
Activated STAT3 Is a Novel Regulator of the XRCC1 Promoter and Selectively Increases XRCC1 Protein Levels in Triple Negative Breast Cancer


Abstract

1. Introduction
2. Results
2.1. XRCC1 Promoter Has Active Regions Driving Reporter Expression Not Associated with Known Transcription Factors
2.2. ChIP Confirmed the STAT3 Binding Site within the XRCC1 Promoter
2.3. STAT3 Expression Attenuated XRCC1 Expression
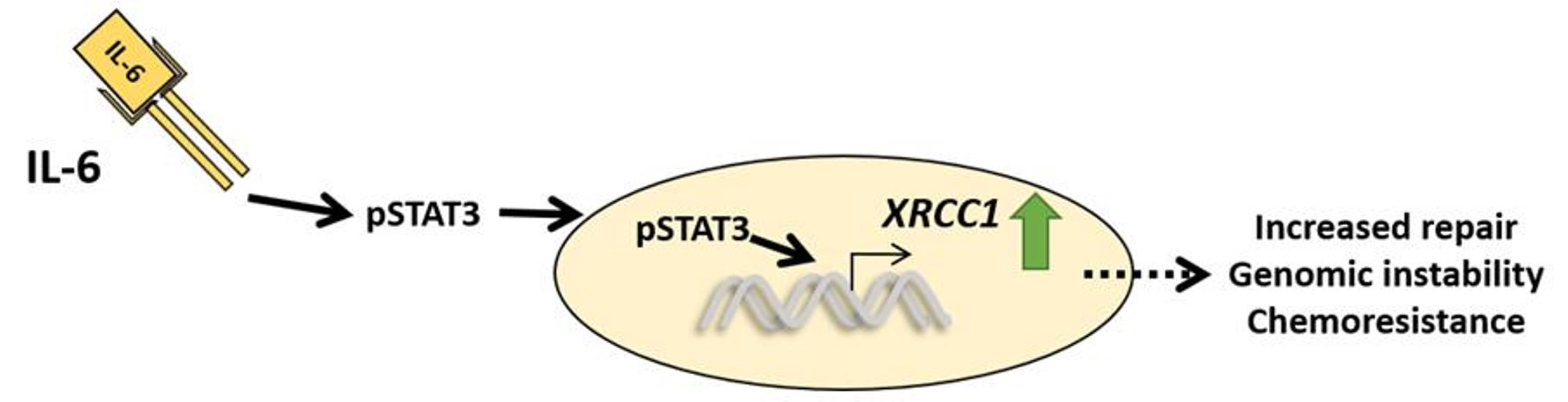
2.4. STAT3 Regulation Is Prevalent in TNBC
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Promoter Luciferase Assay
4.3. Promoter Binding ELISA
4.4. Chromatin Immunoprecipitation (ChIP)
4.5. Modulated Expression of STAT3
4.6. Gene Expression and qPCR
4.7. Cytokine Exposure
4.8. Immunoblot
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caldecott, K.W. DNA single-strand break repair. Exp. Cell Res. 2014, 329, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.H.; Kunkel, T.A. Passing the baton in base excision repair. Nat. Genet. 2000, 7, 176–178. [Google Scholar] [CrossRef]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of Poly(ADP-ribose) Polymerase-1 and XRCC1/DNA Ligase III in an Alternative Route for DNA Double-strand Breaks Rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef]
- Frit, P.; Barboule, N.; Yuan, Y.; Gomez, D.; Calsou, P. Alternative end-joining pathway(s): Bricolage at DNA breaks. DNA Repair 2014, 17, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. Mol. Mech. Mutagen. 2011, 711, 61–72. [Google Scholar] [CrossRef]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.; Fousteri, M.I. Sealing of Chromosomal DNA Nicks during Nucleotide Excision Repair Requires XRCC1 and DNA Ligase IIIα in a Cell-Cycle-Specific Manner. Mol. Cell 2007, 27, 311–323. [Google Scholar] [CrossRef]
- Narciso, L.; Fortini, P.; Pajalunga, D.; Franchitto, A.; Liu, P.; Degan, P.; Frechet, M.; Demple, B.; Crescenzi, M.; Dogliotti, E. Terminally differentiated muscle cells are defective in base excision DNA repair and hypersensitive to oxygen injury. Proc. Natl. Acad. Sci. USA 2007, 104, 17010–17015. [Google Scholar] [CrossRef]
- Sykora, P.; Yang, J.-L.; Ferrarelli, L.K.; Tian, J.; Tadokoro, T.; Kulkarni, A.; Weissman, L.; Keijzers, G.; Wilson, D.M.; Mattson, M.P.; et al. Modulation of DNA base excision repair during neuronal differentiation. Neurobiol. Aging 2013, 34, 1717–1727. [Google Scholar] [CrossRef]
- Tebbs, R.S.; Flannery, M.L.; Meneses, J.J.; Hartmannc, A.; Tucker, J.D.; Thompson, L.H.; Cleaver, J.E.; Pedersen, R.A. Requirement for theXrcc1DNA Base Excision Repair Gene during Early Mouse Development. Dev. Biol. 1999, 208, 513–529. [Google Scholar] [CrossRef]
- Horton, J.K.; Gassman, N.R.; Dunigan, B.D.; Stefanick, D.F.; Wilson, S.H. DNA polymerase β-dependent cell survival independent of XRCC1 expression. DNA Repair 2015, 26, 23–29. [Google Scholar] [CrossRef]
- Lee, K.J.; Piett, C.G.; Andrews, J.F.; Mann, E.; Nagel, Z.D.; Gassman, N.R. Defective base excision repair in the response to DNA damaging agents in triple negative breast cancer. PLoS ONE 2019, 14, e0223725. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Alabdullah, M.; Alblihy, A.; Miligy, I.; Mesquita, K.A.; Chan, S.Y.; Moseley, P.; Rakha, E.A.; Madhusudan, S. PARP1 blockade is synthetically lethal in XRCC1 deficient sporadic epithelial ovarian cancers. Cancer Lett. 2020, 469, 124–133. [Google Scholar] [CrossRef]
