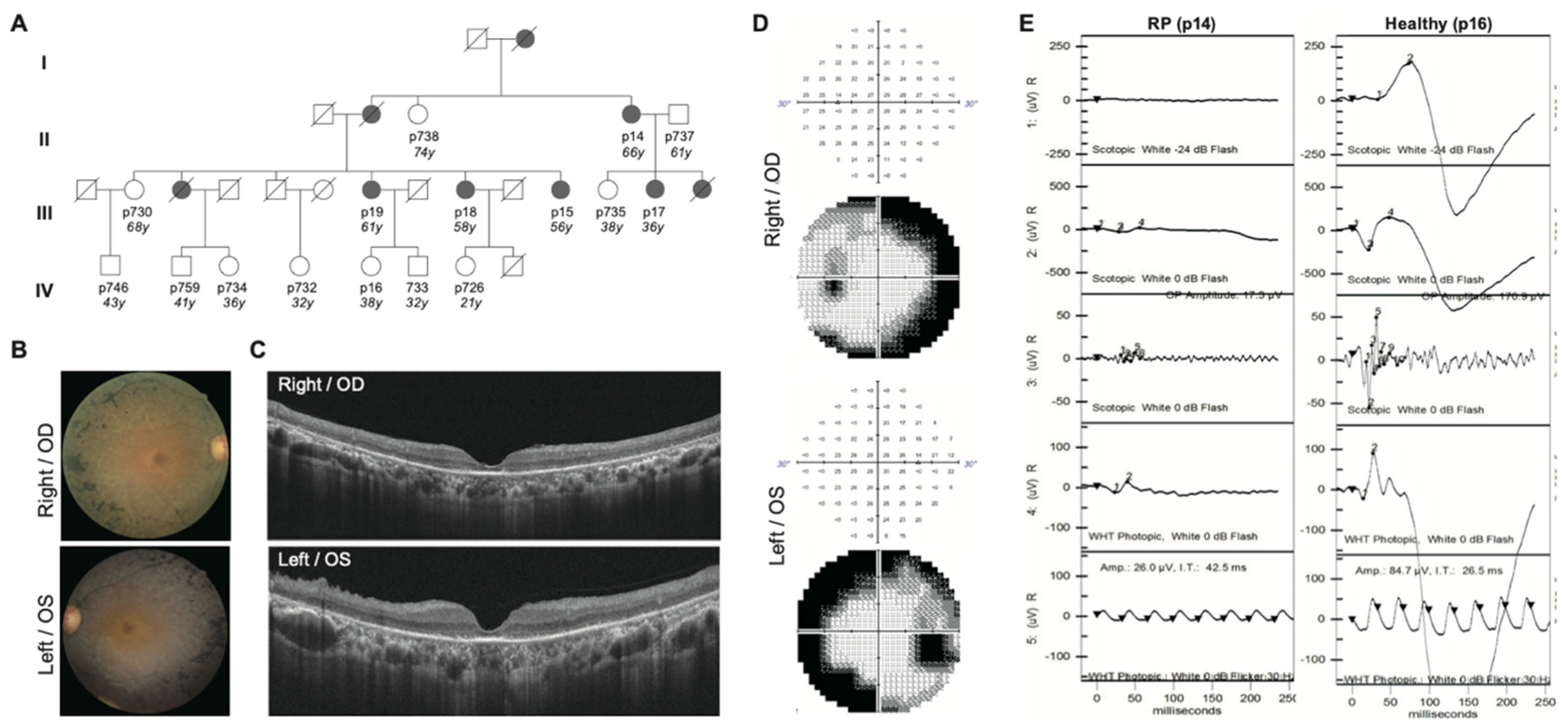
2.1. Clinical Diagnosis Identifies Five RP Patients Out of a Family of Fourteen
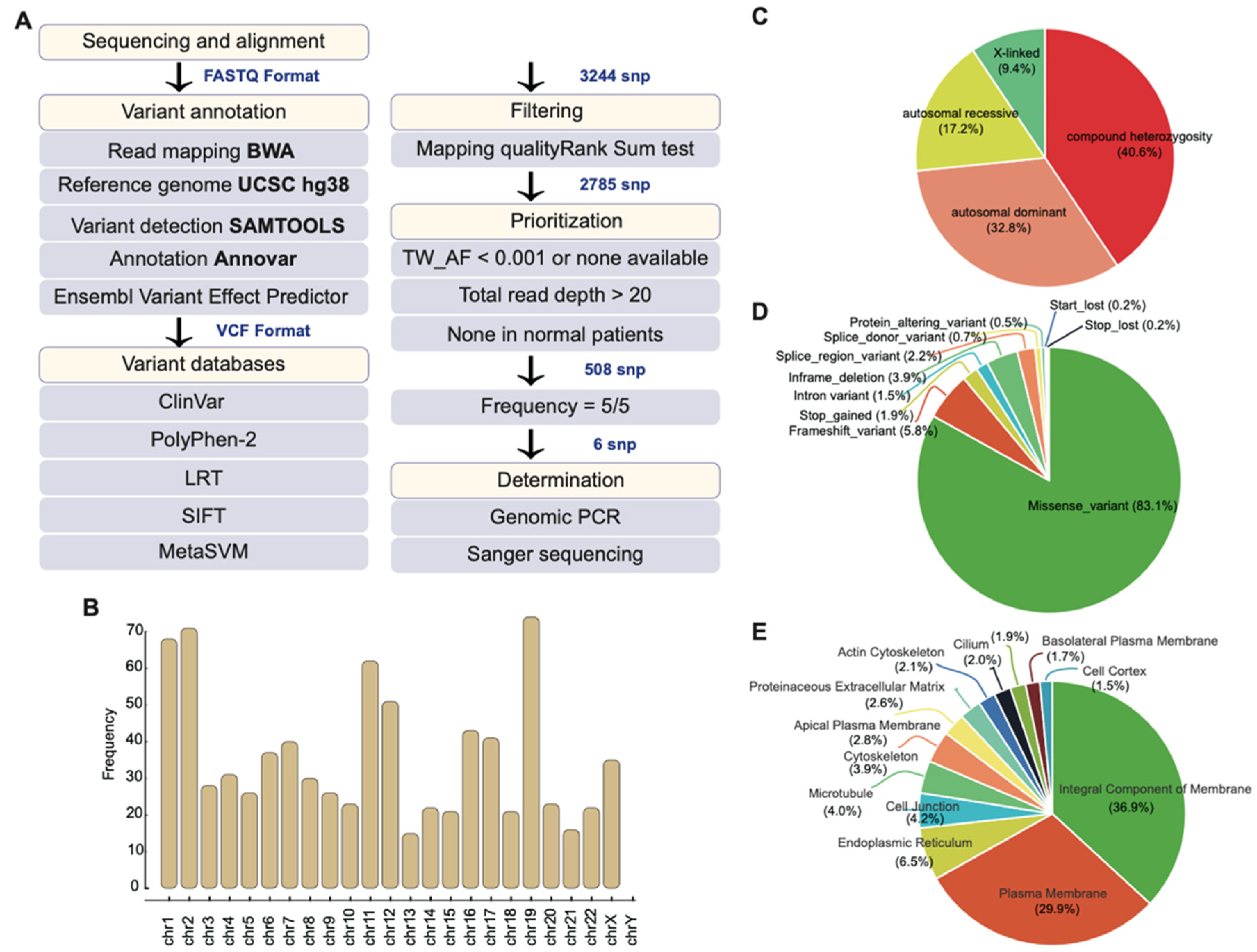
2.2. Whole-Exome Sequencing Identifies Potential Disease-Causing Variants
2.3. Mutational Profile of the Family
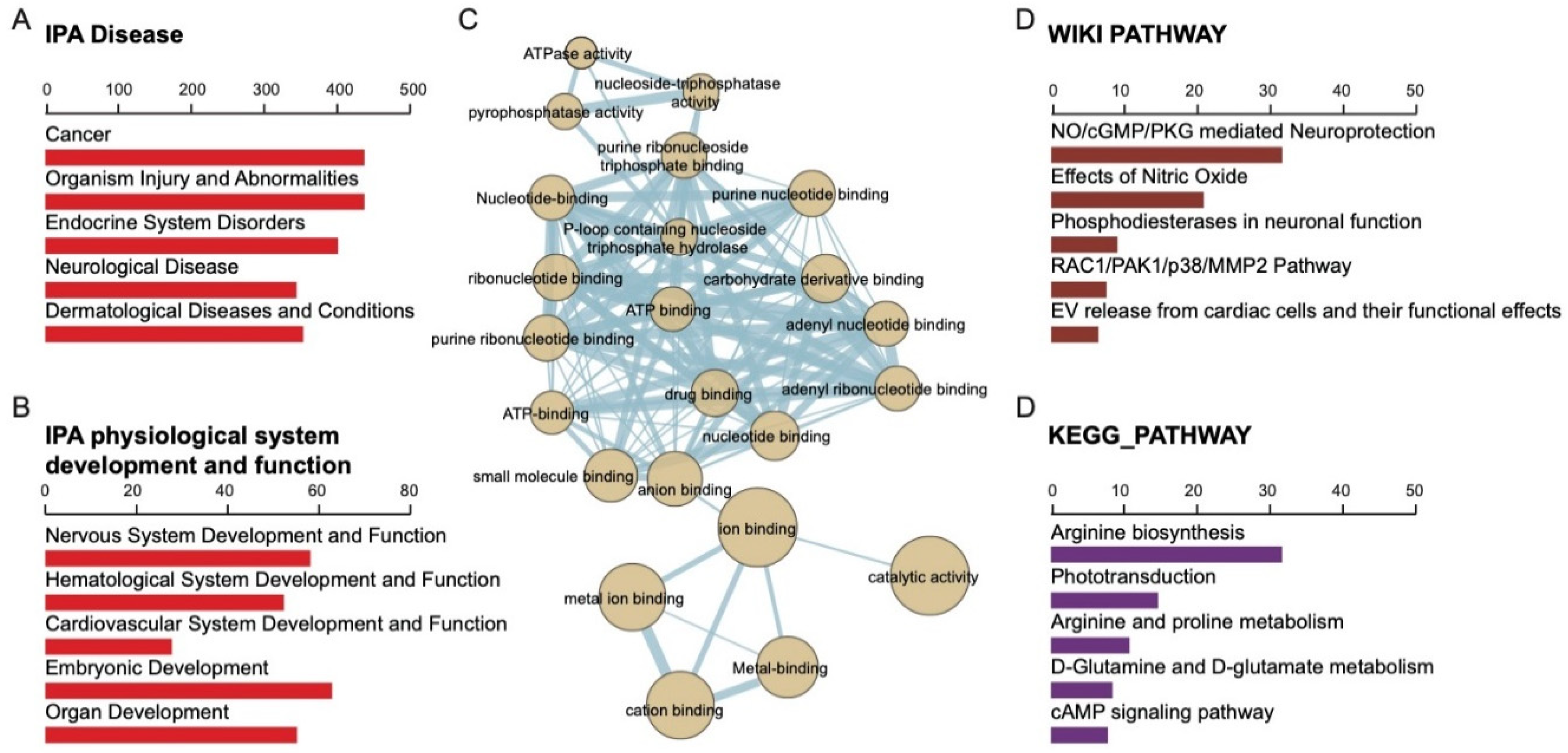
2.4. Gene Ontology of 508 Candidate SNPs and Their Associated Biological Function
2.5. Highly Penetrant Disease-Causing Variants with High Frequency
3. Discussion
4. Materials and Methods
4.1. Samples Collection, Clinical Examination, and Genomic DNA extraction
4.2. Whole-Exome Sequencing
4.3. Variant Calling and Variant Annotation
4.4. IPA Analysis, WIKI Pathway, and Gene Ontology
4.5. Sanger Sequencing
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Marigo, V. Programmed cell death in retinal degeneration: Targeting apoptosis in photoreceptors as potential therapy for retinal degeneration. Cell Cycle 2007, 6, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Haim, M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol. Scand. 2002, 80, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Sharon, D.; Banin, E. Nonsyndromic retinitis pigmentosa is highly prevalent in the Jerusalem region with a high frequency of founder mutations. Mol. Vis. 2015, 21, 783. [Google Scholar]
- Aramian, L.; Chaudhary, K.; Bronson, B. Psychiatric Evaluation of a Patient for a Bionic Eye Implant. Psychiatr. Ann. 2020, 50, 230–232. [Google Scholar] [CrossRef]
- Iwanami, M.; Oshikawa, M.; Nishida, T.; Nakadomari, S.; Kato, S. High prevalence of mutations in the EYS gene in Japanese patients with autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1033–1040. [Google Scholar] [CrossRef]
- Daiger, S.; Rossiter, B.; Greenberg, J.; Christoffels, A.; Hide, W. Data services and software for identifying genes and mutations causing retinal degeneration. Investig. Ophthalmol. Vis. Sci. 1998, 39, 103–105. [Google Scholar]
