
Transmembrane TNF and Its Receptors TNFR1 and TNFR2 in Mycobacterial Infections



Abstract
1. Tumor Necrosis Factor (TNF) and Tumor Necrosis Factor-α Converting Enzyme (TACE)
2. Tumor Necrosis Factor Receptors
2.1. Tumor Necrosis Factor Receptor 1
2.2. Tumor Necrosis Factor Receptor 2
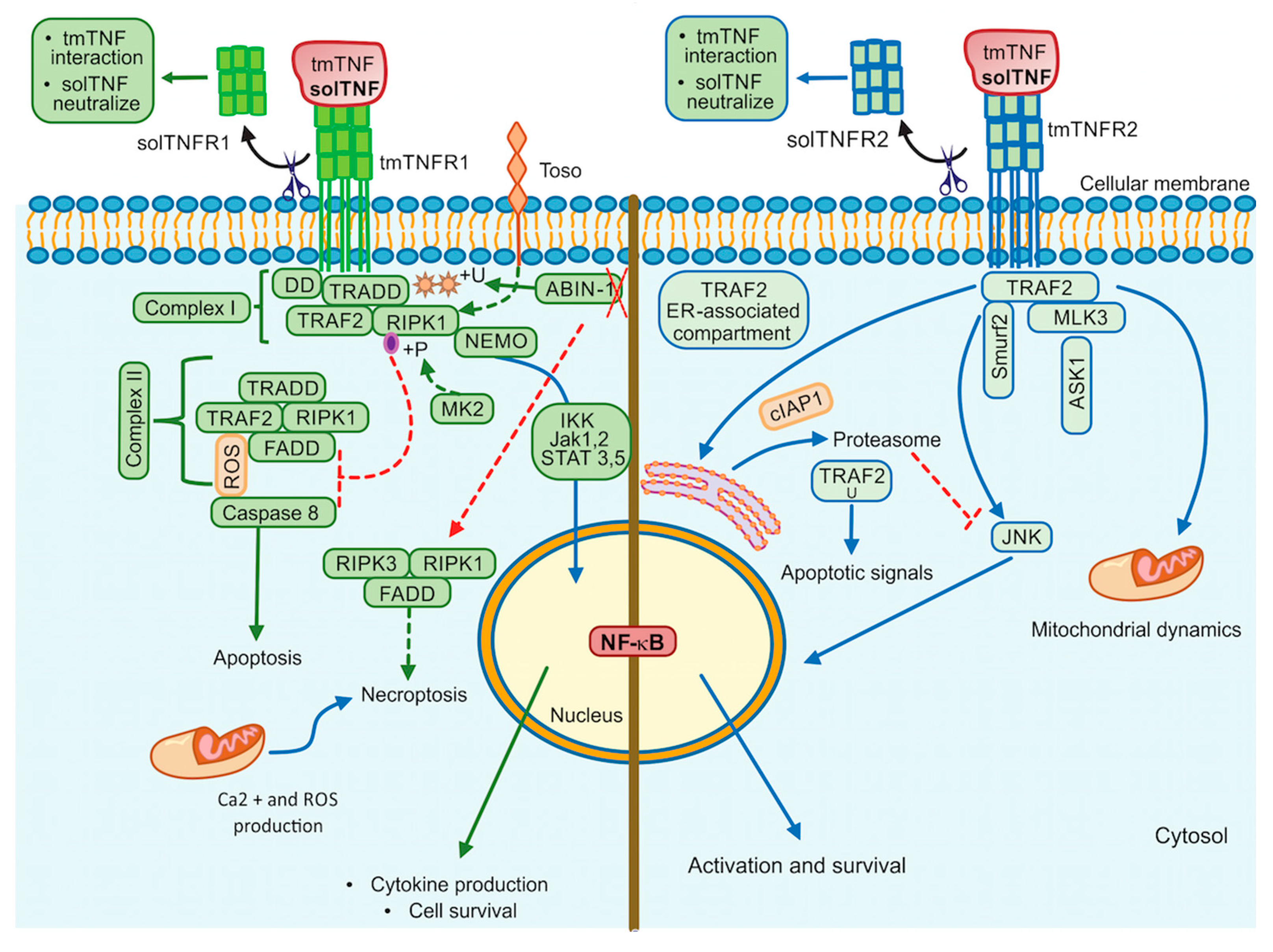
3. tmTNF Signalling Pathway
4. TNF/TNFR1/TNFR2 Inhibitors
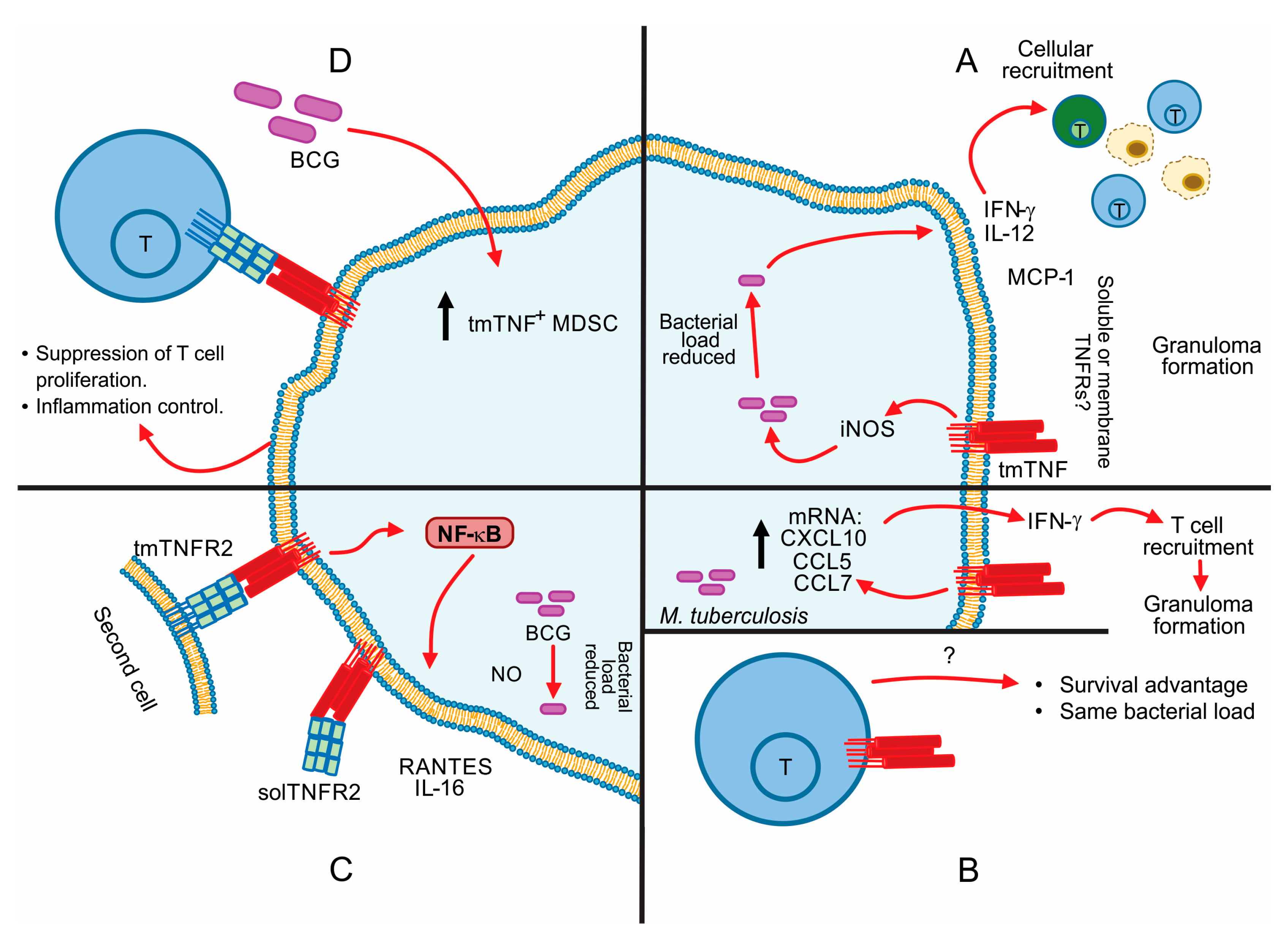
5. Contribution of tmTNF in the Control of Mycobacterial Infections
6. TNF Apoptosis Inhibition in Macrophage-Mycobacterial Infection
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin-Converting Enzyme-2 |
| ADAM17 | A Disintegrin and Metalloproteinase (ADAM) domain 17 |
| AICD | Activation-Induced Cell Death |
| ASK1 | Apoptosis Signal-Regulating Kinase 1 |
| CAV1 | Caveolin-1 |
| DD | Death Domain |
| DEDs | Death Effector Domains |
| DISC | Death-Inducing Signalling Complex |
| ER | Endoplasmic Reticulum |
| FADD | Fas-Associated Death Domain |
| FasL | Fas ligand or CD95L |
| FcγR | Fc region of IgG |
| HIF-1α) | Hypoxic-Inducible Factor-1 subunit α |
| IKK | Ikappa B kinase |
| IKK-α | Subunit α del IκB kinase complex (also IKK1) |
| IKK-β | Subunit β del IκB kinase complex (also IKK2) |
| IKK-γ | Subunit γ (also NEMO) |
| iNOS | Inducible Nitric Oxide Synthase |
| iRHOM2 | Rhomboid 5 homolog 2 |
| JNK | c-Jun-N-terminal kinase |
| LS | Leader Sequence |
| MAP3K | Mitogen-Activated Protein Kinase Kinase Kinase |
| MAPK | Mitogen-Activated Protein Kinases |
| MAPKAP kinase-2 | MAP-kinase-activated protein kinase-2 |
| MDSC | Myeloid-Derived Suppressor Cells |
| NDH-1 | Type I NADH Dehydrogenase |
| NF-κB | Nuclear Factor kappa B |
| NRD | Nardilysin |
| PGRN | Progranulin |
| PI3K | Phosphatidylinositol 3-kinase |
| PKC | Protein kinase C |
| RANTES | Regulated on Activation Normal T Cell Expressed and Secreted chemokine |
| RIPK1 | Receptor Interacting Protein Kinase 1 |
| RIPK1U | Ubiquitination of RIPK1 |
| ROS | reactive oxygen species |
| solTNF | Soluble TNF |
| Src | Sarcoma kinase |
| TACE | TNF Converting Enzyme |
| TGF-β | Transforming growth factor-beta |
| tmTNF | Cell membrane anchored TNF or transmembrane TNF |
| TNF | Tumor Necrosis Factor |
| TNFR1 | TNF receptor 1 |
| TNFR2 | TNF receptor 2 |
| TRADD | TNFR1-Associated Death Domain |
| TRAF2 | TNFR-Associated Factor 2 |
| Uev1A | Ubiquitin (Ub)-conjugating enzyme variant 1A |
| ZBG | Zinc Binding Groups |
References
- Müller, U.; Jongeneel, C.V.; Nedospasov, S.A.; Lindahl, K.F.; Steinmetz, M. Tumour Necrosis Factor and Lymphotoxin Genes Map Close to H-2D in the Mouse Major Histocompatibility Complex. Nature 1987, 325, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Spies, T.; Morton, C.C.; Nedospasov, S.A.; Fiers, W.; Pious, D.; Strominger, J.L. Genes for the Tumor Necrosis Factors Alpha and Beta Are Linked to the Human Major Histocompatibility Complex. Proc. Natl. Acad. Sci. USA 1986, 83, 8699–8702. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.A.; Daniels, K.A.; Welsh, R.M. Rapid Production of TNF-Alpha Following TCR Engagement of Naive CD8 T Cells. J. Immunol. 2005, 175, 5043–5049. [Google Scholar] [CrossRef]
- Wingfield, P.; Pain, R.H.; Craig, S. Tumour Necrosis Factor Is a Compact Trimer. FEBS Lett. 1987, 211, 179–184. [Google Scholar] [CrossRef]
- Chensue, S.W.; Remick, D.G.; Shmyr-Forsch, C.; Beals, T.F.; Kunkel, S.L. Immunohistochemical Demonstration of Cytoplasmic and Membrane-Associated Tumor Necrosis Factor in Murine Macrophages. Am. J. Pathol. 1988, 133, 564–572. [Google Scholar] [PubMed]
- Kriegler, M.; Perez, C.; DeFay, K.; Albert, I.; Lu, S.D. A Novel Form of TNF/Cachectin Is a Cell Surface Cytotoxic Transmembrane Protein: Ramifications for the Complex Physiology of TNF. Cell 1988, 53, 45–53. [Google Scholar] [CrossRef]
- Schlöndorff, J.; Becherer, J.D.; Blobel, C.P. Intracellular Maturation and Localization of the Tumour Necrosis Factor Alpha Convertase (TACE). Biochem. J. 2000, 347 Pt 1, 131–138. [Google Scholar] [CrossRef]
- Zheng, Y.; Saftig, P.; Hartmann, D.; Blobel, C. Evaluation of the Contribution of Different ADAMs to Tumor Necrosis Factor Alpha (TNFalpha) Shedding and of the Function of the TNFalpha Ectodomain in Ensuring Selective Stimulated Shedding by the TNFalpha Convertase (TACE/ADAM17). J. Biol. Chem. 2004, 279, 42898–42906. [Google Scholar] [CrossRef]
- Reddy, P.; Slack, J.L.; Davis, R.; Cerretti, D.P.; Kozlosky, C.J.; Blanton, R.A.; Shows, D.; Peschon, J.J.; Black, R.A. Functional Analysis of the Domain Structure of Tumor Necrosis Factor-Alpha Converting Enzyme. J. Biol. Chem. 2000, 275, 14608–14614. [Google Scholar] [CrossRef]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor Necrosis Factor-Alpha Convertase (ADAM17) Mediates Regulated Ectodomain Shedding of the Severe-Acute Respiratory Syndrome-Coronavirus (SARS-CoV) Receptor, Angiotensin-Converting Enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-Alpha-Converting Enzyme by the Spike Protein of SARS-CoV and ACE2 Induces TNF-Alpha Production and Facilitates Viral Entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef] [PubMed]