- Ali, R.; Alblihy, A.; Toss, M.S.; Algethami, M.; Al Sunni, R.; Green, A.R.; Rakha, E.A.; Madhusudan, S. XRCC1 deficient triple negative breast cancers are sensitive to ATR, ATM and Wee1 inhibitor either alone or in combination with olaparib. Ther. Adv. Med. Oncol. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Al-Kawaz, A.; Toss, M.S.; Green, A.R.; Miligy, I.M.; Mesquita, K.A.; Seedhouse, C.; Mirza, S.; Band, V.A.; Rakha, E.; et al. Targeting PARP1 in XRCC1-Deficient Sporadic Invasive Breast Cancer or Preinvasive Ductal Carcinoma In Situ Induces Synthetic Lethality and Chemoprevention. Cancer Res. 2018, 78, 6818–6827. [Google Scholar] [CrossRef] [PubMed]
- Sizova, D.V.; Keh, A.; Taylor, B.F.; Sweasy, J.B. The R280H X-ray cross-complementing 1 germline variant induces genomic instability and cellular transformation. DNA Repair 2015, 31, 73–79. [Google Scholar] [CrossRef] [PubMed]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef]
- Hoch, N.C.; Hanzlikova, H.; Rulten, S.L.; Tétreault, M.; Koumulainen, E.; Ju, L.; Hornyak, P.; Zeng, Z.; Gittens, W.; Rey, S.A.; et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nat. Cell Biol. 2017, 541, 87–91. [Google Scholar] [CrossRef]
- Taylor, R.M.; Thistlethwaite, A.; Caldecott, K.W. Central Role for the XRCC1 BRCT I Domain in Mammalian DNA Single-Strand Break Repair. Mol. Cell. Biol. 2002, 22, 2556–2563. [Google Scholar] [CrossRef]
- Brem, R.; Hall, J. XRCC1 is required for DNA single-strand break repair in human cells. Nucleic Acids Res. 2005, 33, 2512–2520. [Google Scholar] [CrossRef]
- Horton, J.K.; Watson, M.; Stefanick, D.F.; Shaughnessy, D.T.; Taylor, A.J.; Wilson, S.H. XRCC1 and DNA polymerase β in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008, 18, 48–63. [Google Scholar] [CrossRef]
- Horton, J.K.; Stefanick, D.F.; Gassman, N.R.; Williams, J.G.; Gabel, S.A.; Cuneo, M.J.; Prasad, R.; Kedar, P.S.; Derose, E.F.; Hou, E.W.; et al. Preventing oxidation of cellular XRCC1 affects PARP-mediated DNA damage responses. DNA Repair 2013, 12, 774–785. [Google Scholar] [CrossRef]
- Gassman, N.R.; Wilson, S.H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair 2015, 31, 52–63. [Google Scholar] [CrossRef]
- Abdel-Fatah, T.; Sultana, R.; Abbotts, R.; Hawkes, C.; Seedhouse, C.; Chan, S.; Madhusudan, S. Clinicopathological and functional significance of XRCC1 expression in ovarian cancer. Int. J. Cancer 2012, 132, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Chen, Q.; Wang, Q.; Sun, Y.; Wang, S.; Li, A.; Xu, S.; Røe, O.D.; Wang, M.; Zhang, R.; et al. JWA reverses cisplatin resistance via the CK2-XRCC1 pathway in human gastric cancer cells. Cell Death Dis. 2014, 5, e1551. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Miao, X.; Zhang, Y.; Li, D.; Zou, Q.; Yuan, Y.; Liu, R.; Yang, Z. XRCC1 Is a Promising Predictive Biomarker and Facilitates Chemo-Resistance in Gallbladder Cancer. Front. Mol. Biosci. 