
Autophagy Induction by Trichodermic Acid Attenuates Endoplasmic Reticulum Stress-Mediated Apoptosis in Colon Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
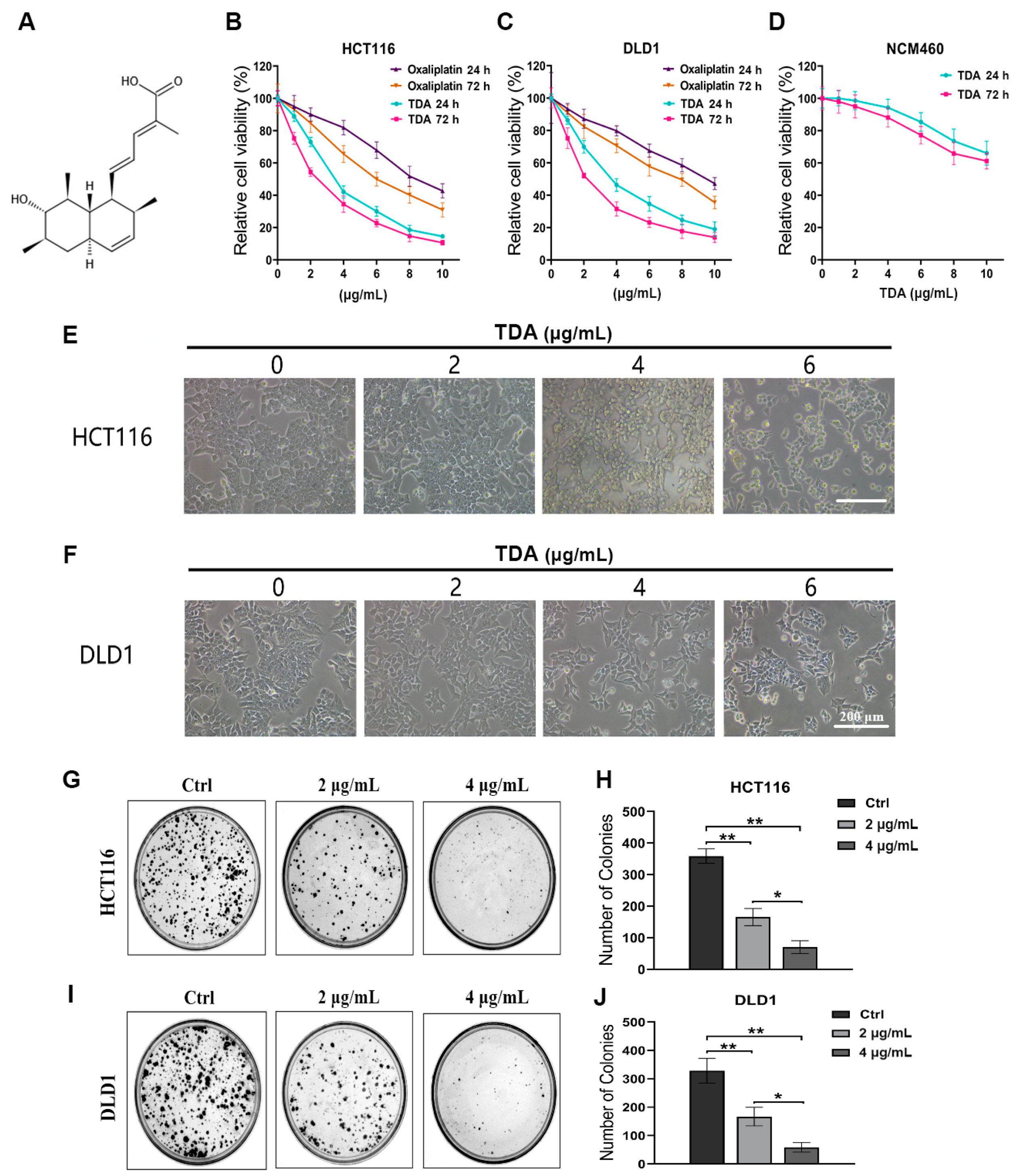
2.1. TDA Inhibits Colon Cancer Cell Viability In Vitro
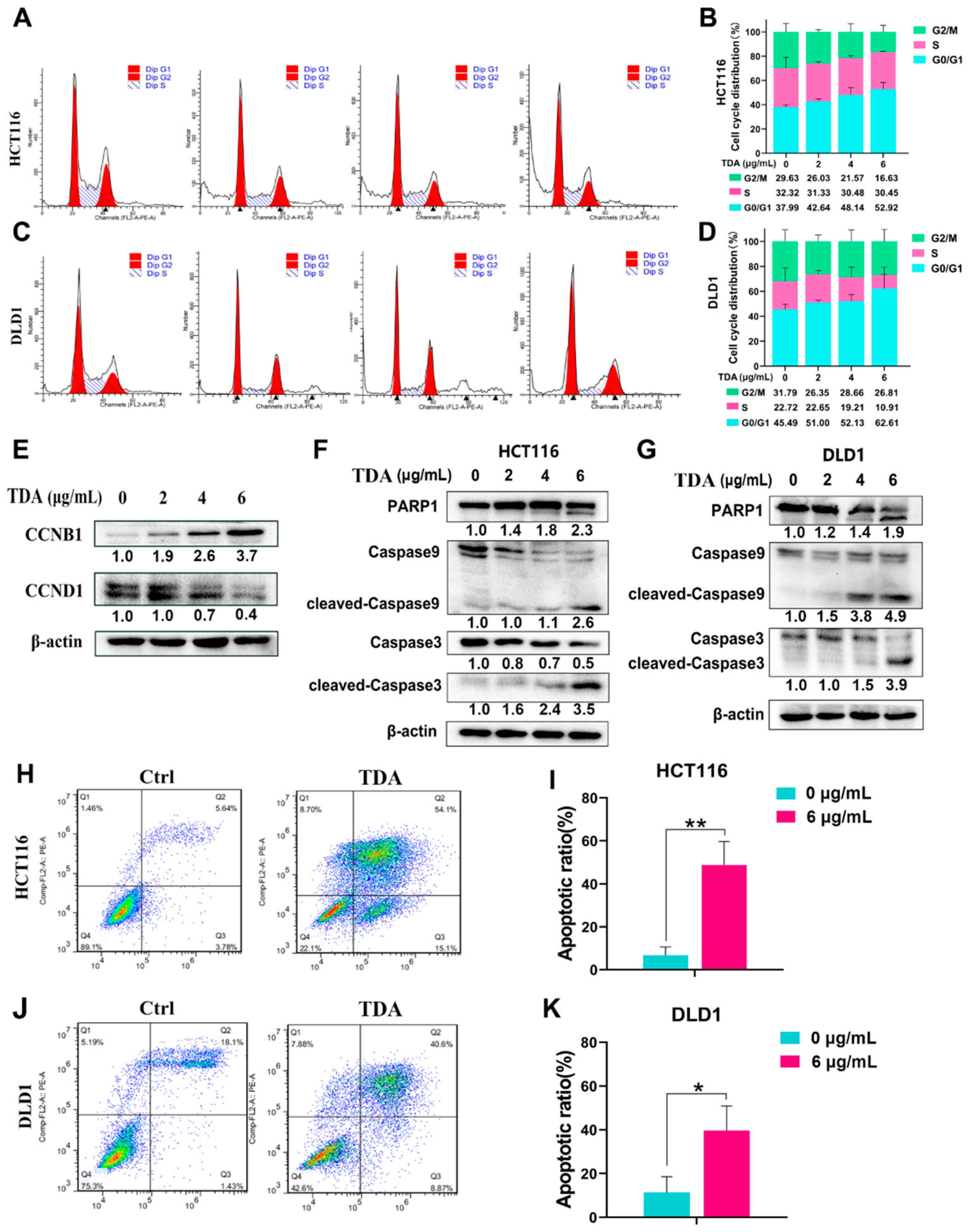
2.2. TDA Treatment Induces Cell Cycle Arrest and Apoptosis in Colon Cancer Cells
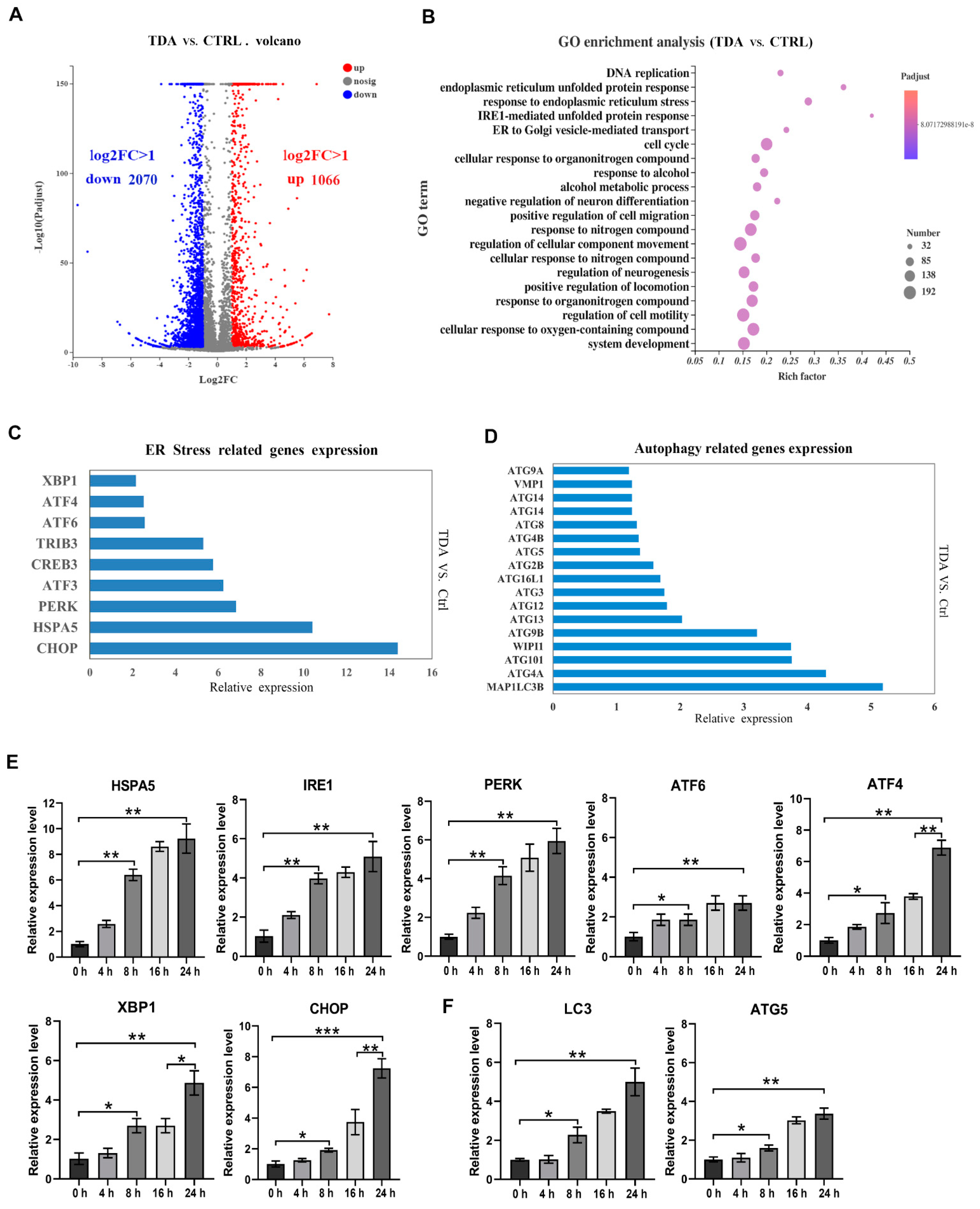
2.3. Transcriptome Analysis Identified the Apoptotic- and Autophagy-Related Genes Contributive to the TDA-Induced Cell Death
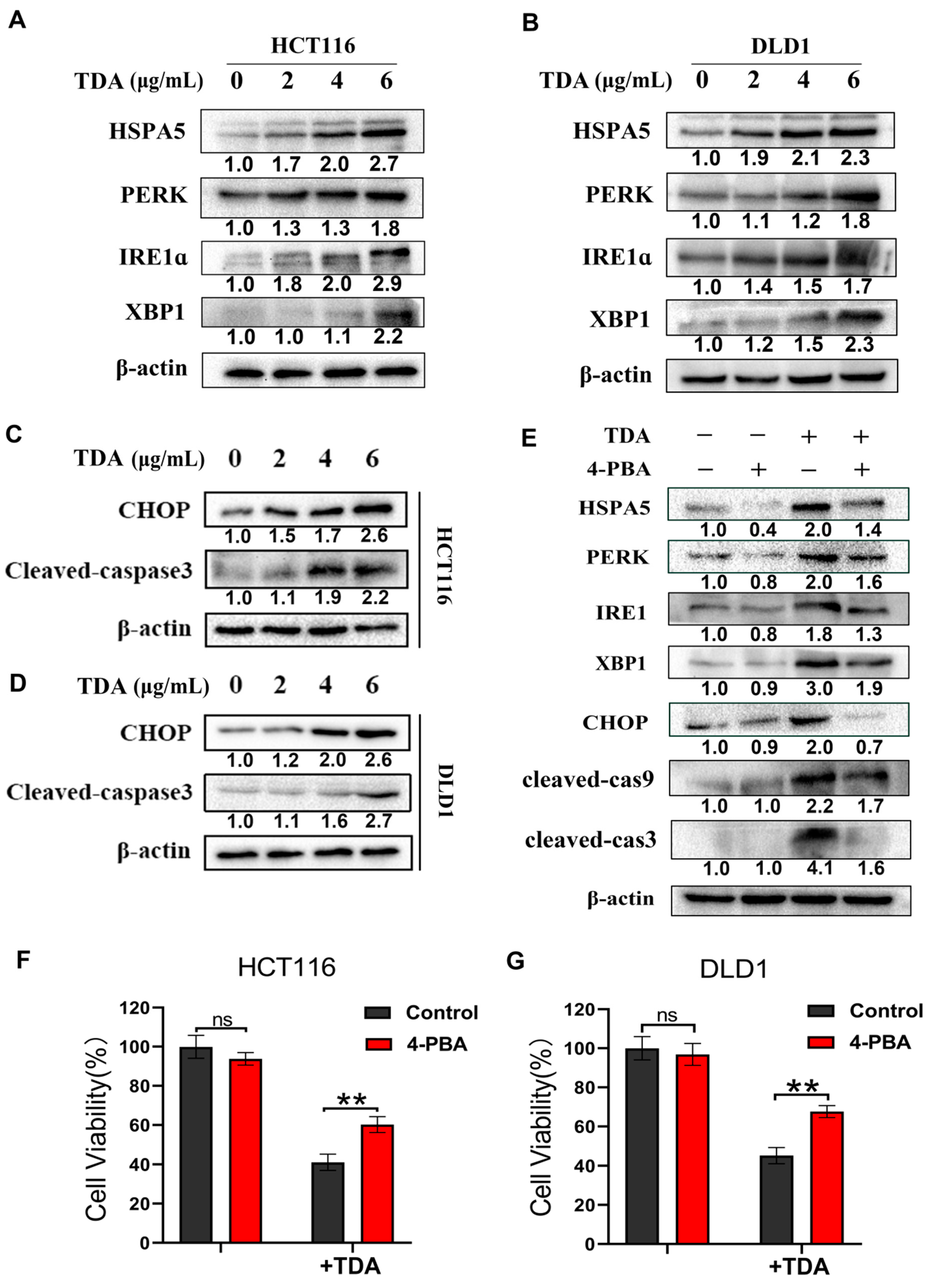
2.4. TDA Induced Apoptosis through IRE1α and PERK Pathways
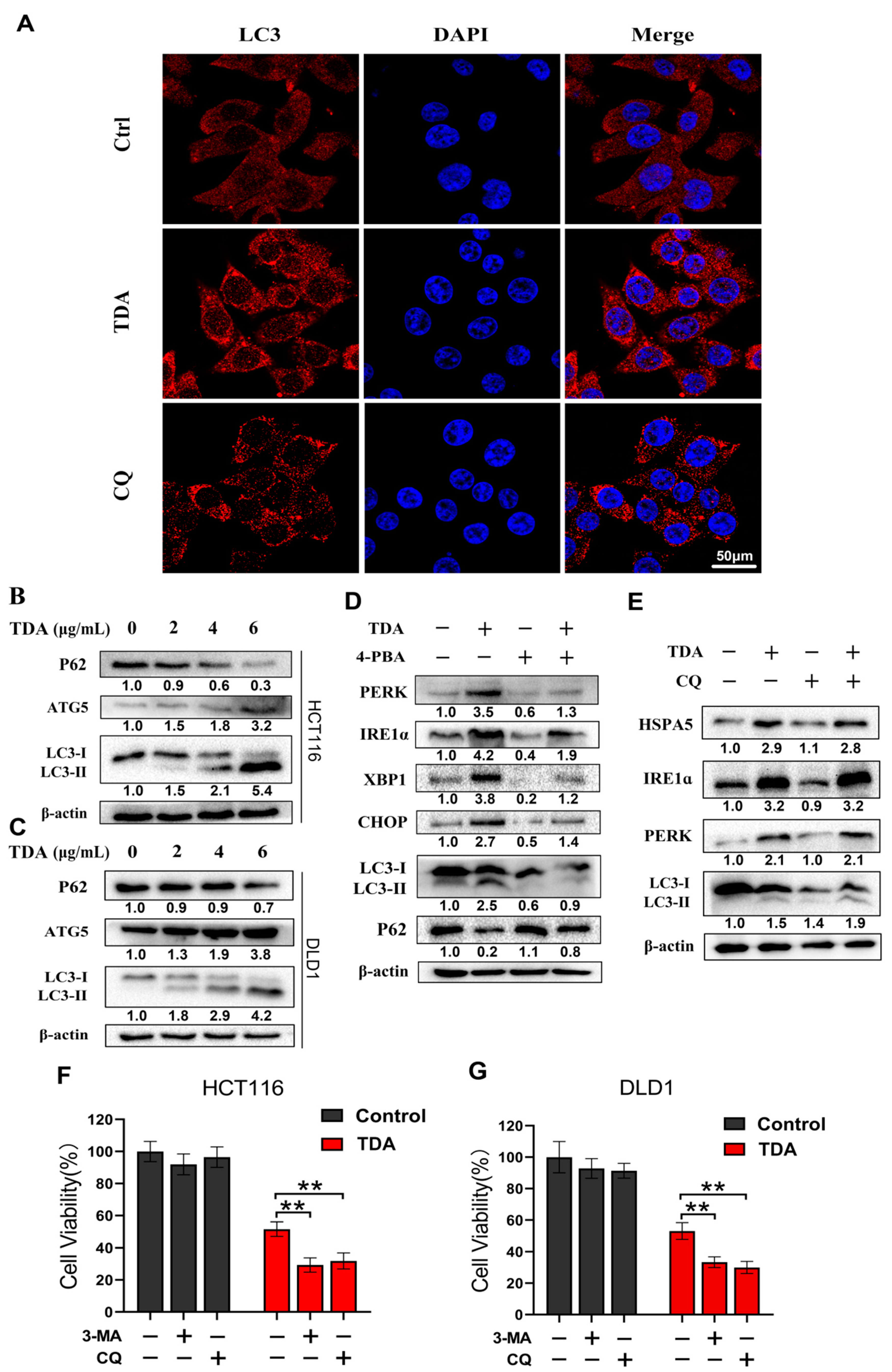
2.5. Endoplasmic Reticulum Stress Induces Protective Autophagy
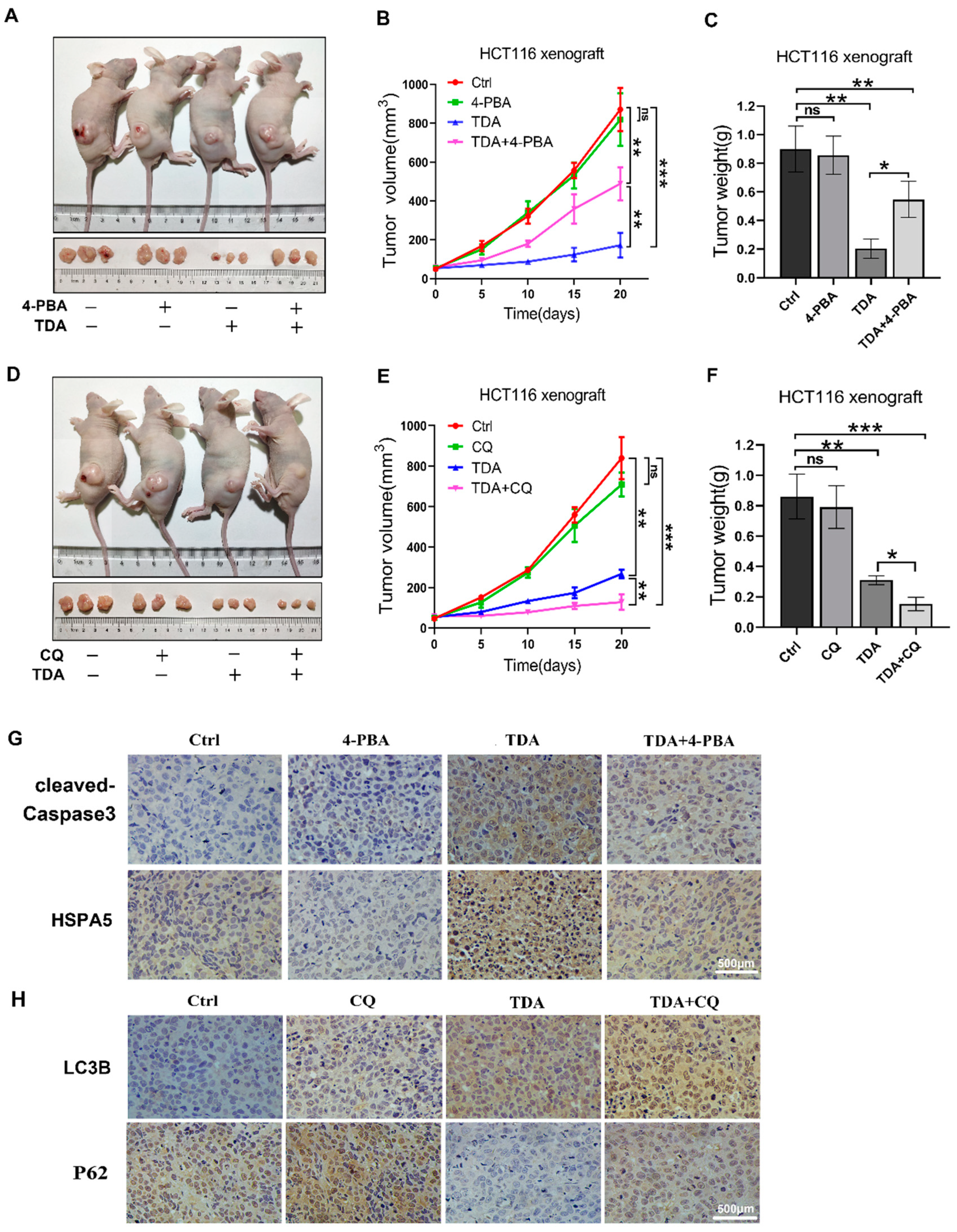
2.6. Treatment of TDA Retards the Growth of Transplanted Tumors
3. Discussion
4. Materials and Methods
4.1. Animal Study
4.2. Cell Culture and Reagents
4.3. Cell Viability and Colony Formation Assay
4.4. Flow Cytometric Analysis
4.5. Western Blotting Analysis
4.6. Real-Time PCR Analysis
4.7. Confocal Microscopy Imaging
4.8. Immunohistochemistry
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| ER | Endoplasmic Reticulum |
| UPR | Unfolded protein response |
| HSPA5 | Heat shock protein A5 |
| IRE1α | Inositol-requiring enzyme 1α |
| PERK | Protein kinase R-like ER kinase |
| ATF6 | Activating transcription factor 6 |
| XBP1 | X-box binding protein 1 |
| CHOP | C/EBP homologous protein |
| ATF4 | Activating transcription factor 4 |
| eIF2α | Eukaryotic translation initiation factor 2α |
| PARP1 | Poly (ADP-ribose) polymerase family, member 1 |
| Caspase9/3 | Cysteinyl aspartate specific proteinase 9/3 |
| JNK | The c-Jun N-terminal kinases |
| TRAF2 | Tumor necrosis factor alpha (TNFa) receptor associated factor 2 |
| mTOR | The mammalian target of rapamycin |
| PI3K | Phosphatidylinositol 3 kinase |
| AKT | Protein kinase B |
| MET | Hepatocyte growth factor receptor |
| LC3B | Microtubule-associated protein 1 light chain 3 |
| ATG5 | Autophagy related 5 |
| P62 | Sequestosome 1 |
| 4-PBA | 4-phenylbutyrate |
| CQ | Chloroquine |
| 3-MA | 3-methyladenine |