- Farber, D.B.; Danciger, M. Identification of genes causing photoreceptor degenerations leading to blindness. Curr. Opin. Neurobiol. 1997, 7, 666–673. [Google Scholar] [CrossRef]
- Inglehearn, C.F. Molecular genetics of human retinal dystrophies. Eye 1998, 12, 571–579. [Google Scholar] [CrossRef]
- Van Soest, S.; Westerveld, A.; De Jong, P.T.; Bleeker-Wagemakers, E.M.; Bergen, A.A. Retinitis pigmentosa: Defined from a molecular point of view. Surv. Ophthalmol. 1999, 43, 321–334. [Google Scholar] [CrossRef]
- Arai, Y.; Maeda, A.; Hirami, Y.; Ishigami, C.; Kosugi, S.; Mandai, M.; Kurimoto, Y.; Takahashi, M. Retinitis pigmentosa with EYS mutations is the most prevalent inherited retinal dystrophy in Japanese populations. J. Ophthalmol. 2015. [Google Scholar] [CrossRef]
- Albarry, M.A.; Hashmi, J.A.; Alreheli, A.Q.; Albalawi, A.M.; Khan, B.; Ramzan, K.; Basit, S. Novel homozygous loss-of-function mutations in RP1 and RP1L1 genes in retinitis pigmentosa patients. Ophthalmic Genet. 2019, 40, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Hata, M.; Ooto, S.; Ogino, K.; Gotoh, N.; Morooka, S.; Hasegawa, T.; Hirashima, T.; Sugahara, M.; Kuroda, Y. Choroidal and retinal atrophy of Bietti crystalline dystrophy patients with CYP4V2 mutations compared to retinitis pigmentosa patients with EYS mutations. Retina 2017, 37, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Yoshida, A.; Kawai, K.; Arai, Y.; Akiba, R.; Inaba, A.; Takagi, S.; Fujiki, R.; Hirami, Y.; Kurimoto, Y. Development of a molecular diagnostic test for Retinitis Pigmentosa in the Japanese population. Jpn. J. Ophthalmol. 2018, 62, 451–457. [Google Scholar] [CrossRef]
- Daiger, S.P.; Shankar, S.P.; Schindler, A.B.; Sullivan, L.S.; Bowne, S.J.; King, T.M.; Daw, E.W.; Stone, E.M.; Heckenlively, J.R. Genetic factors modifying clinical expression of autosomal dominant RP. Retin. Degener. Dis. 2006, 572, 3–8. [Google Scholar]