2020, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- De Summa, S.; Pinto, R.; Pilato, B.; Sambiasi, D.; Porcelli, L.; Guida, G.; Mattioli, E.; Paradiso, A.; Merla, G.; Micale, L.; et al. Expression of base excision repair key factors and miR17 in familial and sporadic breast cancer. Cell Death Dis. 2014, 5, e1076. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Abdel-Fatah, T.; Abbotts, R.; Hawkes, C.; Albarakati, N.; Seedhouse, C.; Ball, G.; Chan, S.; Rakha, E.A.; Ellis, I.O.; et al. Targeting XRCC1 Deficiency in Breast Cancer for Personalized Therapy. Cancer Res. 2013, 73, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Karahalil, B.; Bohr, V.; Wilson, I.D. Impact of DNA polymorphisms in key DNA base excision repair proteins on cancer risk. Hum. Exp. Toxicol. 2012, 31, 981–1005. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Lee, K.J.; Mann, E.; Wright, G.; Piett, C.G.; Nagel, Z.D.; Gassman, N.R. Exploiting DNA repair defects in triple negative breast cancer to improve cell killing. Ther. Adv. Med. Oncol. 2020, 12. [Google Scholar] [CrossRef]
- Chavez, K.J.; Garimella, S.V.; Lipkowitz, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2011, 32, 35–48. [Google Scholar] [CrossRef]
- Chen, D.; Yu, Z.; Zhu, Z.; Lopez, C.D. E2F1 Regulates the Base Excision Repair Gene XRCC1 and Promotes DNA Repair. J. Biol. Chem. 2008, 283, 15381–15389. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.C.; Grou, C.P.; Legrand, A.J.; Chen, X.; Söderström, K.; Poletto, M.; Dianov, G.L. Sp1 phosphorylation by ATM downregulates BER and promotes cell elimination in response to persistent DNA damage. Nucleic Acids Res. 2017, 46, 1834–1846. [Google Scholar] [CrossRef] [PubMed]
- Alli, E.; Sharma, V.B.; Sunderesakumar, P.; Ford, J.M. Defective Repair of Oxidative DNA Damage in Triple-Negative Breast Cancer Confers Sensitivity to Inhibition of Poly(ADP-Ribose) Polymerase. Cancer Res. 2009, 69, 3589–3596. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.M.; Yau, C.; Sanil, A.; Glas, A.; Petricoin, E.; Wulfkuhle, J.; Severson, T.M.; Linn, S.; Brown-Swigart, L.; Hirst, G.; et al. DNA repair deficiency biomarkers and the 70-gene ultra-high risk signature as predictors of veliparib/carboplatin response in the I-SPY 2 breast cancer trial. npj Breast Cancer 2017, 3, 1–9. [Google Scholar] [CrossRef]
- Xu, W.; Wang, S.; Chen, Q.; Zhang, Y.; Ni, P.; Wu, X.; Zhang, J.; Qiang, F.; Li, A.; Røe, O.D.; et al. TXNL1-XRCC1 pathway regulates cisplatin-induced cell death and contributes to resistance in human gastric cancer. Cell Death Dis. 2014, 5, e1055. [Google Scholar] [CrossRef] [PubMed]
- Sak, S.C.; Harnden, P.; Johnston, C.F.; Paul, A.B.; Kiltie, A.E. APE1 and XRCC1 Protein Expression Levels Predict Cancer-Specific Survival Following Radical Radiotherapy in Bladder Cancer. Clin. Cancer Res. 2005, 11, 6205–6211. [Google Scholar] [CrossRef]
- Christmann, M.; Kaina, B. Transcriptional regulation of human DNA repair genes following genotoxic stress: Trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res. 2013, 41, 8403–8420. [Google Scholar] [CrossRef]