| CCK-8 | Cell Counting Kit-8 |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Tan, Z.; Hollis-Hansen, K.; Zhang, Y.; Yu, C.; Li, Y. Epidemiological Trends in Colorectal Cancer in China: An Ecological Study. Dig. Dis. Sci. 2016, 62, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Center, M.M.; Jemal, A.; Ward, E. International trends in colorectal cancer incidence rates. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1688–1694. [Google Scholar] [CrossRef] [PubMed]
- Franzese, C.; Comito, T.; Toska, E.; Tozzi, A.; Clerici, E.; De Rose, F.; Franceschini, D.; Navarria, P.; Reggiori, G.; Tomatis, S.; et al. Predictive factors for survival of oligometastatic colorectal cancer treated with Stereotactic body radiation therapy. Radiother. Oncol. 2019, 133, 220–226. [Google Scholar] [CrossRef]
- Hadjipetrou, A.; Anyfantakis, D.; Galanakis, C.G.; Kastanakis, M.; Kastanakis, S. Colorectal cancer, screening and primary care: A mini literature review. World J. Gastroenterol. 2017, 23, 6049–6058. [Google Scholar] [CrossRef]
- Valentini, V.; Morganti, A.G.; Gambacorta, M.A.; Mohiuddin, M.; Doglietto, G.B.; Coco, C.; De Paoli, A.; Rossi, C.; Di Russo, A.; Valvo, F.; et al. Preoperative hyperfractionated chemoradiation for locally recurrent rectal cancer in patients previously irradiated to the pelvis: A multicentric phase II study. Int. J. Radiat. Oncol. Biol. Phys. 2006, 64, 1129–1139. [Google Scholar] [CrossRef]
- Banerjee, A.; Pathak, S.; Subramanium, V.D.; Dharanivasan, G.; Murugesan, R.; Verma, R.S. Strategies for targeted drug delivery in treatment of colon cancer: Current trends and future perspectives. Drug Discov. Today 2017, 22, 1224–1232. [Google Scholar] [CrossRef]
- Jardim, D.L.; Rodrigues, C.A.; Novis, Y.A.S.; Rocha, V.G.; Hoff, P.M. Oxaliplatin-related thrombocytopenia. Ann. Oncol. 2012, 23, 1937–1942. [Google Scholar] [CrossRef]
- Lee, C.S.; Ryan, E.J.; Doherty, G.A. Gastro-intestinal toxicity of chemotherapeutics in colorectal cancer: The role of inflammation. World J. Gastroenterol. 2014, 20, 3751–3761. [Google Scholar] [CrossRef]
- Zhao, T.; Xu, L.-L.; Zhang, Y.; Lin, Z.-H.; Xia, T.; Yang, D.-F.; Chen, Y.-M.; Yang, X.-L. Three new α-pyrone derivatives from the plant endophytic fungus Penicillium ochrochloronthe and their antibacterial, antifungal, and cytotoxic activities. J. Asian Nat. Prod. Res. 2018, 21, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-L.; Chen, Y.-C.; Tao, M.-H.; Li, H.-H.; Zhang, W.-M. Two new octahydronaphthalene derivatives from Trichoderma spirale, an endophytic fungus derived from Aquilaria sinensis. Helv. Chim. Acta 2012, 95, 805–809. [Google Scholar] [CrossRef]