- Dryja, T.P.; McGee, T.L.; Hahn, L.B.; Cowley, G.S.; Olsson, J.E.; Reichel, E.; Sandberg, M.A.; Berson, E.L. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N. Engl. J. Med. 1990, 323, 1302–1307. [Google Scholar] [CrossRef]
- Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Lewis, R.A.; Garcia, C.A.; Ruiz, R.S.; Blanton, S.H.; Northrup, H. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: A screen of known genes in 200 families. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3052–3064. [Google Scholar] [CrossRef] [PubMed]
- Seyedahmadi, B.J.; Rivolta, C.; Keene, J.A.; Berson, E.L.; Dryja, T.P. Comprehensive screening of the USH2A gene in Usher syndrome type II and non-syndromic recessive retinitis pigmentosa. Exp. Eye Res. 2004, 79, 167–173. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 1–9. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Black, A.; Bennett, J.; Cremers, F.P. Lighting a candle in the dark: Advances in genetics and gene therapy of recessive retinal dystrophies. J. Clin. Investig. 2010, 120, 3042–3053. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Fadaie, Z.; Cornelis, S.S.; Cremers, F.P.; Roosing, S. Identification and analysis of genes associated with inherited retinal diseases. Retin. Degener. 2019, 3–27. [Google Scholar] [CrossRef]
- Roosing, S.; Thiadens, A.A.; Hoyng, C.B.; Klaver, C.C.; den Hollander, A.I.; Cremers, F.P. Causes and consequences of inherited cone disorders. Prog. Retin. Eye Res. 2014, 42, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Eppig, J.T. The mammalian phenotype ontology: Enabling robust annotation and comparative analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.A.; Miller, R.; Digles, D.; Lopes, E.N.; Ehrhart, F. WikiPathways: Connecting communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Adamo, H.P.; Filoteo, A.G.; Enyedi, A.; Penniston, J.T. Mutants in the Putative Nucleotide-binding Region of the Plasma Membrane Ca2+-Pump: A reduction in activity due to slow dephosphorylation. J. Biol. Chem. 1995, 270, 30111–30114. [Google Scholar] [CrossRef]