- Wright, G.; Gassman, N.R. Transcriptional dysregulation of base excision repair proteins in breast cancer. DNA Repair 2020, 93, 102922. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Arya, A.; Naema, A.F.; Wong, W.F.; Sethi, G.; Looi, C.Y. Signal Transducer and Activator of Transcription (STATs) Proteins in Cancer and Inflammation: Functions and Therapeutic Implication. Front. Oncol. 2019, 9, 48. [Google Scholar] [CrossRef]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A multifaceted oncoprotein. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef]
- Qin, J.-J.; Yan, L.; Zhang, J.; Zhang, W.-D. STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef]
- Gearing, L.J.; Cumming, H.E.; Chapman, R.; Finkel, A.M.; Woodhouse, I.B.; Luu, K.; Gould, J.A.; Forster, S.C.; Hertzog, P.J. CiiiDER: A tool for predicting and analysing transcription factor binding sites. PLoS ONE 2019, 14, e0215495. [Google Scholar] [CrossRef]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Anderson, A.; Harrison, A.; Lange, A.M.; Jin, G.; Watabe, K.; Lo, H.-W. Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene 2018, 37, 2502–2514. [Google Scholar] [CrossRef] [PubMed]
- Knüpfer, H.; Preiss, R. Significance of interleukin-6 (IL-6) in breast cancer (review). Breast Cancer Res. Treat. 2006, 102, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Dong, Z.; Chen, Y.; Wang, F.; Wang, C.J.; Peng, H.; He, Y.; Hangoc, G.; Pollok, K.; Sandusky, G.; et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene 2015, 35, 783–792. [Google Scholar] [CrossRef]
- Heo, T.-H.; Wahler, J.; Suh, N. Potential therapeutic implications of IL-6/IL-6R/gp130-targeting agents in breast cancer. Oncotarget 2016, 7, 15460–15473. [Google Scholar] [CrossRef]
- Weng, Y.-S.; Tseng, H.-Y.; Chen, Y.-A.; Shen, P.-C.; Al Haq, A.T.; Chen, L.-M.; Tung, Y.-C.; Hsu, H.-L. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef]
- Conway, M.E.; McDaniel, J.M.; Graham, J.M.; Guillen, K.P.; Oliver, P.G.; Parker, S.L.; Yue, P.; Turkson, J.; Buchsbaum, D.J.; Welm, B.E.; et al. STAT3 and GR Cooperate to Drive Gene Expression and Growth of Basal-Like Triple-Negative Breast Cancer. Cancer Res. 2020, 80, 4355–4370. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, J.M.; Varley, K.E.; Gertz, J.; Savic, D.S.; Roberts, B.S.; Bailey, S.K.; Shevde, L.A.; Ramaker, R.C.; Lasseigne, B.N.; Kirby, M.K.; et al. Genomic regulation of invasion by STAT3 in triple negative breast cancer. Oncotarget 2016, 8, 8226–8238. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hu, B.; Ni, J.; Wu, J.; Jiang, W.; Chen, C.; Yang, L.; Zeng, Y.; Wan, R.; Hu, G.; et al. Transcriptional repression of SOCS3 mediated by IL-6/STAT3 signaling via DNMT1 promotes pancreatic cancer growth and metastasis. J. Exp. Clin. Cancer Res. 2016, 35, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Centurione, L.; Aiello, F.B. DNA Repair and Cytokines: TGF-β, IL-6, and Thrombopoietin as Different Biomarkers of Radioresistance. Front. Oncol. 2016, 6, 175. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.K.; Srivastava, S.K.; Bhardwaj, A.; Singh, A.P.; Tyagi, N.; Marimuthu, S.; Dyess, D.L.; Zotto, V.D.; Carter, J.E.; Singh, S. Resistin and interleukin-6 exhibit racially-disparate expression in breast cancer patients, display molecular association and promote growth and aggressiveness of tumor cells through STAT3 activation. Oncotarget 2015, 6, 11231–11241. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Poage, G.M.; Den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of Triple-Negative Breast Cancer Cells Relies upon Coordinate Autocrine Expression of the Proinflammatory Cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef]