- Zipeto, D.; Palmeira, J.d.F.; Argañaraz, G.A.; Argañaraz, E.R. ACE2/ADAM17/TMPRSS2 Interplay May Be the Main Risk Factor for COVID-19. Front. Immunol. 2020, 11, 576745. [Google Scholar] [CrossRef]
- Moreno-Càceres, J.; Mainez, J.; Mayoral, R.; Martín-Sanz, P.; Egea, G.; Fabregat, I. Caveolin-1-Dependent Activation of the Metalloprotease TACE/ADAM17 by TGF-β in Hepatocytes Requires Activation of Src and the NADPH Oxidase NOX1. FEBS J. 2016, 283, 1300–1310. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Yang, X.; Wang, J.; Wang, R.; Qian, X.; Zhang, W.; Xiao, W. Uev1A-Ubc13 Catalyzes K63-Linked Ubiquitination of RHBDF2 to Promote TACE Maturation. Cell. Signal. 2018, 42, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Nishi, E.; Hiraoka, Y.; Yoshida, K.; Okawa, K.; Kita, T. Nardilysin Enhances Ectodomain Shedding of Heparin-Binding Epidermal Growth Factor-like Growth Factor through Activation of Tumor Necrosis Factor-Alpha-Converting Enzyme. J. Biol. Chem. 2006, 281, 31164–31172. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, Y.; Yoshida, K.; Ohno, M.; Matsuoka, T.; Kita, T.; Nishi, E. Ectodomain Shedding of TNF-Alpha Is Enhanced by Nardilysin via Activation of ADAM Proteases. Biochem. Biophys. Res. Commun. 2008, 370, 154–158. [Google Scholar] [CrossRef]
- Mezyk-Kopeć, R.; Bzowska, M.; Stalińska, K.; Chełmicki, T.; Podkalicki, M.; Jucha, J.; Kowalczyk, K.; Mak, P.; Bereta, J. Identification of ADAM10 as a Major TNF Sheddase in ADAM17-Deficient Fibroblasts. Cytokine 2009, 46, 309–315. [Google Scholar] [CrossRef]
- Cruceriu, D.; Baldasici, O.; Balacescu, O.; Berindan-Neagoe, I. The Dual Role of Tumor Necrosis Factor-alpha (TNF-α) in Breast Cancer: Molecular Insights and Therapeutic Approaches. Cell Oncol. 2020, 43, 1–18. [Google Scholar] [CrossRef]
- Kanda, K.; Komekado, H.; Sawabu, T.; Ishizu, S.; Nakanishi, Y.; Nakatsuji, M.; Akitake-Kawano, R.; Ohno, M.; Hiraoka, Y.; Kawada, M.; et al. Nardilysin and ADAM Proteases Promote Gastric Cancer Cell Growth by Activating Intrinsic Cytokine Signalling via Enhanced Ectodomain Shedding of TNF-α. EMBO Mol. Med. 2012, 4, 396–411. [Google Scholar] [CrossRef]
- Romera, C.; Hurtado, O.; Botella, S.H.; Lizasoain, I.; Cárdenas, A.; Fernández-Tomé, P.; Leza, J.C.; Lorenzo, P.; Moro, M.A. In Vitro Ischemic Tolerance Involves Upregulation of Glutamate Transport Partly Mediated by the TACE/ADAM17-Tumor Necrosis Factor-Alpha Pathway. J. Neurosci. 2004, 24, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, O.; Cárdenas, A.; Lizasoain, I.; Boscá, L.; Leza, J.C.; Lorenzo, P.; Moro, M.A. Up-Regulation of TNF-Alpha Convertase (TACE/ADAM17) after Oxygen-Glucose Deprivation in Rat Forebrain Slices. Neuropharmacology 2001, 40, 1094–1102. [Google Scholar] [CrossRef]
- Charbonneau, M.; Harper, K.; Grondin, F.; Pelmus, M.; McDonald, P.P.; Dubois, C.M. Hypoxia-Inducible Factor Mediates Hypoxic and Tumor Necrosis Factor Alpha-Induced Increases in Tumor Necrosis Factor-Alpha Converting Enzyme/ADAM17 Expression by Synovial Cells. J. Biol. Chem. 2007, 282, 33714–33724. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Vujanovic, N.L. Soluble TNF Regulates TACE via AP-2α Transcription Factor in Mouse Dendritic Cells. J. Immunol. 2017, 198, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Nyormoi, O.; Wang, Z.; Doan, D.; Ruiz, M.; McConkey, D.; Bar-Eli, M. Transcription Factor AP-2alpha Is Preferentially Cleaved by Caspase 6 and Degraded by Proteasome during Tumor Necrosis Factor Alpha-Induced Apoptosis in Breast Cancer Cells. Mol. Cell. Biol. 2001, 21, 4856–4867. [Google Scholar] [CrossRef][Green Version]
- Kucher, B.M.; Neary, J.T. Bi-Functional Effects of ATP/P2 Receptor Activation on Tumor Necrosis Factor-Alpha Release in Lipopolysaccharide-Stimulated Astrocytes. J. Neurochem. 2005, 92, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Barberà-Cremades, M.; Gómez, A.I.; Baroja-Mazo, A.; Martínez-Alarcón, L.; Martínez, C.M.; de Torre-Minguela, C.; Pelegrín, P. P2X7 Receptor Induces Tumor Necrosis Factor-α Converting Enzyme Activation and Release to Boost TNF-α Production. Front. Immunol. 2017, 8, 862. [Google Scholar] [CrossRef] [PubMed]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor Necrosis Factor Signaling Requires IRhom2 to Promote Trafficking and Activation of TACE. Science 2012, 335, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Cavadas, M.; Oikonomidi, I.; Gaspar, C.J.; Burbridge, E.; Badenes, M.; Félix, I.; Bolado, A.; Hu, T.; Bileck, A.; Gerner, C.; et al. Phosphorylation of IRhom2 Controls Stimulated Proteolytic Shedding by the Metalloprotease ADAM17/TACE. Cell Rep. 2017, 21, 745–757. [Google Scholar] [CrossRef]
- Loetscher, H.; Schlaeger, E.J.; Lahm, H.W.; Pan, Y.C.; Lesslauer, W.; Brockhaus, M. Purification and Partial Amino Acid Sequence Analysis of Two Distinct Tumor Necrosis Factor Receptors from HL60 Cells. J. Biol. Chem. 1990, 265, 20131–20138. [Google Scholar] [CrossRef]