- Yuan, E. Republic of China (Taiwan). In The International Compendium of Construction Contracts; De Gruyter: Berlin, Germany, 2013. [Google Scholar]
- Chen, Y.-J.; Roumeliotis, T.I.; Chang, Y.-H.; Chen, C.-T.; Han, C.-L.; Lin, M.-H.; Chen, H.-W.; Chang, G.-C.; Chang, Y.-L.; Wu, C.-T. Proteogenomics of non-smoking lung cancer in East Asia delineates molecular signatures of pathogenesis and progression. Cell 2020, 182, 226–244.e217. [Google Scholar] [CrossRef]
- Lee, M.; Chen, C.; Lee, C.; Chen, C.; Chong, M.; Ouyang, W.; Chiu, N.; Chuo, L.; Chen, C.; Tan, H. Genome-wide association study of bipolar I disorder in the Han Chinese population. Mol. Psychiatry 2011, 16, 548–556. [Google Scholar] [CrossRef]
- Lee, Y.; Kuo, H.; Chang, J.; Chang, L.; Huang, L. Taiwan Pediatric ID Alliance, Chen YT, Tsai FJ, Wu JY. Two new susceptibility loci for Kawasaki disease identified through genome-wide association analysis. Nat. Genet. 2012, 44, 522–525. [Google Scholar] [CrossRef]
- Liou, Y.-J.; Wang, H.-H.; Lee, M.-T.M.; Wang, S.-C.; Chiang, H.-L.; Chen, C.-C.; Lin, C.-H.; Chung, M.-S.; Kuo, C.-C.; Liao, D.-L. Genome-wide association study of treatment refractory schizophrenia in Han Chinese. PLoS ONE 2012, 7, e33598. [Google Scholar] [CrossRef]
- Tsai, F.-J.; Lee, Y.-C.; Chang, J.-S.; Huang, L.-M.; Huang, F.-Y.; Chiu, N.-C.; Chen, M.-R.; Chi, H.; Lee, Y.-J.; Chang, L.-C. Identification of novel susceptibility Loci for kawasaki disease in a Han chinese population by a genome-wide association study. PLoS ONE 2011, 6, e16853. [Google Scholar] [CrossRef]
- Tsai, F.-J.; Yang, C.-F.; Chen, C.-C.; Chuang, L.-M.; Lu, C.-H.; Chang, C.-T.; Wang, T.-Y.; Chen, R.-H.; Shiu, C.-F.; Liu, Y.-M. A genome-wide association study identifies susceptibility variants for type 2 diabetes in Han Chinese. PLoS Genet. 2010, 6, e1000847. [Google Scholar] [CrossRef] [PubMed]
- Kazma, R.; Bailey, J.N. Population-based and family-based designs to analyze rare variants in complex diseases. Genet. Epidemiol. 2011, 35, S41–S47. [Google Scholar] [CrossRef]