- Shiina, I.; Umezaki, Y.; Ohashi, Y.; Yamazaki, Y.; Dan, S.; Yamori, T. Total Synthesis of AMF-26, an Antitumor Agent for Inhibition of the Golgi System, Targeting ADP-Ribosylation Factor 1. J. Med. Chem. 2012, 56, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Ignashkova, T.I.; Gendarme, M.; Peschk, K.; Eggenweiler, H.M.; Lindemann, R.K.; Reiling, J.H. Cell survival and protein secretion associated with Golgi integrity in response to Golgi stress-inducing agents. Traffic 2017, 18, 530–544. [Google Scholar] [CrossRef]
- Obata, Y.; Horikawa, K.; Shiina, I.; Takahashi, T.; Murata, T.; Tasaki, Y.; Suzuki, K.; Yonekura, K.; Esumi, H.; Nishida, T.; et al. Oncogenic Kit signalling on the Golgi is suppressed by blocking secretory trafficking with M-COPA in gastrointestinal stromal tumours. Cancer Lett. 2018, 415, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Watari, K.; Nakamura, M.; Fukunaga, Y.; Furuno, A.; Shibata, T.; Kawahara, A.; Hosoi, F.; Kuwano, T.; Kuwano, M.; Ono, M. The antitumor effect of a novel angiogenesis inhibitor (an octahydronaphthalene derivative) targeting both VEGF receptor and NF-kappaB pathway. Int. J. Cancer 2012, 131, 310–321. [Google Scholar] [CrossRef]
- Hattori, T.; Watanabe-Takahashi, M.; Shiina, I.; Ohashi, Y.; Dan, S.; Nishikawa, K.; Yamori, T.; Naito, M. M-COPA, a novel Golgi system disruptor, suppresses apoptosis induced by Shiga toxin. Genes Cells 2016, 21, 901–906. [Google Scholar] [CrossRef]
- Ohashi, Y.; Okamura, M.; Hirosawa, A.; Tamaki, N.; Akatsuka, A.; Wu, K.M.; Choi, H.W.; Yoshimatsu, K.; Shiina, I.; Yamori, T.; et al. M-COPA, a Golgi Disruptor, Inhibits Cell Surface Expression of MET Protein and Exhibits Antitumor Activity against MET-Addicted Gastric Cancers. Cancer Res. 2016, 76, 3895–3903. [Google Scholar] [CrossRef]
- Ohashi, Y.; Iijima, H.; Yamaotsu, N.; Yamazaki, K.; Sato, S.; Okamura, M.; Sugimoto, K.; Dan, S.; Hirono, S.; Yamori, T. AMF-26, a novel inhibitor of the Golgi system, targeting ADP-ribosylation factor 1 (Arf1) with potential for cancer therapy. J. Biol. Chem. 2012, 287, 3885–3897. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.; Huang, E.Y.; Olzmann, J.A. Endoplasmic Reticulum-Associated Degradation and Lipid Homeostasis. Ann. Rev. Nutr. 2016, 36, 511–542. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ackerman, S.L. Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 2006, 18, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.W.; Chen, Q.; Tay, A.S.; Shenolikar, S.; Nicchitta, C.V. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell 2014, 158, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, N.; Chiang, W.C.; Kurt, T.D.; Sigurdson, C.J.; Lin, J.H. Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death. Am. J. Pathol. 2015, 185, 1800–1808. [Google Scholar] [CrossRef]
- Bhat, T.A.; Chaudhary, A.K.; Kumar, S.; O’Malley, J.; Inigo, J.R.; Kumar, R.; Yadav, N.; Chandra, D. Endoplasmic reticulum-mediated unfolded protein response and mitochondrial apoptosis in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 58–66. [Google Scholar] [CrossRef]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Nomura, Y. ER signaling in unfolded protein response. Life Sci. 2003, 74, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression during Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef]
- Schonthal, A.H. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem. Pharmacol. 2013, 85, 653–666. [Google Scholar] [CrossRef]
- Lim, E.J.; Heo, J.; Kim, Y.H. Tunicamycin promotes apoptosis in leukemia cells through ROS generation and downregulation of survivin expression. Apoptosis 2015, 20, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Negron, J.E.; Zhang, Z.; Rivera-Ruiz, A.P.; Banerjee, A.; Romero-Nutz, E.C.; Sanchez-Torres, N.; Baksi, K.; Banerjee, D.K. Tunicamycin-induced ER stress in breast cancer cells neither expresses GRP78 on the surface nor secretes it into the media. Glycobiology 2018, 28, 61–68. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Li, W.; Guan, Y. Tunicamycin inhibits colon carcinoma growth and aggressiveness via modulation of the ERK-JNK-mediated AKT/mTOR signaling pathway. Mol. Med. Rep. 2018, 17, 4203–4212. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, D.; Doganlar, O.; Erdogan, S.; Merakli, M.; Dogan, A.; Turker, P.; Bostanci, A.; Doganlar, Z.B. Naringin Combined with NF-kappaB Inhibition and Endoplasmic Reticulum Stress Induces Apoptotic Cell Death via Oxidative Stress and the PERK/eIF2alpha/ATF4/CHOP Axis in HT29 Colon Cancer Cells. Biochem. Genet. 