- Garcia, I.; Olleros, M.L. The Roles of Tumor Necrosis Factor and Other Macrophage-Derived Cytokines in Host Defense Mechanisms During the Course of Mycobacterium tuberculosis Infection. In Current Topics on The Profiles of Host Immunological Response to Mycobacterial Infections; Research Signpost: Thiruvananthapuram, Kerala, India, 2009; pp. 1–46. [Google Scholar]
- Dembic, Z.; Loetscher, H.; Gubler, U.; Pan, Y.C.; Lahm, H.W.; Gentz, R.; Brockhaus, M.; Lesslauer, W. Two Human TNF Receptors Have Similar Extracellular, but Distinct Intracellular, Domain Sequences. Cytokine 1990, 2, 231–237. [Google Scholar] [CrossRef]
- Jacobsen, F.W.; Rothe, M.; Rusten, L.; Goeddel, D.V.; Smeland, E.B.; Veiby, O.P.; Slørdal, L.; Jacobsen, S.E. Role of the 75-KDa Tumor Necrosis Factor Receptor: Inhibition of Early Hematopoiesis. Proc. Natl. Acad. Sci. USA 1994, 91, 10695–10699. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Ayres, T.M.; Wong, G.H.; Goeddel, D.V. A Novel Domain within the 55 Kd TNF Receptor Signals Cell Death. Cell 1993, 74, 845–853. [Google Scholar] [CrossRef]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef]
- Idress, M.; Milne, B.F.; Thompson, G.S.; Trembleau, L.; Jaspars, M.; Houssen, W.E. Structure-Based Design, Synthesis and Bioactivity of a New Anti-TNFα Cyclopeptide. Molecules 2020, 25, 922. [Google Scholar] [CrossRef]
- Guo, D.; Dunbar, J.D.; Yang, C.H.; Pfeffer, L.M.; Donner, D.B. Induction of Jak/STAT Signaling by Activation of the Type 1 TNF Receptor. J. Immunol. 1998, 160, 2742–2750. [Google Scholar] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Ea, C.-K.; Deng, L.; Xia, Z.-P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha Requires Site-Specific Ubiquitination of RIP1 and Polyubiquitin Binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Aguileta, M.A.; Goossens, V.; Dubuisson, C.; Grootjans, S.; Dejardin, E.; Vandenabeele, P.; Bertrand, M.J.M. RIPK3 Contributes to TNFR1-Mediated RIPK1 Kinase-Dependent Apoptosis in Conditions of CIAP1/2 Depletion or TAK1 Kinase Inhibition. Cell Death Differ. 2013, 20, 1381–1392. [Google Scholar] [CrossRef]
- Jiang, Y.; Yu, M.; Hu, X.; Han, L.; Yang, K.; Ba, H.; Zhang, Z.; Yin, B.; Yang, X.-P.; Li, Z.; et al. STAT1 Mediates Transmembrane TNF-Alpha-Induced Formation of Death-Inducing Signaling Complex and Apoptotic Signaling via TNFR1. Cell Death Differ. 2017, 24, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Jouan-Lanhouet, S.; Divert, T.; Theatre, E.; Bertin, J.; Gough, P.J.; Giansanti, P.; Heck, A.J.R.; Dejardin, E.; Vandenabeele, P.; et al. NF-ΚB-Independent Role of IKKα/IKKβ in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 2015, 60, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.H.; Lang, P.A.; Lang, K.S.; Adam, D.; Fattakhova, G.; Föger, N.; Kamal, M.A.; Prilla, P.; Mathieu, S.; Wagner, C.; et al. Toso Regulates the Balance between Apoptotic and Nonapoptotic Death Receptor Signaling by Facilitating RIP1 Ubiquitination. Blood 2011, 118, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Jacob, C.O. The Mouse Cell Surface Protein TOSO Regulates Fas/Fas Ligand-Induced Apoptosis Through its Binding to Fas-Associated Death Domain. J. Biol. Chem. 2005, 280, 9618–9626. [Google Scholar] [CrossRef]
- Jaco, I.; Annibaldi, A.; Lalaoui, N.; Wilson, R.; Tenev, T.; Laurien, L.; Kim, C.; Jamal, K.; Wicky John, S.; Liccardi, G.; et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol. Cell 2017, 66, 698–710.e5. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Delanghe, T.; Rojas-Rivera, D.; Priem, D.; Delvaeye, T.; Bruggeman, I.; Van Herreweghe, F.; Vandenabeele, P.; Bertrand, M.J.M. MK2 Phosphorylation of RIPK1 Regulates TNF-Mediated Cell Death. Nat. Cell Biol. 2017, 19, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-Alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef]
- Roca, F.J.; Whitworth, L.J.; Redmond, S.; Jones, A.A.; Ramakrishnan, L. TNF Induces Pathogenic Programmed Macrophage Necrosis in Tuberculosis through a Mitochondrial-Lysosomal-Endoplasmic Reticulum Circuit. Cell 2019, 178, 1344–1361.e11. [Google Scholar] [CrossRef]
- Roca, F.J.; Ramakrishnan, L. TNF Dually Mediates Resistance and Susceptibility to Mycobacteria Through Mitochondrial Reactive Oxygen Species. Cell 2013, 153, 521–534. [Google Scholar] [CrossRef]
- Dziedzic, S.A.; Su, Z.; Jean Barrett, V.; Najafov, A.; Mookhtiar, A.K.; Amin, P.; Pan, H.; Sun, L.; Zhu, H.; Ma, A.; et al. ABIN-1 Regulates RIPK1 Activation by Linking Met1 Ubiquitylation with Lys63 Deubiquitylation in TNF-RSC. Nat. Cell Biol. 2018, 20, 58–68. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Weber, R.F.; Figari, I.S.; Reynolds, C.; Palladino, M.A.; Goeddel, D.V. The Two Different Receptors for Tumor Necrosis Factor Mediate Distinct Cellular Responses. Proc. Natl. Acad. Sci. USA 1991, 88, 9292–9296. [Google Scholar] [CrossRef]
- Chan, F.K.; Lenardo, M.J. A Crucial Role for P80 TNF-R2 in Amplifying P60 TNF-R1 Apoptosis Signals in T Lymphocytes. Eur. J. Immunol. 2000, 30, 652–660. [Google Scholar] [CrossRef]