- Pandey, J.P. Genomewide association studies and assessment of risk of disease. N. Engl. J. Med. 2010, 363, 2076–2077. [Google Scholar] [PubMed]
- Bansal, V.; Libiger, O.; Torkamani, A.; Schork, N.J. Statistical analysis strategies for association studies involving rare variants. Nat. Rev. Genet. 2010, 11, 773–785. [Google Scholar] [CrossRef]
- Eichler, E.E.; Flint, J.; Gibson, G.; Kong, A.; Leal, S.M.; Moore, J.H.; Nadeau, J.H. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 2010, 11, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Maher, B. Personal genomes: The case of the missing heritability. Nat. News 2008, 456, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Momozawa, Y.; Mizukami, K. Unique roles of rare variants in the genetics of complex diseases in humans. J. Hum. Genet. 2021, 66, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Altshuler, D.; Rader, D.J. From Darwin’s finches to canaries in the coal mine—mining the genome for new biology. N. Engl. J. Med. 2008, 358, 2760–2763. [Google Scholar] [CrossRef] [PubMed]
- King, E.A.; Davis, J.W.; Degner, J.F. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019, 15, e1008489. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Plenge, R.M.; Scolnick, E.M.; Altshuler, D. Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov. 2013, 12, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Tolone, A.; Belhadj, S.; Rentsch, A.; Schwede, F.; Paquet-Durand, F. The cGMP pathway and inherited photoreceptor degeneration: Targets, compounds, and biomarkers. Genes 2019, 10, 453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Beuve, A.; Townes-Anderson, E. The nitric oxide-cGMP signaling pathway differentially regulates presynaptic structural plasticity in cone and rod cells. J. Neurosci. 2005, 25, 2761–2770. [Google Scholar] [CrossRef][Green Version]