- Marsden, C.G.; Dragon, J.A.; Wallace, S.S.; Sweasy, J.B. Base Excision Repair Variants in Cancer. Methods Enzymol. 2017, 591, 119–157. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| XRCC1 Full Length Forward | CTTACGCGTGCTAGCGGACGCAGAACCC |
| XRCC1 Full Length Reverse | GCGTCTTCCATGGTCACCGAGTCCTGGCTGC |
| XRCC1 ∆766 Forward | CTTACGCGTGCTAGCGCAAGGGGACAGAGAGAAGAG |
| XRCC1 ∆612 Forward | CTTACGCGTGCTAGCGAGGCCGAGGCAGGTGGATC |
| XRCC1 ∆310 Forward | CTTACGCGTGCTAGCGGATTTGCTTTCTCGGCTTC |
| XRCC1 ∆35 Forward | CTTACGCGTGCTAGCGGCCGGGGTTTGAAAGGC |
| Primer | Sequence |
|---|---|
| XRCC1 -564 to -457 Forward | TGGGCAACATGGCAAGA |
| XRCC1 -564 to -457 Reverse | CTCCTAAGTAGCTGGGATTACAC |
| XRCC1 -452 to -358 Forward | AGTGGGAGGATCCCTTGG |
| XRCC1 -452 to -358 Reverse | ACAGGGTCTTGCTCTCTCA |
| XRCC1 -312 to -236 Forward | AAAGATTTGCTTTCTCGGCTTC |
| XRCC1 -312 to -236 Reverse | CAGTCGCGCCTCTCTTC |
| XRCC1 -251 to -110 Forward | TTTCTTCCAGACACCAATCCC |
| XRCC1 -251 to -110 Reverse | TAGCAACGAGCGTTTCCTC |
| XRCC1 -127 to -16 Forward | AGGAAACGCTCGTTGCTAA |
| XRCC1 -127 to -16 Reverse | TCGGGCCTTTCAAACCC |
| XRCC1 SP1 Site Forward [33] | ATTGGGAGGCGAGGCTA |
| XRCC1 SP1 Site Reverse [33] | TCTCCAGAGCGGGAAGAG |
| Gene | Primer |
|---|---|
| XRCC1 | Hs00959834_m1 FAM |
| STAT3 | Hs00374280_m1 FAM |
| ACTIN | Hs01060665_g1 VIC |
| Antibody | Dilution |
|---|---|
| XRCC1 (Fisher Scientific #MS434P1) | 1:1000 |
| STAT3 (Cell Signaling Technology #9139) | 1:1000 |
| Alpha-Tubulin (Sigma Aldrich #T9026) | 1:5000 |
| pSTAT3 Y705 (Cell Signaling Technology #9131) | 1:500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, G.; Sonavane, M.; Gassman, N.R. Activated STAT3 Is a Novel Regulator of the XRCC1 Promoter and Selectively Increases XRCC1 Protein Levels in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 5475. https://doi.org/10.3390/ijms22115475
Wright G, Sonavane M, Gassman NR. Activated STAT3 Is a Novel Regulator of the XRCC1 Promoter and Selectively Increases XRCC1 Protein Levels in Triple Negative Breast Cancer. International Journal of Molecular Sciences. 2021; 22(11):5475. https://doi.org/10.3390/ijms22115475
Chicago/Turabian StyleWright, Griffin, Manoj Sonavane, and Natalie R. Gassman. 2021. "Activated STAT3 Is a Novel Regulator of the XRCC1 Promoter and Selectively Increases XRCC1 Protein Levels in Triple Negative Breast Cancer" International Journal of Molecular Sciences 22, no. 11: 5475. https://doi.org/10.3390/ijms22115475
APA StyleWright, G., Sonavane, M., & Gassman, N. R. (2021). Activated STAT3 Is a Novel Regulator of the XRCC1 Promoter and Selectively Increases XRCC1 Protein Levels in Triple Negative Breast Cancer. International Journal of Molecular Sciences, 22(11), 5475. https://doi.org/10.3390/ijms22115475