- Rothe, M.; Wong, S.C.; Henzel, W.J.; Goeddel, D.V. A Novel Family of Putative Signal Transducers Associated with the Cytoplasmic Domain of the 75 KDa Tumor Necrosis Factor Receptor. Cell 1994, 78, 681–692. [Google Scholar] [CrossRef]
- Nishitoh, H.; Saitoh, M.; Mochida, Y.; Takeda, K.; Nakano, H.; Rothe, M.; Miyazono, K.; Ichijo, H. ASK1 Is Essential for JNK/SAPK Activation by TRAF2. Mol. Cell 1998, 2, 389–395. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Ashwell, J.D. TNF-RII and c-IAP1 Mediate Ubiquitination and Degradation of TRAF2. Nature 2002, 416, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-J.; Conze, D.B.; Li, X.; Ying, S.-X.; Hanover, J.A.; Ashwell, J.D. TNF-Alpha Induced c-IAP1/TRAF2 Complex Translocation to a Ubc6-Containing Compartment and TRAF2 Ubiquitination. EMBO J. 2005, 24, 1886–1898. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Long, J.; Liu, L.; He, T.; Jiang, L.; Zhao, C.; Li, Z. A Review of Endoplasmic Reticulum (ER) Stress and Nanoparticle (NP) Exposure. Life Sci. 2017, 186, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of Caspase-12, an Endoplastic Reticulum (ER) Resident Caspase, through Tumor Necrosis Factor Receptor-Associated Factor 2-Dependent Mechanism in Response to the ER Stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [CrossRef]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine Tumor Necrosis Factor Alpha Links Endoplasmic Reticulum Stress to the Membrane Death Receptor Pathway through IRE1alpha-Mediated NF-KappaB Activation and down-Regulation of TRAF2 Expression. Mol. Cell. Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, I.; Coornaert, B.; Beyaert, R. Smurf2 Is a TRAF2 Binding Protein That Triggers TNF-R2 Ubiquitination and TNF-R2-Induced JNK Activation. Biochem. Biophys. Res. Commun. 2008, 374, 752–757. [Google Scholar] [CrossRef]
- Korchnak, A.C.; Zhan, Y.; Aguilar, M.T.; Chadee, D.N. Cytokine-Induced Activation of Mixed Lineage Kinase 3 Requires TRAF2 and TRAF6. Cell. Signal. 2009, 21, 1620–1625. [Google Scholar] [CrossRef]
- Sondarva, G.; Kundu, C.N.; Mehrotra, S.; Mishra, R.; Rangasamy, V.; Sathyanarayana, P.; Ray, R.S.; Rana, B.; Rana, A. TRAF2-MLK3 Interaction Is Essential for TNF-Alpha-Induced MLK3 Activation. Cell Res. 2010, 20, 89–98. [Google Scholar] [CrossRef]
- Sente, T.; Van Berendoncks, A.M.; Fransen, E.; Vrints, C.J.; Hoymans, V.Y. Tumor Necrosis Factor-α Impairs Adiponectin Signalling, Mitochondrial Biogenesis, and Myogenesis in Primary Human Myotubes Cultures. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1164–H1175. [Google Scholar] [CrossRef] [PubMed]
- Drabarek, B.; Dymkowska, D.; Szczepanowska, J.; Zabłocki, K. TNFα Affects Energy Metabolism and Stimulates Biogenesis of Mitochondria in EA.Hy926 Endothelial Cells. Int. J. Biochem. Cell Biol. 2012, 44, 1390–1397. [Google Scholar] [CrossRef]
- Aravamudan, B.; Kiel, A.; Freeman, M.; Delmotte, P.; Thompson, M.; Vassallo, R.; Sieck, G.C.; Pabelick, C.M.; Prakash, Y.S. Cigarette Smoke-Induced Mitochondrial Fragmentation and Dysfunction in Human Airway Smooth Muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L840–L854. [Google Scholar] [CrossRef]
- Delmotte, P.; Marin Mathieu, N.; Sieck, G.C. TNFα Induces Mitochondrial Fragmentation and Biogenesis in Human Airway Smooth Muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L137–L151. [Google Scholar] [CrossRef] [PubMed]
- Nan, J.; Hu, H.; Sun, Y.; Zhu, L.; Wang, Y.; Zhong, Z.; Zhao, J.; Zhang, N.; Wang, Y.; Wang, Y.; et al. TNFR2 Stimulation Promotes Mitochondrial Fusion via Stat3- and NF-KB-Dependent Activation of OPA1 Expression. Circ. Res. 2017, 121, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Grell, M.; Douni, E.; Wajant, H.; Löhden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The Transmembrane Form of Tumor Necrosis Factor Is the Prime Activating Ligand of the 80 KDa Tumor Necrosis Factor Receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef]
- Grell, M.; Wajant, H.; Zimmermann, G.; Scheurich, P. The Type 1 Receptor (CD120a) Is the High-Affinity Receptor for Soluble Tumor Necrosis Factor. Proc. Natl. Acad. Sci. USA 1998, 95, 570–575. [Google Scholar] [CrossRef]
- Olleros, M.L.; Vesin, D.; Bisig, R.; Santiago-Raber, M.L.; Schuepbach-Mallepell, S.; Kollias, G.; Gaide, O.; Garcia, I. Membrane-Bound TNF Induces Protective Immune Responses to M. bovis BCG Infection: Regulation of memTNF and TNF Receptors Comparing two memTNF molecules. PLoS ONE 2012, 7, e31469. [Google Scholar] [CrossRef]
- Shi, W.; Li, L.; Shi, X.; Zheng, F.; Zeng, J.; Jiang, X.; Gong, F.; Zhou, M.; Li, Z. Inhibition of Nuclear Factor-KappaB Activation Is Essential for Membrane-Associated TNF-Alpha-Induced Apoptosis in HL-60 Cells. Immunol. Cell Biol. 2006, 84, 366–373. [Google Scholar] [CrossRef]
- Zhang, H.; Yan, D.; Shi, X.; Liang, H.; Pang, Y.; Qin, N.; Chen, H.; Wang, J.; Yin, B.; Jiang, X.; et al. Transmembrane TNF-Alpha Mediates “Forward” and “Reverse” Signaling, Inducing Cell Death or Survival via the NF-KappaB Pathway in Raji Burkitt Lymphoma Cells. J. Leukoc. Biol. 2008, 84, 789–797. [Google Scholar] [CrossRef]