- Paquet-Durand, F.; Hauck, S.M.; Van Veen, T.; Ueffing, M.; Ekström, P. PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J. Neurochem. 2009, 108, 796–810. [Google Scholar] [CrossRef]
- Stephenson, J.D.; Laskowski, R.A.; Nightingale, A.; Hurles, M.E.; Thornton, J.M. VarMap: A web tool for mapping genomic coordinates to protein sequence and structure and retrieving protein structural annotations. Bioinformatics 2019, 35, 4854–4856. [Google Scholar] [CrossRef]
- Meyer, S.; Yilmaz, U.; Kim, Y.-J.; Steinfeld, R.; Meyberg-Solomayer, G.; Oehl-Jaschkowitz, B.; Tzschach, A.; Gortner, L.; Igel, J.; Schofer, O. Congenital CLN disease in two siblings. Wien. Med. Wochenschr. 2015, 165, 210–213. [Google Scholar] [CrossRef]
- Wei, C.-Y.; Yang, J.-H.; Yeh, E.-C.; Tsai, M.-F.; Kao, H.-J.; Lo, C.-Z.; Chang, L.-P.; Lin, W.-J.; Hsieh, F.-J.; Belsare, S. Genetic profiles of 103,106 individuals in the Taiwan Biobank provide insights into the health and history of Han Chinese. NPJ Genom. Med. 2021, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, D.P. PANTHER Version 11: Expanded Annotation Data from Gene Ontology and Reactome Pathways, and Data Analysis Tool Enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gender | Diagnosis | Age | Age of Onset | RP Fundus Feature | Visual Acuity OD OS | Macular Involvement OD OS | Visual Field OD OS | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| p14 | Female | RP | 66 | 43 | Yes | 0.4 | 0.1 | Yes | Yes | - | - |
| p15 | Female | RP | 56 | 40 | Yes | 0.6 | 0.6 | Yes | Yes | - | - |
| p17 | Female | RP | 36 | 29 | Yes | 0.8 | 0.6 | Mild | Mild | - | - |
| p18 | Female | RP | 58 | 55 | Yes | 0.5 | 0.3 | Mild | Mild | + | + |
| P19 | Female | RP | 61 | 47 | Yes | 0.7 | 0.9 | Yes | Yes | - | - |
| P16 | Female | Healthy | 38 | ND | No | 1.0 | 1.0 | No | No | + | + |
| P726 | Male | Healthy | 21 | ND | No | 1.0 | 1.0 | No | No | ++ | ++ |
| P730 | Female | Healthy | 68 | ND | No | 0.8 | 0.7 | No | No | ++ | ++ |
| P732 | Female | Healthy | 32 | ND | No | 1.0 | 1.0 | No | No | +++ | +++ |