2021, 59, 159–184. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, H.; Li, R.; Xin, J.; Wu, S.; Lan, J.; Xue, K.; Li, X.; Zuo, C.; Jiang, W.; et al. Cucurbitacin I induces cancer cell death through the endoplasmic reticulum stress pathway. J. Cell Biochem. 2018, 120, 2391–2403. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, J.; Kim, Y.S. Celecoxib induces cell death on non-small cell lung cancer cells through endoplasmic reticulum stress. Anat. Cell Biol. 2017, 50, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Jiang, F.; Zhou, J.Y.; Zhang, D.; Liu, M.H.; Chen, Y.G. Artesunate induces apoptosis and autophagy in HCT116 colon cancer cells, and autophagy inhibition enhances the artesunateinduced apoptosis. Int. J. Mol. Med. 2018, 42, 1295–1304. [Google Scholar]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, J.; Zhang, L.; Zhu, Z.; Fan, J.; Chen, L.; Zhuang, L.; Luo, J.; Chen, H.; Liu, L.; et al. MicroRNA 23b regulates autophagy associated with radioresistance of pancreatic cancer cells. Gastroenterology 2013, 145, 1133–1143 e12. [Google Scholar] [CrossRef]
- Shi, T.T.; Yu, X.X.; Yan, L.J.; Xiao, H.T. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother. Pharmacol. 2017, 79, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Burada, F.; Nicoli, E.R.; Ciurea, M.E.; Uscatu, D.C.; Ioana, M.; Gheonea, D.I. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Devenport, S.N.; Shah, Y.M. Functions and Implications of Autophagy in Colon Cancer. Cells 2019, 8, 1349. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yao, J.; Xie, J.; Liu, Z.; Zhou, Y.; Pan, H.; Han, W. The role of autophagy in colitis-associated colorectal cancer. Signal Transduct. Target. Ther. 2018, 3, 31. [Google Scholar] [CrossRef]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R.; et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef]
- Verfaillie, T.; Salazar, M.; Velasco, G.; Agostinis, P. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int. J. Cell Biol. 2010, 2010, 930590. [Google Scholar] [CrossRef]
- Bejcek, J.; Spiwok, V.; Kmonickova, E.; Rimpelova, S. Na(+)/K(+)-ATPase Revisited: On Its Mechanism of Action, Role in Cancer, and Activity Modulation. Molecules 2021, 26, 1905. [Google Scholar] [CrossRef] [PubMed]