- Yan, D.; Qin, N.; Zhang, H.; Liu, T.; Yu, M.; Jiang, X.; Feng, W.; Wang, J.; Yin, B.; Zhang, T.; et al. Expression of TNF-Alpha Leader Sequence Renders MCF-7 Tumor Cells Resistant to the Cytotoxicity of Soluble TNF-Alpha. Breast Cancer Res. Treat. 2009, 116, 91–102. [Google Scholar] [CrossRef]
- Zheng, F.; Liu, N.; Chen, Q.; Yang, L.; Liu, L.; Xiong, P.; Feng, W.; Jiang, X.; Gong, F.; Li, Z. Leader Sequence Is Required for Activity of Transmembrane Tumor Necrosis Factor-Alpha. Mol. Immunol. 2009, 46, 3336–3344. [Google Scholar] [CrossRef]
- Uysal, H.; Chavez-Galan, L.; Vesin, D.; Blaser, G.; Benkhoucha, M.; Ryffel, B.; Quesniaux, V.F.J.; Garcia, I. Transmembrane TNF and Partially TNFR1 Regulate TNFR2 Expression and Control Inflammation in Mycobacterial-Induced Pleurisy. Int. J. Mol. Sci. 2018, 19, 1959. [Google Scholar] [CrossRef]
- Chavez-Galan, L.; Vesin, D.; Segueni, N.; Prasad, P.; Buser-Llinares, R.; Blaser, G.; Pache, J.C.; Ryffel, B.; Quesniaux, V.F.; Garcia, I. Tumor Necrosis Factor and Its Receptors Are Crucial to Control Mycobacterium bovis Bacillus Calmette-Guerin Pleural Infection in a Murine Model. Am. J. Pathol. 2016, 186, 2364–2377. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell. Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Olleros, M.L.; Vesin, D.; Fotio, A.L.; Santiago-Raber, M.L.; Tauzin, S.; Szymkowski, D.E.; Garcia, I. Soluble TNF, but not membrane TNF is critical in LPS-induced hepatitis. J. Hepatol. 2010, 53, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Galan, L.; Vesin, D.; Blaser, G.; Uysal, H.; Benmerzoug, S.; Rose, S.; Ryffel, B.; Quesniaux, V.F.J.; Garcia, I. Myeloid Cell TNFR1 Signaling Dependent Liver Injury and Inflammation upon BCG Infection. Sci. Rep. 2019, 9, 5297. [Google Scholar] [CrossRef]
- Rodriguez-Cruz, A.; Vesin, D.; Ramon-Luing, L.; Zuñiga, J.; Quesniaux, V.; Ryffel, B.; Lascurain, R.; Garcia, I.; Chávez-Galán, L. CD3+ Macrophages Deliver Pro-Inflammatory Cytokines by a CD3- and Transmembrane TNF-Dependent Pathway and Are Increased at the BCG-Infection Site. Front. Immunol. 2019, 10, 2550. [Google Scholar] [CrossRef]
- Cabado, A.G.; Leira, F.; Vieytes, M.R.; Vieites, J.M.; Botana, L.M. Cytoskeletal Disruption Is the Key Factor That Triggers Apoptosis in Okadaic Acid-Treated Neuroblastoma Cells. Arch. Toxicol. 2004, 78, 74–85. [Google Scholar] [CrossRef]
- Kutsuna, H.; Suzuki, K.; Kamata, N.; Kato, T.; Hato, F.; Mizuno, K.; Kobayashi, H.; Ishii, M.; Kitagawa, S. Actin Reorganization and Morphological Changes in Human Neutrophils Stimulated by TNF, GM-CSF, and G-CSF: The Role of MAP Kinases. Am. J. Physiol. Cell Physiol. 2004, 286, C55–C64. [Google Scholar] [CrossRef]
- Chen, H.; Xiao, L.; Zhang, H.; Liu, N.; Liu, T.; Liu, L.; Hu, X.; Yan, D.; Yang, K.; Yin, B.; et al. The Involvement of β-Actin in the Signaling of Transmembrane TNF-α-Mediated Cytotoxicity. J. Leukoc. Biol. 2011, 89, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Kasibhatla, S.; Genestier, L.; Green, D.R. Regulation of Fas-Ligand Expression during Activation-Induced Cell Death in T Lymphocytes via Nuclear Factor KappaB. J. Biol. Chem. 1999, 274, 987–992. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, J.; Jia, L.; Huang, J.; He, C.; Hu, F.; Yuan, L.; Wang, G.; Yu, M.; Li, Z. Transmembrane TNF-α Promotes Activation-Induced Cell Death by Forward and Reverse signaling. Oncotarget 2017, 8, 63799–63812. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zheng, Y.; Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Antoni, C.; Braun, J. Side Effects of Anti-TNF Therapy: Current Knowledge. Clin. Exp. Rheumatol. 2002, 20 (Suppl. 28), S152–S157. [Google Scholar]
- Lee, H.; Park, H.Y.; Jeon, K.; Jeong, B.-H.; Hwang, J.-W.; Lee, J.; Cha, H.-S.; Koh, E.-M.; Kang, E.-S.; Koh, W.-J. QuantiFERON-TB Gold In-Tube Assay for Screening Arthritis Patients for Latent Tuberculosis Infection before Starting Anti-Tumor Necrosis Factor Treatment. PLoS ONE 2015, 10, e0119260. [Google Scholar] [CrossRef] [PubMed]
- Bloemendaal, F.M.; Levin, A.D.; Wildenberg, M.E.; Koelink, P.J.; McRae, B.L.; Salfeld, J.; Lum, J.; van der Neut Kolfschoten, M.; Claassens, J.W.; Visser, R.; et al. Anti-Tumor Necrosis Factor with a Glyco-Engineered Fc-Region Has Increased Efficacy in Mice with Colitis. Gastroenterology 2017, 153, 1351–1362.e4. [Google Scholar] [CrossRef]
- Sultana, S.; Bishayi, B. Neutralization of TNFR-1 and TNFR-2 Modulates, S. Aureus Induced Septic Arthritis by Regulating the Levels of pro Inflammatory and Anti-Inflammatory Cytokines during the Progression of the Disease. Immunol. Lett. 2018, 196, 33–51. [Google Scholar] [CrossRef]
- Yin, B.; Hu, X.; Wang, J.; Liang, H.; Li, X.; Niu, N.; Li, B.; Jiang, X.; Li, Z. Blocking TNF-α by Combination of TNF-α- and TNFR-Binding Cyclic Peptide Ameliorates the Severity of TNBS-Induced Colitis in Rats. Eur. J. Pharmacol. 2011, 656, 119–124. [Google Scholar] [CrossRef]