| P733 | Male | Healthy | 323 | ND | No | 1.0 | 1.0 | No | No | +++ | +++ |
| P734 | Male | Healthy | 36 | ND | No | 1.0 | 1.0 | No | No | +++ | +++ |
| P735 | Female | Healthy | 38 | ND | No | 1.0 | 1.0 | No | No | +++ | +++ |
| P737 | Male | Healthy | 61 | ND | No | 0.8 | 0.9 | No | No | ++ | ++ |
| P738 | Female | Healthy | 74 | ND | No | 0.8 | 0.8 | No | No | ++ | ++ |
| Symbol | SnpDB | Chrom | Pos | Ref | Alt | Hgvsc | Consequence | Impact | Inheri-tance | Clin_ Sig | Polyphen | Lrt | Sift | Metasvm |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR56A5 | Rs116908319 | Chr11 | 5968052 | G | A | NM_001146033.1:c.443C > T | missense variant | Mid | -- | -- | B | -- | T | -- |
| OR52L1 | Rs762369133 | Chr11 | 5986614 | T | C | NM 001005173.3:c.317A > G | missense variant | Mid | -- | -- | B | N | T | T |
| CTSD | Rs752612332 | Chr11 | 1759599 | T | TG | NM_001909.5:c.268dup | Frameshift variant | High | AR | P | -- | -- | -- | -- |
| PRF1 | Novel | Chr10 | 70600774 | CAGCCA | C | NM 005041.5:c.124_128del | Frameshift variant | High | -- | -- | -- | -- | -- | -- |
| KBTBD13 | Rs367648853 | Chr15 | 65077773 | G | A | NM 001101362.2:c.958G*A | Missense_variant | Mid | AD | -- | B | -- | D | T |
| ATP2B4 | Rs769793412 | Chr1 | 203707074 | A | G | NM_001001396.2:c.1165A > G | Missense_ variant | Mid | -- | -- | PD | N | T | T |
| Consequence | Gene | Snp | p14 | p15 | p17 | p18 | p19 | Retinitis Pigmentosa |
|---|---|---|---|---|---|---|---|---|
| Missense variant | OR56A5 | Rs116908319 | o | o | o | o | o | none |
| Missense variant | OR52L1 | Rs762369133 | o | o | o | o | o | None |
| Frameshift variant | CTSD | Rs752612332 | o | o | o | o | o | 10.1002/mds.28106 |
| Frameshift variant | PRF1 | Novel | o | o | o | o | o | None |
| Missense variant | KBTBD13 | Rs367648853 | o | o | o | o | o | 10.1016/j.ajhg.2020.10.020 |
| Missense variant | ATP2B4 | Rs769793412 | o | o | o | o | o | 10.1080/13816810.2019.1703014 |
| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
|---|---|---|
| ATP2B4 | CCACTGTCTGTTCCCTATGC | CAGGGACTTCTGCTCTTGTG |
| CTSD | CCCTGCTGAGAGCAAGGACC | GACAGAAGCCAGGGGTCTAGA |
| KBTBD13 | CTTCTGCTACGACCCCGAC | CCTCGATGGCGTAGAGCAG |
| OR52L1 | GCCTATGATGGTGGCTTGG | GGAGGCTATCCCAGCCTTC |
| OR56A5 | GGCCATATCTCCCTCACC C | GGAAGCCTCTCTGCACCAG |
| PRF1 | CTTCAGTGGAGCTGACTTTG | GGGAAGGGAGCAGTCATC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-Y.; Chang, Y.-C.; Hsiao, Y.-J.; Chien, Y.; Jheng, Y.-C.; Wu, J.-R.; Ching, L.-J.; Hwang, D.-K.; Hsu, C.-C.; Lin, T.-C.; et al. Identification of Novel Genomic-Variant Patterns of OR56A5, OR52L1, and CTSD in Retinitis Pigmentosa Patients by Whole-Exome Sequencing. Int. J. Mol. Sci. 2021, 22, 5594. https://doi.org/10.3390/ijms22115594
Lin T-Y, Chang Y-C, Hsiao Y-J, Chien Y, Jheng Y-C, Wu J-R, Ching L-J, Hwang D-K, Hsu C-C, Lin T-C, et al. Identification of Novel Genomic-Variant Patterns of OR56A5, OR52L1, and CTSD in Retinitis Pigmentosa Patients by Whole-Exome Sequencing. International Journal of Molecular Sciences. 2021; 22(11):5594. https://doi.org/10.3390/ijms22115594
Chicago/Turabian StyleLin, Ting-Yi, Yun-Chia Chang, Yu-Jer Hsiao, Yueh Chien, Ying-Chun Jheng, Jing-Rong Wu, Lo-Jei Ching, De-Kuang Hwang, Chih-Chien Hsu, Tai-Chi Lin, and et al. 2021. "Identification of Novel Genomic-Variant Patterns of OR56A5, OR52L1, and CTSD in Retinitis Pigmentosa Patients by Whole-Exome Sequencing" International Journal of Molecular Sciences 22, no. 11: 5594. https://doi.org/10.3390/ijms22115594
APA StyleLin, T.-Y., Chang, Y.-C., Hsiao, Y.-J., Chien, Y., Jheng, Y.-C., Wu, J.-R., Ching, L.-J., Hwang, D.-K., Hsu, C.-C., Lin, T.-C., Chou, Y.-B., Huang, Y.-M., Chen, S.-J., Yang, Y.-P., & Tsai, P.-H. (2021). Identification of Novel Genomic-Variant Patterns of OR56A5, OR52L1, and CTSD in Retinitis Pigmentosa Patients by Whole-Exome Sequencing. International Journal of Molecular Sciences, 22(11), 5594. https://doi.org/10.3390/ijms22115594