- Rimpelova, S.; Zimmermann, T.; Drasar, P.B.; Dolensky, B.; Bejcek, J.; Kmonickova, E.; Cihlarova, P.; Gurska, S.; Kuklikova, L.; Hajduch, M.; et al. Steroid Glycosides Hyrcanoside and Deglucohyrcanoside: On Isolation, Structural Identification, and Anticancer Activity. Foods 2021, 10, 136. [Google Scholar] [CrossRef]
- Skubnik, J.; Pavlickova, V.; Ruml, T.; Rimpelova, S. Current Perspectives on Taxanes: Focus on Their Bioactivity, Delivery and Combination Therapy. Plants (Basel) 2021, 10, 569. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2008, 9, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. The influence of natural products upon drug discovery. Nat. Prod. Rep. 2000, 17, 215–234. [Google Scholar] [CrossRef]
- Qu, J.; Zou, T.; Lin, Z. The Roles of the Ubiquitin-Proteasome System in the Endoplasmic Reticulum Stress Pathway. Int. J. Mol. Sci. 2021, 22, 1526. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. GRP78 Induction in Cancer: Therapeutic and Prognostic Implications: Figure 1. Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef] [PubMed]
- Jager, R.; Bertrand, M.J.; Gorman, A.M.; Vandenabeele, P.; Samali, A. The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biol. Cell 2012, 104, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2011, 151, 217–219. [Google Scholar] [CrossRef]
- Ma, Y.; Brewer, J.W.; Alan Diehl, J.; Hendershot, L.M. Two Distinct Stress Signaling Pathways Converge Upon the CHOP Promoter During the Mammalian Unfolded Protein Response. J. Mol. Biol. 2002, 318, 1351–1365. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Thomas, S.; Sharma, N.; Golden, E.B.; Cho, H.; Agarwal, P.; Gaffney, K.J.; Petasis, N.A.; Chen, T.C.; Hofman, F.M.; Louie, S.G.; et al. Preferential killing of triple-negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors. Cancer Lett. 2012, 325, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol. Rev. 1999, 79, 683–701. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Jakobsen, C.M.; Janssen, S.; Khan, S.R.; Garrett, E.S.; Lilja, H.; Christensen, S.B.; Isaacs, J.T. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J. Natl. Cancer Inst. 2003, 95, 990–1000. [Google Scholar] [CrossRef]
- Banerjee, A.; Lang, J.-Y.; Hung, M.-C.; Sengupta, K.; Banerjee, S.K.; Baksi, K.; Banerjee, D.K. Unfolded Protein Response Is Required in nu/nu Mice Microvasculature for Treating Breast Tumor with Tunicamycin. J. Biol. Chem. 2011, 286, 29127–29138. [Google Scholar] [CrossRef] [PubMed]