- Jian, J.; Li, G.; Hettinghouse, A.; Liu, C. Progranulin: A Key Player in Autoimmune Diseases. Cytokine 2018, 101, 48–55. [Google Scholar] [CrossRef]
- Steed, P.M.; Tansey, M.G.; Zalevsky, J.; Zhukovsky, E.A.; Desjarlais, J.R.; Szymkowski, D.E.; Abbott, C.; Carmichael, D.; Chan, C.; Cherry, L.; et al. Inactivation of TNF Signaling by Rationally Designed Dominant-Negative TNF Variants. Science 2003, 301, 1895–1898. [Google Scholar] [CrossRef]
- Olleros, M.L.; Vesin, D.; Lambou, A.F.; Janssens, J.P.; Ryffel, B.; Rose, S.; Frémond, C.; Quesniaux, V.F.; Szymkowski, D.E.; Garcia, I. Dominant-negative tumor necrosis factor protects from Mycobacterium bovis Bacillus Calmette Guérin (BCG) and endotoxin-induced liver injury without compromising host immunity to BCG and Mycobacterium tuberculosis. J. Infect. Dis. 2009, 199, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Garcia, I.; Olleros, M.L.; Quesniaux, V.F.; Jacobs, M.; Allie, N.; Nedospasov, S.A.; Szymkowski, D.E.; Ryffel, B. Roles of soluble and membrane TNF and related ligands in mycobacterial infections: Effects of selective and non-selective TNF inhibitors during infection. Adv. Exp. Med. Biol. 2011, 691, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Spohn, G.; Guler, R.; Johansen, P.; Keller, I.; Jacobs, M.; Beck, M.; Rohner, F.; Bauer, M.; Dietmeier, K.; Kündig, T.M.; et al. A virus-like particle-based vaccine selectively targeting soluble TNF-a protects from arthritis without inducing reactivation of latent tuberculosis. J. Immunol. 2007, 178, 7450–7457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, Z.; Zhao, Y. Selective Inhibition of Tumor Necrosis Factor Receptor-1 (TNFR1) for the Treatment of Autoimmune Diseases. Cytokine Growth Factor Rev. 2020, 55, 80–85. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, J.; Xu, A.; Zhong, C.; Lu, W.; Deng, L.; Li, R. A Rationally Designed TNF-α Epitope-Scaffold Immunogen Induces Sustained Antibody Response and Alleviates Collagen-Induced Arthritis in Mice. PLoS ONE 2016, 11, e0163080. [Google Scholar] [CrossRef] [PubMed]
- Murumkar, P.R.; Ghuge, R.B.; Chauhan, M.; Barot, R.R.; Sorathiya, S.; Choudhary, K.M.; Joshi, K.D.; Yadav, M.R. Recent Developments and Strategies for the Discovery of TACE Inhibitors. Expert Opin. Drug Discov. 2020, 15, 779–801. [Google Scholar] [CrossRef]
- Olleros, M.L.; Chavez-Galan, L.; Segueni, N.; Bourigault, M.L.; Vesin, D.; Kruglov, A.A.; Drutskaya, M.S.; Bisig, R.; Ehlers, S.; Aly, S.; et al. Control of Mycobacterial Infections in Mice Expressing Human Tumor Necrosis Factor (TNF) but Not Mouse TNF. Infect. Immun. 2015, 83, 3612–3623. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Münkel, S.; Neumeyer, J.; Müller, D.; Branschädel, M.; Scheurich, P.; Pfizenmaier, K. A Humanized Tumor Necrosis Factor Receptor 1 (TNFR1)-Specific Antagonistic Antibody for Selective Inhibition of Tumor Necrosis Factor (TNF) Action. J. Immunother. 2008, 31, 225–234. [Google Scholar] [CrossRef]
- Shibata, H.; Yoshioka, Y.; Ohkawa, A.; Abe, Y.; Nomura, T.; Mukai, Y.; Nakagawa, S.; Taniai, M.; Ohta, T.; Mayumi, T.; et al. The Therapeutic Effect of TNFR1-Selective Antagonistic Mutant TNF-Alpha in Murine Hepatitis Models. Cytokine 2008, 44, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Shibata, H.; Nakamura, T.; Yoshioka, Y.; Abe, Y.; Nomura, T.; Taniai, M.; Ohta, T.; Ikemizu, S.; Nakagawa, S.; et al. Structure-Function Relationship of Tumor Necrosis Factor (TNF) and Its Receptor Interaction Based on 3D Structural Analysis of a Fully Active TNFR1-Selective TNF Mutant. J. Mol. Biol. 2009, 385, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Ando, D.; Kamada, H.; Taki, S.; Niiyama, M.; Mukai, Y.; Tadokoro, T.; Maenaka, K.; Nakayama, T.; Kado, Y.; et al. A Trimeric Structural Fusion of an Antagonistic Tumor Necrosis Factor-α Mutant Enhances Molecular Stability and Enables Facile Modification. J. Biol. Chem. 2017, 292, 6438–6451. [Google Scholar] [CrossRef]
- Goodall, L.J.; Ovecka, M.; Rycroft, D.; Friel, S.L.; Sanderson, A.; Mistry, P.; Davies, M.L.; Stoop, A.A. Pharmacokinetic and Pharmacodynamic Characterisation of an Anti-Mouse TNF Receptor 1 Domain Antibody Formatted for In Vivo Half-Life Extension. PLoS ONE 2015, 10, e0137065. [Google Scholar] [CrossRef][Green Version]
- Schmidt, E.M.; Davies, M.; Mistry, P.; Green, P.; Giddins, G.; Feldmann, M.; Stoop, A.A.; Brennan, F.M. Selective Blockade of Tumor Necrosis Factor Receptor I Inhibits Proinflammatory Cytokine and Chemokine Production in Human Rheumatoid Arthritis Synovial Membrane Cell Cultures. Arthritis Rheum. 2013, 65, 2262–2273. [Google Scholar] [CrossRef]
- Steeland, S.; Puimège, L.; Vandenbroucke, R.E.; Van Hauwermeiren, F.; Haustraete, J.; Devoogdt, N.; Hulpiau, P.; Leroux-Roels, G.; Laukens, D.; Meuleman, P.; et al. Generation and Characterization of Small Single Domain Antibodies Inhibiting Human Tumor Necrosis Factor Receptor 1. J. Biol. Chem. 2015, 290, 4022–4037. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, Y.-H.; Lv, D.-Y.; Chen, X.-F.; Chen, L.-D.; Zhu, Z.-Y.; Chai, Y.-F.; Zhang, J.-P. Identification of a Ligand for Tumor Necrosis Factor Receptor from Chinese Herbs by Combination of Surface Plasmon Resonance Biosensor and UPLC-MS. Anal. Bioanal. Chem. 2016, 408, 5359–5367. [Google Scholar] [CrossRef]