- Gorska, M.; Popowska, U.; Sielicka-Dudzin, A.; Kuban-Jankowska, A.; Sawczuk, W.; Knap, N.; Cicero, G.; Wozniak, F. Geldanamycin and its derivatives as Hsp90 inhibitors. Front. Biosci. 2012, 17, 2269–2277. [Google Scholar] [CrossRef]
- Carew, J.S.; Nawrocki, S.T.; Krupnik, Y.V.; Dunner, K., Jr.; McConkey, D.J.; Keating, M.J.; Huang, P. Targeting endoplasmic reticulum protein transport: A novel strategy to kill malignant B cells and overcome fludarabine resistance in CLL. Blood 2006, 107, 222–231. [Google Scholar] [CrossRef]
- Casanova, J.E. Regulation of Arf activation: The Sec7 family of guanine nucleotide exchange factors. Traffic 2007, 8, 1476–1485. [Google Scholar] [CrossRef]
- Ohuchi, K.; Takahashi, C.; Hirasawa, N.; Watanabe, M.; Fujiki, H.; Tsurufuji, S. Stimulation of histamine release and arachidonic acid metabolism in rat peritoneal mast cells by thapsigargin, a non-TPA-type tumor promoter. Biochim. Biophys. Acta 1989, 1003, 9–14. [Google Scholar] [CrossRef]
- Lee, J.-S.; Zheng, Z.; Mendez, R.; Ha, S.-W.; Xie, Y.; Zhang, K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol. Lett. 2012, 211, 29–38. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Dunner, K., Jr.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005, 65, 11658–11666. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Dunner, K., Jr.; Boise, L.H.; Chiao, P.J.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005, 65, 11510–11519. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Livesey, K.M.; Tang, D.; Zeh, H.J.; Lotze, M.T. Autophagy inhibition in combination cancer treatment. Curr. Opin. Investig. Drugs 2009, 10, 1269–1279. [Google Scholar] [PubMed]
- Garrido, W.; Rocha, J.D.; Jaramillo, C.; Fernandez, K.; Oyarzun, C.; San Martin, R.; Quezada, C. Chemoresistance in high-grade gliomas: Relevance of adenosine signalling in stem-like cells of glioblastoma multiforme. Curr. Drug Targets 2014, 15, 931–942. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, S.Y.; Kim, M.; Cheon, C.; Ko, S.G. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis. 2018, 9, 875. [Google Scholar] [CrossRef]
- Tsai, D.H.; Chung, C.H.; Lee, K.T. Antrodia cinnamomea induces autophagic cell death via the CHOP/TRB3/Akt/mTOR pathway in colorectal cancer cells. Sci. Rep. 2018, 8, 17424. [Google Scholar] [CrossRef]
- Jiang, X.; Overholtzer, M.; Thompson, C.B. Autophagy in cellular metabolism and cancer. J. Clin. Investig. 2015, 125, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Harris-White, M.E. Redox regulation of autophagy in healthy brain and neurodegeneration. Neurobiol. Dis. 2015, 84, 50–59. [Google Scholar] [CrossRef]
- Guo, M.L.; Liao, K.; Periyasamy, P.; Yang, L.; Cai, Y.; Callen, S.E.; Buch, S. Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy 2015, 11, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Yu, L.; Bai, C.; Liu, L.; Long, H.; Shi, L.; Lin, Z. USP27-mediated Cyclin E stabilization drives cell cycle progression and hepatocellular tumorigenesis. Oncogene 2018, 37, 2702–2713. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Dong, L.; Li, H.; Liu, Z.; Luo, Z.; Duan, G.; Dai, X.; Lin, Z. Ubiquitination-mediated degradation of SIRT1 by SMURF2 suppresses CRC cell proliferation and tumorigenesis. Oncogene 2020, 39, 4450–4464. [Google Scholar] [CrossRef]
- Lin, Z.; Yang, H.; Tan, C.; Li, J.; Liu, Z.; Quan, Q.; Kong, S.; Ye, J.; Gao, B.; Fang, D. USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep. 2013, 5, 1639–1649. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, J.; Zeng, C.; Zou, T.; Chen, X.; Yang, X.; Lin, Z. Autophagy Induction by Trichodermic Acid Attenuates Endoplasmic Reticulum Stress-Mediated Apoptosis in Colon Cancer Cells. Int. J. Mol. Sci. 2021, 22, 5566. https://doi.org/10.3390/ijms22115566
Qu J, Zeng C, Zou T, Chen X, Yang X, Lin Z. Autophagy Induction by Trichodermic Acid Attenuates Endoplasmic Reticulum Stress-Mediated Apoptosis in Colon Cancer Cells. International Journal of Molecular Sciences. 2021; 22(11):5566. https://doi.org/10.3390/ijms22115566
Chicago/Turabian StyleQu, Junyan, Cheng Zeng, Tingting Zou, Xu Chen, Xiaolong Yang, and Zhenghong Lin. 2021. "Autophagy Induction by Trichodermic Acid Attenuates Endoplasmic Reticulum Stress-Mediated Apoptosis in Colon Cancer Cells" International Journal of Molecular Sciences 22, no. 11: 5566. https://doi.org/10.3390/ijms22115566
APA StyleQu, J., Zeng, C., Zou, T., Chen, X., Yang, X., & Lin, Z. (2021). Autophagy Induction by Trichodermic Acid Attenuates Endoplasmic Reticulum Stress-Mediated Apoptosis in Colon Cancer Cells. International Journal of Molecular Sciences, 22(11), 5566. https://doi.org/10.3390/ijms22115566