- Lo, C.H.; Vunnam, N.; Lewis, A.K.; Chiu, T.-L.; Brummel, B.E.; Schaaf, T.M.; Grant, B.D.; Bawaskar, P.; Thomas, D.D.; Sachs, J.N. An Innovative High-Throughput Screening Approach for Discovery of Small Molecules That Inhibit TNF Receptors. SLAS Discov. 2017, 22, 950–961. [Google Scholar] [CrossRef]
- Proudfoot, A.; Bayliffe, A.; O’Kane, C.M.; Wright, T.; Serone, A.; Bareille, P.J.; Brown, V.; Hamid, U.I.; Chen, Y.; Wilson, R.; et al. Novel Anti-Tumour Necrosis Factor Receptor-1 (TNFR1) Domain Antibody Prevents Pulmonary Inflammation in Experimental Acute Lung Injury. Thorax 2018, 73, 723–730. [Google Scholar] [CrossRef]
- Richter, F.; Seifert, O.; Herrmann, A.; Pfizenmaier, K.; Kontermann, R.E. Improved Monovalent TNF Receptor 1-Selective Inhibitor with Novel Heterodimerizing Fc. mAbs 2019, 11, 653–665. [Google Scholar] [CrossRef]
- Lo, C.H.; Schaaf, T.M.; Grant, B.D.; Lim, C.K.-W.; Bawaskar, P.; Aldrich, C.C.; Thomas, D.D.; Sachs, J.N. Noncompetitive Inhibitors of TNFR1 Probe Conformational Activation States. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Torrey, H.; Butterworth, J.; Mera, T.; Okubo, Y.; Wang, L.; Baum, D.; Defusco, A.; Plager, S.; Warden, S.; Huang, D.; et al. Targeting TNFR2 with Antagonistic Antibodies Inhibits Proliferation of Ovarian Cancer Cells and Tumor-Associated Tregs. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Torrey, H.; Khodadoust, M.; Tran, L.; Baum, D.; Defusco, A.; Kim, Y.H.; Faustman, D.L. Targeted Killing of TNFR2-Expressing Tumor Cells and Tregs by TNFR2 Antagonistic Antibodies in Advanced Sézary Syndrome. Leukemia 2019, 33, 1206–1218. [Google Scholar] [CrossRef]
- WHO. Global Tuberculosis Report 2020; Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2020; ISBN 978-92-4-001313-1. [Google Scholar]
- Gernez-Rieux, C.; Gervois, M. Protection conferred by BCG during the twenty years following vaccination. Bull. World Health Organ. 1973, 48, 139–154. [Google Scholar]
- Chávez-Galán, L.; Vesin, D.; Martinvalet, D.; Garcia, I. Low Dose BCG Infection as a Model for Macrophage Activation Maintaining Cell Viability. J. Immunol. Res. 2016, 2016, 4048235. [Google Scholar] [CrossRef]
- Ruiz, A.; Guzmán-Beltrán, S.; Carreto-Binaghi, L.E.; Gonzalez, Y.; Juárez, E. DNA from Virulent M. tuberculosis Induces TNF-α Production and Autophagy in M1 Polarized Macrophages. Microb. Pathog. 2019, 132, 166–177. [Google Scholar] [CrossRef]
- Harris, J.; Hanrahan, O.; De Haro, S.A. Measuring Autophagy in Macrophages. Curr. Protoc. Immunol. 2009. [Google Scholar] [CrossRef]
- Das, S.; Ghosh, A.K.; Singh, S.; Saha, B.; Ganguly, A.; Das, P. Unmethylated CpG Motifs in the L. Donovani DNA Regulate TLR9-Dependent Delay of Programmed Cell Death in Macrophages. J. Leukoc. Biol. 2015, 97, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Olleros, M.L.; Guler, R.; Corazza, N.; Vesin, D.; Eugster, H.-P.; Marchal, G.; Chavarot, P.; Mueller, C.; Garcia, I. Transmembrane TNF Induces an Efficient Cell-Mediated Immunity and Resistance to Mycobacterium Bovis Bacillus Calmette-Guérin Infection in the Absence of Secreted TNF and Lymphotoxin-Alpha. J. Immunol. 2002, 168, 3394–3401. [Google Scholar] [CrossRef] [PubMed]
- Olleros, M.L.; Guler, R.; Vesin, D.; Parapanov, R.; Marchal, G.; Martinez-Soria, E.; Corazza, N.; Pache, J.-C.; Mueller, C.; Garcia, I. Contribution of Transmembrane Tumor Necrosis Factor to Host Defense against Mycobacterium Bovis Bacillus Calmette-Guerin and Mycobacterium Tuberculosis Infections. Am. J. Pathol. 2005, 166, 1109–1120. [Google Scholar] [CrossRef]
- Saunders, B.M.; Tran, S.; Ruuls, S.; Sedgwick, J.D.; Briscoe, H.; Britton, W.J. Transmembrane TNF Is Sufficient to Initiate Cell Migration and Granuloma Formation and Provide Acute, but Not Long-Term, Control of Mycobacterium Tuberculosis Infection. J. Immunol. 2005, 174, 4852–4859. [Google Scholar] [CrossRef] [PubMed]
- Dambuza, I.; Keeton, R.; Allie, N.; Hsu, N.-J.; Randall, P.; Sebesho, B.; Fick, L.; Quesniaux, V.J.F.; Jacobs, M. Reactivation of M. tuberculosis Infection in Trans-Membrane Tumour Necrosis Factor Mice. PLoS ONE 2011, 6, e25121. [Google Scholar] [CrossRef] [PubMed]
- Keeton, R.; Allie, N.; Dambuza, I.; Abel, B.; Hsu, N.J.; Sebesho, B.; Randall, P.; Burger, P.; Fick, E.; Quesniaux, V.F.; et al. Soluble TNFRp75 regulates host protective immunity against Mycobacterium tuberculosis. J. Clin. Investig. 2014, 124, 1537–1551. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Galan, L.; Vesin, D.; Uysal, H.; Blaser, G.; Benkhoucha, M.; Ryffel, B.; Quesniaux, V.F.J.; Garcia, I. Transmembrane Tumor Necrosis Factor Controls Myeloid-Derived Suppressor Cell Activity via TNF Receptor 2 and Protects from Excessive Inflammation during BCG-Induced Pleurisy. Front. Immunol. 2017, 8, 999. [Google Scholar] [CrossRef]
- Hu, X.; Li, B.; Li, X.; Zhao, X.; Wan, L.; Lin, G.; Yu, M.; Wang, J.; Jiang, X.; Feng, W.; et al. Transmembrane TNF-α Promotes Suppressive Activities of Myeloid-Derived Suppressor Cells via TNFR2. J. Immunol. 2014, 192, 1320–1331. [Google Scholar] [CrossRef]
- Beg, A.A.; Baltimore, D. An Essential Role for NF-KappaB in Preventing TNF-Alpha-Induced Cell Death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, T.; Liu, Z.; Zhang, G.; Wang, J.; Feng, S.; Liang, J. Inhibition of Autophagy by MiR-30A Induced by Mycobacterium tuberculosis as a Possible Mechanism of Immune Escape in Human Macrophages. Jpn. J. Infect. Dis. 2015, 68, 420–424. [Google Scholar] [CrossRef]
- Liu, M.; Li, W.; Xiang, X.; Xie, J. Mycobacterium tuberculosis Effectors Interfering Host Apoptosis Signaling. Apoptosis 2015, 20, 883–891. [Google Scholar] [CrossRef]
- Walters, A.; Keeton, R.; Labuschagné, A.; Hsu, N.J.; Jacobs, M. TNFRp75-dependent immune regulation of alveolar macrophages and neutrophils during early Mycobacterium tuberculosis and Mycobacterium bovis BCG infection. J. Immunol. 2021, 162, 220–234. [Google Scholar] [CrossRef]
- Balcewicz-Sablinska, M.K.; Keane, J.; Kornfeld, H.; Remold, H.G. Pathogenic Mycobacterium tuberculosis Evades Apoptosis of Host Macrophages by Release of TNF-R2, Resulting in Inactivation of TNF-Alpha. J. Immunol. 1998, 161, 2636–2641. [Google Scholar]
- Oddo, M.; Renno, T.; Attinger, A.; Bakker, T.; MacDonald, H.R.; Meylan, P.R. Fas Ligand-Induced Apoptosis of Infected Human Macrophages Reduces the Viability of Intracellular Mycobacterium tuberculosis. J. Immunol. 1998, 160, 5448–5454. [Google Scholar] [PubMed]
- Danelishvili, L.; Yamazaki, Y.; Selker, J.; Bermudez, L.E. Secreted Mycobacterium tuberculosis Rv3654c and Rv3655c Proteins Participate in the Suppression of Macrophage Apoptosis. PLoS ONE 2010, 5, e10474. [Google Scholar] [CrossRef]
- Ouyang, T.; Bai, R.-Y.; Bassermann, F.; von Klitzing, C.; Klumpen, S.; Miething, C.; Morris, S.W.; Peschel, C.; Duyster, J. Identification and Characterization of a Nuclear Interacting Partner of Anaplastic Lymphoma Kinase (NIPA). J. Biol. Chem. 2003, 278, 30028–30036. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gan, H.; Remold, H.G. A Mechanism of Virulence: Virulent Mycobacterium tuberculosis Strain H37Rv, but Not Attenuated H37Ra, Causes Significant Mitochondrial Inner Membrane Disruption in Macrophages Leading to Necrosis. J. Immunol. 2006, 176, 3707–3716. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Velmurugan, K.; Cowan, M.J.; Briken, V. The Type I NADH Dehydrogenase of Mycobacterium tuberculosis Counters Phagosomal NOX2 Activity to Inhibit TNF-Alpha-Mediated Host Cell Apoptosis. PLoS Pathog. 2010, 6, e1000864. [Google Scholar] [CrossRef]
- Kundu, M.; Pathak, S.K.; Kumawat, K.; Basu, S.; Chatterjee, G.; Pathak, S.; Noguchi, T.; Takeda, K.; Ichijo, H.; Thien, C.B.F.; et al. A TNF- and c-Cbl-Dependent FLIP(S)-Degradation Pathway and Its Function in Mycobacterium tuberculosis -Induced Macrophage Apoptosis. Nat. Immunol. 2009, 10, 918–926. [Google Scholar] [CrossRef]
- Sanchez, A.; Espinosa, P.; Esparza, M.A.; Colon, M.; Bernal, G.; Mancilla, R. Mycobacterium tuberculosis 38-KDa Lipoprotein Is Apoptogenic for Human Monocyte-Derived Macrophages. Scand. J. Immunol. 2009, 69, 20–28. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Inhibitor | Class | Reference |
|---|---|---|---|
| TNFR1 | XPro1595 | TNF mutein | [93] |
| Atrosab | Humanized IgG1 | [101] | |
| R1antTNF R1antTNF-T8 scR1antTNF | TNF muteins | [102,103,104] | |
| DMS5540 | Bispecific fusion protein | [105] | |
| AlbudAb | Antibody | [106] | |
| TROS (TNF Receptor- One Silencer) | Nanobody (Nb) technology | [107] | |
| PMG (physcion-8-O-β-D-monoglucoside) | Small molecule | [108] | |
| PLAD (pre-ligand assembly domain) | Small molecule | [109] | |
| ASO | Antisense oligonucleotides | [109] | |
| GSK1995057 | Antibody | [110] | |
| Atrosimab | Atrosab monovalent form | [111] | |
| Zafirlukast, DS42 | Small molecule | [112] | |
| TNFR2 | Antagonist Ab | Antibodies | [113,114] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz, A.; Palacios, Y.; Garcia, I.; Chavez-Galan, L. Transmembrane TNF and Its Receptors TNFR1 and TNFR2 in Mycobacterial Infections. Int. J. Mol. Sci. 2021, 22, 5461. https://doi.org/10.3390/ijms22115461
Ruiz A, Palacios Y, Garcia I, Chavez-Galan L. Transmembrane TNF and Its Receptors TNFR1 and TNFR2 in Mycobacterial Infections. International Journal of Molecular Sciences. 2021; 22(11):5461. https://doi.org/10.3390/ijms22115461
Chicago/Turabian StyleRuiz, Andy, Yadira Palacios, Irene Garcia, and Leslie Chavez-Galan. 2021. "Transmembrane TNF and Its Receptors TNFR1 and TNFR2 in Mycobacterial Infections" International Journal of Molecular Sciences 22, no. 11: 5461. https://doi.org/10.3390/ijms22115461
APA StyleRuiz, A., Palacios, Y., Garcia, I., & Chavez-Galan, L. (2021). Transmembrane TNF and Its Receptors TNFR1 and TNFR2 in Mycobacterial Infections. International Journal of Molecular Sciences, 22(11), 5461. https://doi.org/10.3390/ijms22115461